第二章 紫外吸收光谱
紫外吸收光谱法

④K带吸收是共轭分子的特征吸收带,是紫外光谱中应用最多的吸 收带
例如:CH2=CH-CH=CH2 * λmax=217nm εmax=104
CH3-CH=CH-CHO(巴豆醛)*
λmax=217.5nm εmax=1.5×104 -CH=CH2
λmax=248nmεmax=1.4×104
第二章 紫外可见吸收光谱法
紫外吸收光谱法(ultraviolet and Visble Spectroscopy,UV-Vis)
是研究物质在紫外区(10--400nm)和可见光区(400-750nm) 的分子吸收光谱法。 §2-1分子吸收光谱 一、分子吸收光谱的产生 各种能级差之间的关系: △E电子> △E振动> △E转动
m ax
21, 000 43, 000 — 121, 000 138, 000
1 ,3 ,5 ,7 -辛 四 烯 环 己 烷
1 ,3 ,5 ,7 ,9 -癸 四 烯 1 ,3 ,5 ,7 ,9 ,11 - 十 二 烷基六烯
异辛烷 异辛烷
23
3、醛和酮 醛和酮中均含有羰基(C=O)。 能实现n* 跃迁 (λmax.270-300nm附近,ε=10-20); n* 跃迁 (λmax.180nm左右); *跃迁(λmax.150nm左右) 。
12
表9-1 某些常见发色团的吸收特性
生色团 烯 溶剂 正庚烷 /nm 177
max
13000
跃迁类型
*
炔
羧基 酰胺基 羰基 偶氮基 硝基 亚硝基 硝酸酯
正庚烷
乙醇 水 正己烷 乙醇 异辛酯 乙醚
178
204 214 186 339,665 280 300,665
10000
第二章 紫外吸收光谱(共85张PPT)
max (己烷) =114+5M+nnR环内-10R环外 当苯环上有助色团时,向长波方向移至200 ~ 220nm。
-卤代酮的构象: -卤代环已酮有以下两个构象(A) (竖键)和(B)。 RCOOH及RCOOR的n → *比RCHO 的 小,即紫移*称为 *跃迁 ,实现 *跃迁需要吸收很多能量,约为185 千卡/克分子。
v=频率 用 周/秒(Cps)或赫兹(Hz) E=能量 单位为尔格,电子伏特eV或卡/摩尔
二、紫外光谱的特征
符合朗伯-比尔定律(Lambert-Beer’s Law),这是 吸收光谱的基本定律,用数学公式表示为:
A= ㏒(I0/I)=abc
式中:A:吸光度 I0:入射光强度 I:透射光强度 a:吸光系数 b:吸收池厚度(cm) c:被测物质浓度g/L I0/I:透射比,用T表示
CH3 CH3
N max =227nm( 900)
CH3
CH3Cl CH3OH
max =173nm( 200) max =183nm
3. *跃迁
电子由轨道跃迁到*轨道称→*跃迁,所吸收的能量比n → *小,峰位约在200nm附近,这种跃迁是强吸收, >104
例:CH2 CH2 max =162nm
近紫外区(200~400nm):在此波长范围内,玻璃有吸收,一般用石 英比色器,因此称近紫外区为石英紫外区,近紫外区最为有用,通常
所谓的紫外光谱就是指近紫外区的光谱。
2. 紫外光谱 以波长10~400nm的电磁波照射物质分子,即以紫外光照
射物质分子,由分子的电子能级跃迁而产生的光谱叫紫外光 谱。紫外光谱是电子光谱的一部分,可见光谱也是电子光谱 ,电子光谱是由电子跃迁而产生的吸收光谱的总称。
第二章 紫外光谱(UV)

苯环最重要的吸收带是B带,虽然强度不高但具有 精细结构很典型。
当苯环上有羟基、氨基等取代基时,吸收峰红移, 吸收强度增大,取代基不同,变化程度不同,可 由此鉴定各种取代基。 例: λmax B带 λmax E 2带
苯
甲苯
254
262
204
208
苯酚
苯甲酸
271
272
213
230
六、推测不饱和化合物λmax峰位的经验规 则
一、饱和烃
饱和烃只能产生σ→ σ* 跃迁,吸收带的位置
在150 nm左右,在远紫外区,超出仪器测定能力。
例如: 甲烷的C—H σ→ σ* 跃迁入max为125nm 乙烷的C—C σ→ σ*跃迁入max 为135nm
2、脂肪醇、胺和卤化物
当σ键与杂原子在一起时,因杂原子氧、氮、 硫、氯等具有非成键电子(n电子),所以,可发生 σ→σ*和 n → σ*两种跃迁。显然n → σ*跃迁
CH3 OH CH=CH2 E2 ´ ø £ º lmax 208nm( e 2460) E2 ´ ø £ º lmax 210nm( e 6200) K´ ø £ º lmax 244nm( e 12000)
识别上述几种吸收带,对推导有机化合物的结 构将会有很大的帮助。
五、各类有机化合物的紫外吸收
可以跃迁的电子有:电子, 电子和n电子。跃迁
的类型有: *, n *, *, n*。
各类电子跃迁的能量大小见下图:
E * > En > E
*
*
> En *
通常有机分子处于基态,电子填入成键或 非键轨道。但有机分子吸收UV后,则受激发变为
激发态,电子进入反键轨道。
紫外吸收光谱

紫外吸收光谱的基本知识 紫外吸收光谱的基本原理 影响紫外吸收光谱的因素
各类有机化合物的紫外吸收光谱
紫外吸收光谱的应用
一、紫外吸收光谱的基本知识
1.概述
紫外吸收光谱:分子价电子能级跃迁。
分子中价电子经紫外或可见光照射时,电子从低能级跃
迁到高能级,此时电子就吸收了相应波长的光,这样产 生的吸收光谱叫紫外吸收光谱。 波长范围:10-800 nm. (1) 远紫外光区: 10-200nm
C
n<p
O
C
C
p
n >p p
n
n
p p
p
n
p
p
C
O 非极性
C
极性
max(氯仿)
C
p
极性
max(水)
非极性
max(甲醇)
n → p*跃迁:兰移; ;e
max(正己烷)
p → p*跃迁:红移; ;e 237 309 243 305
pp* np*
即: E=Ee+Ev+Er ΔΕe>ΔΕv>ΔΕr
能级跃迁
电子能级间跃迁 的同时,总伴随有振 动和转动能级间的跃 迁。即电子光谱中总 包含有振动能级和转 动能级间跃迁产生的
若干谱线而呈现宽谱
带。
讨论:
(1) 转动能级间的能量差Δ Ε r:0.005~0.050eV,跃 迁产生吸收光谱位于远红外区。远红外光谱或分子转动光 谱; (2) 振动能级的能量差Δ Ε v约为:0.05~1eV,跃迁 产生的吸收光谱位于红外区,红外光谱或分子振动光谱; (3) 电子能级的能量差Δ Ε e较大1~20eV。电子跃迁产
只能被真空紫外分光光度计检测到;
波谱分析第二章有机化合物紫外光谱解析

羰基吸收峰受取代基影响显著位移
醛酮均在270 —300nm有R吸收带,但略有差别。 酮: 270 —280nm, 醛: 280—300nm附近 酮比醛多一个烃基,由于超共轭效应π轨道能级降低, π*轨道能级升高, n→π* 跃迁需要较高的能量。
n→ * /nm n→π* /nm
到π*轨道,完成 n→π*跃迁。
→* 跃迁在120—130nm之间产生吸收 π→π* 跃迁在 —160 nm左右产生吸收
n→* 跃迁在 —180 nm左右产生吸收
孤立羰基化合物研究最多的是 n→π* 跃迁,谱带吸收在 270—300nm附近。低强度的宽谱带。 (=10~20)
R带位置的变化对溶剂很敏感
CH3Cl CH3OH CH3NH2
σ→σ* 164-154
150 173
n →σ* 174 183 213
σ*
E
n σ
波谱分析第二章有机化合物紫外光 谱解析
2.烯类化合物
单烯烃: σ→σ* 和π→π* 两种跃迁。
ΔΕπ→π*<ΔΕσ→σ* , 吸收带在200nm左右。
λmax/nm εmax CH2=CH2 π→π* 162 ~104 CH3CH=CHCH3 π→π* 178 ~104 环己烯 π→π* 176 ~104
λmax =114+5×10+11×(48.0-1.7×11)-16.5×2=453.3nm εmax =1.74 × 104× 11=19.1× 104
波谱分析第二章有机化合物紫外光 谱解析
3.羰基化合物
(1)饱和羰基化合物: →* 、 π→π* 、 n→* 、 n→π*四种跃迁; 常常在发生π→π* 跃迁的同时,n 电子亦被激发而跃迁
第二章--紫外光谱

这组数据对应于下面哪个化合物?
AC2 HC2 H B C 2 C H C H C H C H C H 2H
C
CH CH
D
CC HC HC HH
?4
乙酰乙酸乙酯存在酮式和烯醇式互变异 构体,今有两张紫外光谱图,一张在 204nm有弱吸收,另一张在245nm有强吸 收,请判断哪一张是烯醇式。
优点是仪器普及、操作 简单而且灵敏度高
Chapter 2 紫外光谱
§2-1紫外吸收光谱的基本知识 §2-2常见有机物的紫外吸收光谱 §2-3溶剂对吸收光谱的影响 §2-4紫外光谱的解析与应用
§2-1紫外吸收光谱的基本知识
紫外吸收光谱是由于分子中价电子的跃 迁而产生的。 一、紫外吸收光谱的波长范围 二、紫外光谱图的组成 三、电子跃迁的类型 四、常用术语
σ*
σ*
E
л*
n л
σ
σ
C3C H2C HH=2C C2C H H2C H C3H H CH3OC3H
N(C 2CH 3 H )3
§2-2常见有机物的紫外吸收光谱
一、饱和烃及其取代衍生物 二、不饱和烃 三、羰基化合物 四、芳香烃的紫外光谱
一、饱和烃及其取代衍生物
★饱和烃的最大吸收峰一般小于190nm,处于真空紫外 区。如甲烷125nm,乙烷135nm。
甲苯的U图
?
图2-7 a 苯的紫外光谱图 b取代苯与苯紫外光谱的比较
分析:
H C
H H
苯环与甲基的超共轭效应
3、助色团取代苯的紫外光谱
★助色团含有孤电子对,它能与苯环 π 电子 共轭。使 B 带、E 带均移向长波方向。且吸 收强度都增加,精细结构消失。例如苯胺
绪论紫外-可见吸收光谱习题与答案

第二章:紫外可见吸收光谱法1. 紫外-可见光谱的产生是由外层价电子能级跃迁所致,其能级差的大小决定了(3)(1)吸收峰的强度(2)吸收峰的数目(3)吸收峰的位置(4)吸收峰的形状2. 紫外光谱是带状光谱的原因是由于(1)紫外光能量大(2)波长短(3)电子能级差大(4)电子能级跃迁的同时伴随有振动及转动能级跃迁的原因3. 化合物中,下面哪一种跃迁所需的能量最高(1)σ→σ*(2)π→π*(3)n→σ*(4)n→π*4. π→π*跃迁的吸收峰在下列哪种溶剂中测量,其最大吸收波长最大(1)水(2)甲醇(3)乙醇(4)正己烷5. 下列化合物中,在近紫外区(200~400nm)无吸收的是(1)(2)(3)(4)6. 下列化合物,紫外吸收λmax值最大的是(1)(2)(3)(4)二、解答及解析题1.为什么紫外吸收光谱是带状光谱?由于一般紫外可见分光光度计只能提供190-850nm 范围的单色光,因此,我们只能测量n→σ*的跃迁,n→π*跃迁和部分π→π*跃迁的吸收,而对只能产生200nm以下吸收的σ→σ*的跃迁则无法测量. 紫外吸收光谱是带状光谱,分子中在些吸收带已被确认,其中有K带、R带、B带、E1和 h E2带等.2.紫外吸收光谱能提供哪些分子结构信息?紫外光谱在结构分析中有什么用途又有何局限性?(1)如果在200~400nm区间无吸收峰,没该化合物应该无共轭双键系统,或为饱和有机化合物。
(2)如果在270~350nm区间有一个很弱的吸收峰,并且在200nm以上无其他吸收,该化合物含有带孤电子的未共轭的发色轩。
(3)如果在UV光谱中给出许多吸收峰,某些峰甚至出现在可见区,刚该化合物结构中可能具有长链共轭体系或稠环芳香发色团。
如果化合物有颜色,则至少有4~5个相互共轭的发色团。
(4)在UV光谱中,其长波吸收峰的强度在10000~20000之间时,示有α、β不饱和酮或共轭烯烃结构存在。
(5)化合物的长波吸收峰在250nm以上,且波吸收峰的强度在1000~10000之间时,该化合物通常具有芳香结构系统。
第二章+紫外吸收光谱

物质吸收紫外/可见光引起电子能级间的 跃迁而产生的吸收光谱叫紫外/可见光谱。
2.1 紫外吸收光谱的基本概念和原理
一、紫外与可见光波波长范围:
远紫外光区
近紫外光区
可见光区
10 nm 190 nm
400 nm
800 nm
波长10-190 nm范围内的为远紫外区(真空紫外区)
波长190-400 nm范围内的为近紫外区(石英紫外区 )
芳香族化合物的π→π*跃迁。
B带波长230~ 270 nm, 中心在 254 nm, ε ≈204 E带把苯环看成乙烯键和共轭乙烯键π →π* 跃迁引
起的吸收带
2. 2 各类有机化合物的紫外吸收
一、饱和化合物
饱和烷烃 σ→σ*跃迁,λmax〈190 nm 饱和卤代烃、醇、胺等。
化合物 n→σ* εmax
3、选择定则
(1)电子自旋允许跃迁 电子在跃迁过程中,要求自旋方向保持不变。
S0 S1,S0 S2,T1 T2 跃迁允许 S0 T1,S0 T2 禁阻跃迁
(2)对称性允许 允许跃迁要求电子只能在对称性不同性的不同能级间 进行。
g u:σ σ π π 跃迁允许
g g, u u : n π禁阻跃迁
三、紫外光谱的产生和电子跃迁的类型
253 nm
1个延长双键 30
3个环外双键 3 ×5
5个取代基 5×5
323 nm
实测值 320nm
253 + 3 ×5 + 5×5 =293 nm 实测值285nm
2、α,β-不饱和醛、酮最大λmax的计算
注:
(1)环上羰基不作为环外双键。 (2)有两个可供选用的α,β-不饱和羰基母体时,应 优先选择具有波长较长的作母体。例:
紫外吸收

σ*
反键轨道
π*
En
↑↓
π
↑↓
σ
↑↓
非键轨道 成键轨道
2
安徽师范大学
化学与材料科学学院
杨高升
常见有机化合物的紫外光谱吸收带主要有以下几种类型: 1. 远紫外(真空紫外)吸收带 最大吸收波长 < 200 nm,处于真空紫外区。主
2. 溶液酸度的影响 溶液酸度的变化可以改变某些有机化合物的存在形式,并导
致谱带发生位移。最典型的就是酚和芳香胺类化合物。例如
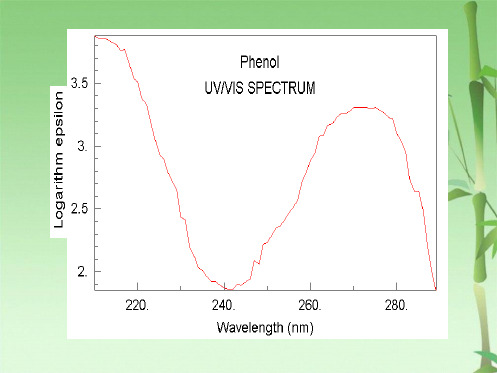
苯酚:
E2带 λmax (ε)
OH
211 nm (6200)
B 带 λmax (ε) 270 nm (1450)
−H+
苯胺:
O-
236 nm (9400)
287 nm (2600)
要是 σ→σ*跃迁引起的,是烷烃的吸收带。 2. 尾端(末端)吸收带 最大吸收波长虽在真空紫外区,但靠近 200 nm,吸收
带的尾部进入近紫外区。主要由 n→σ*跃迁引起,是含杂原子的饱和化合物的 吸收带。如饱和卤代烃、醇、胺等。 3. R 带 最大吸收波长 > 270 nm 的弱吸收带,摩尔吸光系数 ε 很小,一般 < 100。 由 n→π*跃迁引起,是含杂原子的不饱和化合物的吸收带。如醛、酮、硝基及 亚硝基化合物等。 4. K 带 最大吸收波长 > 200 nm 的强吸收带,摩尔吸光系数 ε 很大,一般 > 10000。由共轭体系的 π→π*跃迁引起,是共轭不饱和化合物的吸收带。如共 轭烯烃、共轭的不饱和羰基化合物等。 5. B 带 最大吸收波长 > 200 nm、强度中等且常伴有精细结构的宽吸收带,摩 尔吸光系数 ε 较小,一般 250 ~ 3000 之间。由芳环和芳香杂环化合物 π→π*跃 迁引起的,是芳环和芳香杂环化合物的特征吸收带。如苯的 B 带在 230 ~ 270 nm,为含有多重峰或精细结构的宽吸收带。 6. E带 也是芳香结构的特征吸收带。都是芳香体系中π→π*跃迁引起的。特点 是摩尔吸光系数ε很大,一般 > 10000。E带可分为E1带和E2带。如苯的E带就 有两个:184 nm(E1带,烯带,简单看作是乙烯的π→π*跃迁引起的)、204 nm
第二章紫外吸收光谱

CH3
H+
C OH
CH3
CH3 C OH2+ H2C H
CH3 C CH2 + H2O
A
CH3 C OH CH3
H+ H
CH3 C OH2+ CH3
CH3 C
CH3 B
+ H2O
计算:A max = 214nm + (3×5)nm = 229nm
B max = 214nm + (4×5)nm = 239nm
只能被真空紫外分光光度计检 测到,在紫外区透明,可作为溶 剂使用;
E K
s*
p*
R
n
p
s
n→σ*跃迁
所需能量较大; 吸收波长为150~250nm,大部分在远紫外区, 近紫外区仍不易观察到; 含非键电子的饱和烃衍生物(含N、O、S和卤素 等杂原子)均呈现n→σ* 跃迁。
化合物 H2O
CH3OH CH3CL
s s*
p p*
n s*
n p*
800nm
紫外光谱适用于在200-400nm 区域有吸 收的不饱和分子体系, 特别是具有共轭结构 的化合物。
σ→σ*跃迁
所需能量最大;σ电子只有吸收远紫外光的 能量才能发生跃迁; 饱和烷烃的分子吸收光谱出现在远紫外区; 吸收波长λ<200 nm;
例:甲烷的λmax为125nm , 乙烷 λmax为135nm。
化合物B更接近实验值242nm,产物应为B。
例2. 某化合物分子式为C7H10O,经红外光谱测定含有 酮羰基、甲基及碳碳双键,但不能肯定是六元环酮还
是开链脂肪酮。其紫外吸收max(EtOH)=257nm(>104), 试推测其结构。
有机波谱解析-第二章 紫外光谱

C
Hale Waihona Puke n<pOC
C
p*
n > p p*
n
n C
p* p
p*
n
p n 非极性
p
O 非极性
C C
p
极性
极性
n → p*跃迁:兰移; ; pp np
(4)尽量和文献中所用的溶剂一致。
(5)溶剂挥发性小、不易燃、无毒性、价格便宜。
5. 电子跃迁的类型
紫外吸收光谱是由价电子的能级跃迁而产生的,有机化 合物的紫外—可见吸收光谱是三种电子跃迁的结果:σ电子、 π电子、n电子。 s* n p* H C O
s
p E 分子轨道理论:成键轨道—反键轨道。
各种电子能级的能量及电子跃迁类型如右图
3. 紫 外 光 谱 图
横坐标:波长或频率 纵坐标:吸光度(A) 或 透过率(T)
紫外光谱(图)的特点: 吸收谱带少; 吸收谱带宽; 通常以谱带吸收最强的波长表示谱带位置,称 为最大吸收波长(λmax) ,是分子的特征常数, 与分子电子结构相关,可推测化合物中生色团类 型和共轭大小; 吸收强度以最大吸收波长处的摩尔吸光系数 (εmax)表示,也是分子特征常数和鉴定化合物 的重要依据。
H H c H
取代基 红移距离 -SR 45(nm)
c H
max=162nm 助色基团取代 p
-NR2 40(nm) -OR 30(nm)
p*(K带)发生红移。
-Cl 5(nm) CH3 5(nm)
(2)共轭烯烃中的
p → p*
紫外吸收光谱分析法

23:16:31
21/94
显示器 吸光度与光程的关系 A = abc
0.00 0.10
检测器
参 比
光源
b
0.20
2b
23:16:31
22/94
吸光度与浓度的关系 A = abc
显示器
0.00
检测器
光源
参 比
0.10
c
0.20
2c
23:16:31
23/94
吸光度与波长的关系 A = abc
显示器
光学光谱区
远紫外 近紫外 可见 近红外 中红外
(真空紫外)
远红外
10nm~200nm 200nm 380nm 780 nm
2.5 m
50 m
~380nm ~ 780nm ~ 2.5 m ~ 50 m ~300 m
23:16:31
11/94
物质对光的吸收与发射
物质分子内部3 种运动形式及其对应能级:
23:16:31
17/94
朗伯-比尔定律
A=lg(I0/It)=kbc
意义: 当一束平行单色光通过均匀、透明的吸光介质时,
其吸光度与吸光质点的浓度和吸收层厚度的乘积成正比.
23:16:31
18/94
透光率(透射比)T(Transmittance)
T = It I0
I0 入射光
吸光度A (Absorbance)
23:16:32
31/94
3.电子跃迁与分子吸收光谱
物质分子内部三种运动形式: (1)电子相对于原子核的运动; (2)原子核在其平衡位置附近的相对振动; (3)分子本身绕其重心的转动。
分子具有三种不同能级:电子能级、振动能级和转动能级 三种能级都是量子化的,且各自具有相应的能量。 分子的内能:电子能量Ee 、振动能量Ev 、转动能量Er
紫外吸收光谱.

J:转动量子数 V:振动量子数
二、分子轨道与电子跃迁的类型
1、分子轨道
++
+• •- σs* • + • σs
-+ +-
-+ -ቤተ መጻሕፍቲ ባይዱ
σp*
- + - σp
++
-
-
••
πp*
+
••
πp
分子轨道的能级高低次序是:σ*>π*>n>π>σ
2、电子跃迁的类型: σ →σ*、 n→σ*、 π→π* 、 n→π*
(6)E带 也是芳香结构的特征吸收带,由处于环状共轭的三个乙烯键的 苯型体系中的π→π*的跃迁所产生。E带又可分为E1和E2带。E 带属强吸收,εmax>10000 L·mol-1·cm-1。
三、发色基团、助色基团和吸收
1、发色基团和助色基团 例如,分子中含有π键的C=C、C三C、苯环以及C=O、
红移:由于取代基作用或溶剂效应导致发色基团的吸收峰向长波 移动的现象。
蓝移:由于取代基作用或溶剂效应导致发色基团的吸收峰向短波 移动的现象。
增色效应和减色效应:使吸收带强度增加的作用称助色效应; 反之为减色效应。
表 2-1 一些典型的发色基团及其吸收波长
发色基团
化合物
吸收波长/nm
C=C C三C C=O
近紫外区光能量为609~300kJ/mol,可见光区能量为 300~150 kJ/mol,与化学键的能量相仿,足以导致分子的价 电子由基态跃迁到激发态。
第一节 紫外吸收光谱的基本原理
一、紫外吸收光谱的产生
V3
n2 V2
V1 V0
J0 J1 J2 J3
A E
第二章 紫外吸收光

第二章紫外吸收光(Ultraviolet Absorption Spectroscopy)2.1 波(光)谱分析的一般原理电磁波的分区电子光谱的波长范围在10——800nm远紫外区:10——200近紫外区:200——400可见光区:400——800芳香族化合物或具有共轭体系的物质在近紫外区域有吸收,是紫外光谱研究的主要对象。
测量物质被吸收的电磁波的频率(或波长)和强度,可以得到其特征波谱。
频率(或波长):反映被测物质的结构特征——用于定性分析强度:与物质的含量有关——用于定量分析波(光)谱分析——利用物质对电磁波的选择性吸收对其进行分析的方法2.1.1电磁波的基本性质电磁波能量与波长或频率的关系:E = hυ= h c/λE——光子能量h——Planck常数,等于6.624×10-34j·sυ—频率,以赫兹(Hz)为单位λ—波长,以长度单位表示,纳米(nm),微米(μm)、厘米(cm)、米(m)C—光速,3×1010cm·s-1可知,光子的能量与频率成正比,与波长成反比,频率越低,波长越长,能量越小2.1.2 分子吸收光谱的产生分子吸收电磁波的能量具有量子化的特征∆E = E2-E1= hυ= h c/λ在电子能级跃迁的同时,伴随有多个振动和转动能级的跃迁∆E = ∆Ee +∆Eυ+∆Er所以紫外光谱不是纯电子光谱,而是电子-振动-转动光谱,所以测得的有机物紫外光谱大多是很宽的吸收带。
2.1.3分子吸收光谱仪和分子吸收光谱的表示用于检测紫外或红外等分子吸收光谱的仪器称为分光光度计,工作原理十分相似。
分光光度计由光源、分光系统、样品池、检测器、记录仪等组成。
不同波长的单色光依次透过被测样品,如果某些波长的光能量符合条件,就被吸收,透过光的强度就减弱,产生吸收信号。
吸收光谱的横坐标是波长或频率,纵坐标是吸收强度。
吸收强度用百分透过率(T%)或吸光度(A)表示最大吸收值对应的波长称最大吸收波长(λmax)摩尔吸光系数ε:样品浓度为1mol·L-1的溶液置于1cm样品池中,在一定波长下测得的吸光度值百分(比)吸收系数E1cm1%:溶液浓度为1%(1g/100ml),液层厚度为1cm时,在一定波长下的吸光度值百分吸收系数和摩尔吸收系数的关系ε= E1cm1%×M/10M为摩尔质量2.2 紫外吸收光谱的基本原理2.2.1 紫外吸收光谱与电子跃迁(1)电子跃迁的类型按照分子轨道理论,有成键轨道、非键轨道和反键轨道,分别用σ,π,n, σ*,π*表示σ→σ*激发光的波长在150—160nm范围,落在远紫外区域,π→π* 孤立双键的电子跃迁产生的吸收带在160—180,仍在远紫外区但在共轭体系中,吸收带向长波长方向移动(红移)乙烯的吸收带位于162nm,丁二烯为217nm,1,3,5-己三烯为258nm,这是因为共轭体系的轨道发生了重新组合n→σ*分子中存在-NH2,-OH,-SR,-X等基团时,能发生这种跃迁,一般出现在200nm附近,受杂原子的影响较大。
紫外吸收光谱

三、分子离子化的影响(pH的影响)
若化合物在不同的pH介质中能形成阳离子或阴离 子,则吸收带会随分子的离子化而改变。如苯胺在酸 性介质中会形成苯胺盐阳离子。苯胺形成阳离子之后, 氮原子的未成键电子消失,氨基的助色作用也随之消 失,因此苯胺盐的吸收带从230和280nm移到203和 254nm处。 苯酚分子中OH基团含有两对孤对电子,与苯环上 π电子形成n→π共轭,当形成酚盐阴离子时,氧原子上 孤对电子增加到三对,使n→π共轭作用进一步增强, 从而导致吸收带红移,同时吸收强度也有所增加。
表 2 环状共轭二烯π→π*跃迁的吸收波 长计算方法
π→π*跃迁λ/nm
同环二烯基本值 异环二烯基本值 每一个烷基或环残余取代 每一个环外双键 每一个烷氧基取代 –OR 每一个含硫基团取代 –SR 每一个胺基取代-NRR’ 每一个卤素取代 每一个酰基取代-COOR 增加一个共轭双键
253 214 +5 +5 +6 +30 +60 +5 +0 +30
3、醇、醚、含氮、含硫化合物及卤代物
醇、醚含有未成键电子,能产生n→ζ*跃迁,其 吸收波长都低于200nm。醇的分子间容易形成氢键而 发生缔合,吸收带波长及强度将随缔合的程度而变化。 胺是最简单的含氮有机化合物,胺中氮原子含有 未成键电子,所产生n→ζ*跃迁,其吸收波长处于 200nm附近。 硝基及亚硝基化合物中由于存在氮、氧原子,可 形成n→π共轭体系,所以能产生π→π*和n→π*跃迁, 吸收带位于近紫外区,例如硝基甲烷的吸收波长分别 为210和270nm。
气体 气体 己烷 己烷 水 己烷 水 甲醇 己烷 乙醇 甲醇 甲醇 乙醇 乙醇 己烷 己烷 甲醇 甲醇
第二节 紫外分光光度计光路图
一、单光束分光光度计
光源
光电倍增管
第二章紫外吸收光谱法

第二章:紫外吸收光谱法一、选择1. 频率(MHz)为4.47×108的辐射,其波长数值为(1)670.7nm (2)670.7μ(3)670.7cm (4)670.7m2. 紫外-可见光谱的产生是由外层价电子能级跃迁所致,其能级差的大小决定了(1)吸收峰的强度(2)吸收峰的数目(3)吸收峰的位置(4)吸收峰的形状3. 紫外光谱是带状光谱的原因是由于(1)紫外光能量大(2)波长短(3)电子能级差大(4)电子能级跃迁的同时伴随有振动及转动能级跃迁的原因4. 化合物中,下面哪一种跃迁所需的能量最高(1)ζ→ζ*(2)π→π*(3)n→ζ*(4)n→π*5. π→π*跃迁的吸收峰在下列哪种溶剂中测量,其最大吸收波长最大(1)水(2)甲醇(3)乙醇(4)正己烷6. 下列化合物中,在近紫外区(200~400nm)无吸收的是(1)(2)(3)(4)7. 下列化合物,紫外吸收λ值最大的是max(1)(2)(3)(4)二、解答及解析题1.吸收光谱是怎样产生的?吸收带波长与吸收强度主要由什么因素决定?2.紫外吸收光谱有哪些基本特征?3.为什么紫外吸收光谱是带状光谱?4.紫外吸收光谱能提供哪些分子结构信息?紫外光谱在结构分析中有什么用途又有何局限性?5.分子的价电子跃迁有哪些类型?哪几种类型的跃迁能在紫外吸收光谱中反映出来?6.影响紫外光谱吸收带的主要因素有哪些?7.有机化合物的紫外吸收带有几种类型?它们与分子结构有什么关系?8.溶剂对紫外吸收光谱有什么影响?选择溶剂时应考虑哪些因素?9.什么是发色基团?什么是助色基团?它们具有什么样结构或特征?10.为什么助色基团取代基能使烯双键的n→π*跃迁波长红移?而使羰基n→π*跃迁波长蓝移?11.为什么共轭双键分子中双键数目愈多其π→π*跃迁吸收带波长愈长?请解释其因。
12.芳环化合物都有B吸收带,但当化合物处于气态或在极性溶剂、非极性溶剂中时,B吸收带的形状有明显的差别,解释其原因。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
含杂原子的共轭体系除p-p*跃迁外,还有n-p*跃迁形式!!!
2-7 影响紫外吸收光谱的因素
(1) 溶剂的影响
基态极性大,激发态极性小; 极性越大,则溶剂化作用越强,被 极性溶剂稳定而降低的能量越多; 与极性溶剂的偶极-偶极相互作用 强度以基态大于激发态; 故溶剂极性增加,n-p*跃迁蓝移。
上述六元环酮的计算值与实测值相差较大,未知物的 结构可排除这种可能性。
(4) 如果是开链脂肪酮,则可以多一个双键,并与原有
的共轭体系进一步共轭,max值随之最大。
O
A: CH3 CH CH CH CH C CH3
CH3
O
H2C C CH CH C CH3
max = 215nm + 30nm + 18nm = 263nm
深色位移
C=C 发色基团, 但 p → p*200nm。
H
H
cc
H
H
max=162nm 助色基团取代 p →p* (K带)发生红移。
取代基 -SR
-NR2
红移距离 45(nm) 40(nm)
-OR 30(nm)
-Cl 5(nm)
CH3 5(nm)
2-6 共轭体系与吸收峰波长的关系
---共轭结构分子的变化
s s*
p p*
n s*
n p*nm 区域有吸 收的不饱和分子体系, 特别是具有共轭结构 的化合物。
σ→σ*跃迁
➢所需能量最大;σ电子只有吸收远紫外光的 能量才能发生跃迁; ➢饱和烷烃的分子吸收光谱出现在远紫外区; ➢吸收波长λ<200 nm;
例:甲烷的λmax为125nm , 乙烷 λmax为135nm。
L·mol-1·cm-1。
K带——共轭非封闭体系的p p* 跃迁
举例: 1,3-丁二烯的电子跃迁
LUMO: The Lowest unoccupied molecular orbital (最低非占据轨道) HOMO: The highest occupied molecular orbital(最高占据轨道)
O
C 的p→p*跃迁的吸收波长。
AB
CH3 C O
解:该化合物可以看成是同环二烯(环B)的分子。
共轭二烯的基本值
214nm
同环二烯 烷基取代(5×5)
39nm 25nm
环外双键(5×3)
15nm
增加两个共轭双键(2×30) 60nm
___________________
max计算值 353nm max实测值 355nm
(2)共轭多烯最大吸收波长的计算
Fieser-Kuhn经验公式:
max(己烷溶液)=114+5M+n(48.0-1.7n)-16.5Rendo-10Rexo
max(己烷溶液)=1.74×104n
式中 M --- 取代的烷基数 n --- 共轭双键的数目
Rendo--- 具有环内双键的数目 Rexo --- 具有环外双键的数目
(2) 根据分子式,其不饱和度为3,除羰基和双键外,还 剩余一个不饱和度应为环或者双键;
(3) 若为六元环酮,则可能是以下结构:
O
max = 215nm + 10nm + 12nm = 237nm
CH3
O
CH3 CH3
max = 215nm + 2 x12nm = 239nm
O CH3
CH3
O
O max = 215nm + 12nm = 227nm
(1)饱和的有机化合物:一般无紫外吸收 饱和碳氢化合物: s→s* ,远紫外区 含杂原子化合物:n→s* ,在近紫外区较少
(2) 含非共轭烯、炔基团的化合物 ss*, pp*(~175nm) 若无助色团的作用,则在近紫外区无吸收;
(3) 含杂原子的不饱和化合物 除ss*,pp*(~175nm)外, 还有n→s*,n→p*跃迁。n→p*跃迁一般在紫外区。
解:醇反应失水可经过下面两种途径:
CH3
H+
C OH
CH3
CH3 C OH CH3
H+ H
CH3 C OH2+ H2C H CH3 C OH2+ CH3
CH3 C CH2 + H2O
A
CH3 C
CH3 B
+ H2O
计算:A max = 214nm + (3×5)nm = 229nm
B max = 214nm + (4×5)nm = 239nm
CH3I CH3NH2
max(nm) 167 184 173 258 215
max 1480 150 200 365 600
π→π*跃迁
所需能量较小,吸收波长处于远紫外 区的近紫外端或近紫外区,εmax一般在 104L·mol-1·cm-1以上,属于强吸收。
例如: 乙烯π→π*跃迁的λmax为162nm,εmax为: 1×104
OR 230nm
2-9 紫外光谱提供的结构信息
200-400nm 无吸收峰。饱和化合物,单烯。
270-350 nm有吸收峰(ε=10-100)醛酮 n→* 跃
迁产生的R 带。 250-300 nm 有中等强度的吸收峰(ε=200-2000),
芳环的特征 吸收(具有精细解构的B带)。 200-250 nm有强吸收峰(ε104),表明含有一个
例4. 能有效清除人体内有害氧自由基的b-胡萝卜素是 多烯化合物,计算其p→p*跃迁的max和max。
解:M=10, n=11, Rendo=2, Rexo=0 max= 114 + 5×10 + 11×(48.0-1.7×11) – 16.5×2 - 10×0
= 453.3nm (实测值 452nm) max = 11×1.74×104 = 19.1×104 (实测值 15.2×104)
解:该化合物的基本值为214nm,环C中的双键既是环 B的环外双键,也是环D的环外双键。
共轭二烯的基本值 烷基取代(5×5)
214nm 25nm
环外双键(5×3)
15nm
增加一个共轭双键
30nm
____________________
max计算值 284nm max实测值 283nm
例2. 计算化合物
O-
236nm 287nm
-NH2因共轭作用而成为助 色团; 成胺盐后,孤电子对消失, 失去助色作用,其紫外吸 收与苯无区别。
-OH 也是助色团; 成酚盐后,孤电子对由2 对增加到3对,p-p 共轭作 用进一步增强,紫外吸收 峰红移。
2-8 各类有机化合物的紫外吸收光谱
2-8-1 非共轭体系的简单分子
p p* p p*
苯环上助色基团对吸收带的影响
-O- > -NH2> -OCH3> -OH > -Br > -Cl > CH3
苯环上发色基团对吸收带的影响
-NO2 > -CHO > -COCH3> -COOH > -CN,COO- > -SO2NH2
取代苯甲酰型化合物吸收波长计算
O X
Y
基本值: X=C 246nm X=H 250nm X=OH
CH3
O
B: H2C CH C CH C CH3 max = 215nm + 30nm + 12nm = 257nm
第二章 紫外吸收光谱
Ultraviolet Spectra
2-1 紫外-可见光谱区
2-2 紫外吸收谱带的形成(一)
电子位能曲线 ( 激发态)
III I II 电子跃迁 E
电子位能曲线 (基态)
I (a)
II
III
(b)
(c)
位能曲线上的横线表示振动 能级(转动能级未表示)
(a) 稀薄气体状态(转动能级跃迁谱线); (b)气态压力增加时形成连续曲线; (c) 极性溶剂中,精细结构完全消失.
只能被真空紫外分光光度计检 测到,在紫外区透明,可作为溶 剂使用;
E K
s*
p*
R
n
p
s
n→σ*跃迁
➢所需能量较大; ➢吸收波长为150~250nm,大部分在远紫外区, 近紫外区仍不易观察到; ➢含非键电子的饱和烃衍生物(含N、O、S和卤素 等杂原子)均呈现n→σ* 跃迁。
化合物 H2O
CH3OH CH3CL
激发态极性比基态极性大; 激发态因极性溶剂稳定而降低 的能量比基态能量降低的幅度 大,所以电子跃迁所需的能量 相应减小;故溶剂极性增加, p-p*跃迁红移。
(2) 分子离子化的影响
NH2
230nm 280nm
H+ OHmax
NH3+
203nm 254nm
OH
211nm 270nm
OHH+ max
未成键电子对与羰基p电子相互作用,使基态p 轨道能量降低,而激 发态p*能量提高;电负性杂原子的诱导效应可能降低羰基基态n的能级; 两种作用都使n→p*跃迁能增高,吸收带蓝移。
2-8-2 含有共轭体系的分子
(1)共轭二烯最大吸收波长的计算
例1. 计算化合物
C D 的p→p*跃迁的吸收波长。
AB
例3. 计算全反式番茄红的p→p*跃迁的max和max。
解:M=8, n=11, Rendo=0, Rexo=0 max= 114 + 5×8 + 11×(48.0-1.7×11) – 16.5×0 - 10×0
= 476.6nm (实测值 474nm) max = 11×1.74×104 = 19.1×104 (实测值 18.6×104)
s