2018年药审中心审评通过的进口原研药
《境外已上市境内未上市药品临床技术要求》(征求意见稿)

境外已上市境内未上市药品临床技术要求(征求意见稿)药审中心2020年4月30日目录一、背景 (4)二、适用范围 (4)三、临床评价基本逻辑 (4)(一)临床需求评估 (4)(二)有效性和安全性评价 (5)1、明确临床数据来源 (5)2、评估境外临床试验数据质量 (5)3、了解生物药剂学与临床药理学特征 (5)4、评估总体有效性和安全性 (5)(三)种族敏感性分析 (6)(四)基于中国患者获益风险评估进行决策 (6)四、临床试验要求 (7)(一)境外原研药品 (7)1、安全有效且无种族敏感性 (8)2、安全有效但缺乏种族敏感性数据或存在种族敏感性 (9)3、安全有效性数据不充分或显示无效存在安全性问题 (9)(二)境内外仿制药品 (9)1、确定参比制剂 (9)2、仿制药临床试验要求 (10)(三)特殊情形的考虑 (10)1、国内已上市药品增加境外已批准新剂型、新给药途径、新规格 (10)2、国内已上市药品增加境外已批准新适应症 (11)3、境外已批准的复方药品中单药已在国内上市 (13)4、罕见病及儿童用药 (13)一、背景境外已上市药品进口或仿制,是解决我国患者对临床迫切需求领域药品的可获得性和可及性的重要手段。
为加快境外已上市境内未上市原研药品及仿制药品研发上市进程,加强科学监管,并提高审评审批质量和效率,依据《药品注册管理办法》(总局令第27号)及其配套文件,结合《接受境外临床试验数据的技术指导原则》(2018年52号文),制定对此类药品临床研究和评价的技术要求,为工业界、学术界和监管机构提供技术参考。
二、适用范围本技术要求适用于境外已上市境内未上市的药品,主要包括两类情形:(1)原研进口的化学药品和生物制品;(2)国内外仿制的化学药品和生物制品。
三、临床评价基本逻辑(一)临床需求评估应分析拟申报适应症在我国的疾病流行病学现状、疾病严重程度和预后,现有治疗手段及其局限性,明确该药品与国内现有治疗手段的比较优势,进而对中国患者临床需求的程度做出判断。
国家食品药品监督管理总局关于药品注册审评审批若干政策的公告(2015年第230号)

根据《中华⼈民共和国药品管理法》、《国务院关于改⾰药品医疗器械审评审批制度的意见》(国发〔2015〕44号)等有关规定,为解决药品注册申请积压问题,提⾼药品审评审批质量和效率,经国务院同意,实⾏如下药品注册审评审批政策。
现予以公告: ⼀、提⾼仿制药审批标准 仿制药按与原研药质量和疗效⼀致的原则受理和审评审批。
其中,对已在中国境外上市但尚未在境内上市药品的仿制药注册申请,应与原研药进⾏⽣物等效性研究并按国际通⾏技术要求开展临床试验,所使⽤的原研药由企业⾃⾏采购,向国家⾷品药品监督管理总局申请⼀次性进⼝;未能与原研药进⾏对⽐研究的,应按照创新药的技术要求开展研究。
已经受理的仿制药注册申请,实⾏分类处理: (⼀)中国境内已有批准上市原研药,申请注册的仿制药没有达到与原研药质量和疗效⼀致的,不予批准。
(⼆)中国境外已上市但境内没有批准上市原研药,申请仿制药注册的企业可以选择按原规定进⾏审评审批,但在药品批准上市3年内需按照国发〔2015〕44号⽂件规定进⾏质量和疗效⼀致性评价,未通过⼀致性评价的注销药品批准⽂号;企业也可以选择撤回已申报的注册申请,改按与原研药质量和疗效⼀致的标准完善后重新申报。
对上述重新申报的注册申请实⾏优先审评审批,批准上市后免于进⾏质量和疗效⼀致性评价。
对申报上市的仿制药注册申请,⾸先审查药学研究的⼀致性,药学研究未达到要求的,不再对其他研究资料进⾏审查,直接作出不予批准决定。
⼆、规范改良型新药的审评审批 对改变原研药剂型、酸根、碱基和给药途径等的药品注册申请,申请⼈需证明其技术创新性且临床价值与原品种⽐较具有明显优势;⽆法证明具备上述优势的,不予批准。
改变剂型和规格的⼉童⽤药注册申请除外。
三、优化临床试验申请的审评审批 对新药的临床试验申请,实⾏⼀次性批准,不再采取分期申报、分期审评审批的⽅式;审评时重点审查临床试验⽅案的科学性和对安全性风险的控制,保障受试者的安全。
加强临床试验申请前及过程中审评⼈员与申请⼈的沟通交流,及时解决注册申请和临床试验过程中的问题。
100个还未被仿制的进口独家产品

100个还未被仿制的进口独家产品
2015-03-06医药代表
本期精选了100个还没有仿制品的进口独家产品,并通过咸达数据(V3.0)确认了国内申请仿制最快的企业和进度,个个高大上。
亲爱的民族制药企业,你们如果永远都在研究各部委的政策,钻研招标的空子,忽略药物的本质,中国制药和中国足球还有什么区别!
可喜的是,总有一些企业,能给人些许安慰。
手机药筛利用咸达数据3.0
黄金版,检索出部分产品国内最快仿制进度情况,发现我们me(山) too(寨)的节奏还是挺快的,中国制药还有希望。
这些进度最快的,最有希望将来成为首仿,如果您关心医药股的话,这些家的审评进度您可盯紧了哦!假如您错过了甲磺酸阿帕替尼上市之前买入恒瑞医药、错过了复格列汀批临床前入手复星医药,难道您还要错过下面的品种吗?。
中国药品审评完成、审评通过及注册申请受理情况分析

中国药品审评完成、审评通过及注册申请受理情况分析一、药品注册申请药品注册,是指国家食品药品监督管理局根据药品注册申请人的申请,依照法定程序,对拟上市销售的药品的安全性、有效性、质量可控性等进行系统评价,并决定是否同意其申请的审批过程。
二、药品注册申请审评审批现状1、药品注册申请审评审批相关政策法规新药研发是激发药品产业发展的重要源动力,国家陆续出台了系列药品审评审批制度的改革政策以鼓励药物创新,尤其新版《药品管理法》强调药品全生命周期管理,将研制与注册独立成为专章,建立上市许可持有人制度,极大强化了新药研发环节。
2、审评审批完成情况《2021-2027年中国药品行业发展现状分析及投资策略研究报告》显示:2020年国家药审中心完成中药(包括民族药,下同)、化学药、生物制品各类注册申请审评审批共11582件(含器械组合产品4件,以受理号计,下同),较2019年增长32.67%(如无说明,以注册申请件数计,下同)。
其中,完成需技术审评的注册申请8606件(含5674件需药审中心技术审评和行政审批注册申请),较2019年增长26.24%;完成直接行政审批(无需技术审评,下同)的注册申请2972件。
2020年国家药审中心完成8606件需技术审评的药品注册申请,同比增长26.24%,其中:化学药注册申请为6778件,较2019年增长25.22%;中药注册申请418件,较2019年增长39.33%;生物制品注册申请1410件,较2019年增长27.72%;化学药注册申请约占全部技术审评完成量的78.76%。
3、各类注册申请审评完成情况药审中心完成需技术审评的8606件注册申请中,完成新药临床试验(IND)申请审评1561件,较2019年增长55.94%;完成新药上市申请(NDA)审评289件,完成仿制药上市申请(ANDA)审评1700件;完成仿制药质量和疗效一致性评价(以下简称一致性评价)申请(以补充申请途径申报)1136件,较2019年增长103.22%;完成补充申请技术审评3250件,较2019年增长24.19%;难性临床申请审评126件,较2019年增长0.8%;补充申请审评完成3250件,较2019年增长24.19%;境外生产药品再注册申请审评完成498件,较2019年下降10.91%;复审申请审评完成46件,较2019年增长70.37%。
国家药品监督管理局公告2018年第94号——关于临床试验用生物制品参照药品一次性进口有关事宜的公告

国家药品监督管理局公告2018年第94号——关于临床试验用生物制品参照药品一次性进口有关事宜的公
告
文章属性
•【制定机关】国家药品监督管理局
•【公布日期】2018.11.30
•【文号】国家药品监督管理局公告2018年第94号
•【施行日期】2018.11.30
•【效力等级】部门规范性文件
•【时效性】现行有效
•【主题分类】药政管理
正文
国家药品监督管理局公告
2018年第94号
关于临床试验用生物制品参照药品一次性进口有关事宜的公
告
为落实中共中央办公厅、国务院办公厅《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号)的有关要求,支持和鼓励生物类似药的研发,更好地满足公众用药需求,现决定对符合以下条件、用于临床试验参照药的生物制品,可予以一次性进口。
现将有关事项公告如下:
一、可以申请一次性进口的生物制品范围包括:
(一)国内已经批准注册,但药品研发机构或者生产企业无法及时从国内市场获得的原研生物制品;
(二)国外已上市、国内尚未批准注册但已获批开展临床试验的原研生物制品。
二、国家药品监督管理局委托药品审评中心负责办理临床试验用生物制品参照药品一次性进口的受理、审查及审批。
三、本公告自发布之日起实施。
《关于研制过程中所需研究用对照药品一次性进口有关事宜的公告》(国家食品药品监督管理总局公告2016年第120号)、《生物类似药研发与评价技术指导原则(试行)》(国家食品药品监督管理总局通告2015年第7号)与本公告不一致的,以本公告为准。
特此公告。
国家药监局
2018年11月30日。
《药审中心关于已上市药品说明书增补儿童用药信息的工作流程

第一条(应用范围)为落实《已上市药品说明书增补儿童用药信息工作程序》中药品审评中心(以下简称药审中心)制定品种遴选范围、说明书修订与审核流程,以及品种申报程序等相关配套文件的要求,制定本文件。
第二条(遴选原则)以《ICH R1:用于儿科人群的医学产品的临床研究》《儿科人群药物临床试验技术指导原则》和《成人用药数据外推至儿科人群的技术指导原则》等技术要求为参考。
第三条(审核条件)符合遴选范围的品种应同时满足以下条件:1.该活性成分制剂已在境内上市,包括进口原研药和满足现阶段一致性评价标准的仿制药,并已批准用于我国成人患者和/或部分儿童患者,且安全性和有效性明确。
2.该活性成分制剂在境外(ICH 主要成员国家)已获批儿童适应症,具备较为充分的研究证据,且说明书中儿童用法用量明确。
3.该活性成分制剂已被我国儿童用药相关指南等推荐用于儿童,且推荐的适应症和用法用量信息与境外同活性成分制剂说明书批准的儿童用药信息基本一致。
该活性成分制剂已被用于我国儿童患者,临床用药情况清楚,临床用药数据可查。
4.该活性成分制剂的剂型和规格适用于我国儿童患者。
第四条(申请接收)药审中心接收国家儿科专业协会或国家儿童医学中心提出的申请。
申请以《品种资料信息表》及其附件形式通过公函提交至药审中心。
第五条(初步审查与分类处理)药审中心对《品种资料信息表》及其附件进行初步审查。
对于申请资料符合要求的申请,根据药审中心现行适应症技术审评职能划分,进行分类处理,发送至相应审评部门。
第六条(品种遴选与形成修订建议)审评部门负责对《品种资料信息表》及其附件进行审核,按照已上市药品说明书修订原则与技术审评要求进行评价,形成评价意见。
部门技术委员会负责对评价意见进行审议,审议结果填入《品种名单和说明书修订建议表》。
第七条(专家咨询会议)药审中心组织专家咨询会议对通过遴选的品种及说明书修订建议进行审议。
第八条(公示)药审中心根据专家咨询会议审议结果,修订完善《品种名单和说明书修订建议表》内容,安排公开征求意见。
国务院办公厅关于改革完善仿制药供应保障及使用政策的意见

国务院办公厅关于改革完善仿制药供应保障及使用政策的意见文章属性•【制定机关】国务院办公厅•【公布日期】2018.03.21•【文号】国办发〔2018〕20号•【施行日期】2018.03.21•【效力等级】国务院规范性文件•【时效性】现行有效•【主题分类】药政管理正文国务院办公厅关于改革完善仿制药供应保障及使用政策的意见国办发〔2018〕20号各省、自治区、直辖市人民政府,国务院各部委、各直属机构:为贯彻落实党的十九大精神和党中央、国务院关于推进健康中国建设、深化医改的工作部署,促进仿制药研发,提升仿制药质量疗效,提高药品供应保障能力,更好地满足临床用药及公共卫生安全需求,加快我国由制药大国向制药强国跨越,经国务院同意,现提出如下意见。
一、促进仿制药研发(一)制定鼓励仿制的药品目录。
建立跨部门的药品生产和使用信息共享机制,强化药品供应保障及使用信息监测,及时掌握和发布药品供求情况,引导企业研发、注册和生产。
以需求为导向,鼓励仿制临床必需、疗效确切、供应短缺的药品,鼓励仿制重大传染病防治和罕见病治疗所需药品、处置突发公共卫生事件所需药品、儿童使用药品以及专利到期前一年尚没有提出注册申请的药品。
鼓励仿制的药品目录由国家卫生健康委员会、国家药品监督管理局会同相关部门制定,定期在国家药品供应保障综合管理信息平台等相关平台发布,并实行动态调整。
新批准上市或通过仿制药质量和疗效一致性评价的药品,载入中国上市药品目录集,上市药品目录集内容动态更新并实时公开。
(二)加强仿制药技术攻关。
将鼓励仿制的药品目录内的重点化学药品、生物药品关键共性技术研究列入国家相关科技计划。
健全产学研医用协同创新机制,建立仿制药技术攻关联盟,发挥企业的主导作用和医院、科研机构、高等院校的基础支撑作用,加强药用原辅料、包装材料和制剂研发联动,促进药品研发链和产业链有机衔接。
积极引进国际先进技术,进行消化吸收再提高。
(三)完善药品知识产权保护。
仿制药质量和疗效一致性评价工作介绍

阿托伐他汀钙片 阿莫西林胶囊
苯磺酸氨氯地平片 蒙脱石散
阿法骨化醇片 阿奇霉素片 奥氮平片
厄贝沙坦氢氯噻嗪片 恩替卡韦分散片
富马酸替诺福韦二吡呋酯片 卡托普利片 赖诺普利片
硫酸氢氯吡格雷片 氯沙坦钾片
马来酸依那普利片 恩替卡韦胶囊 福辛普利钠片
富马酸比索洛尔片 格列美脲片 吉非替尼片
BE 试验
受理 立卷审查
基于审 评需要 检查
技术 审评
基于审 评需要 检验
综合 审评
纳入 橙皮书
研究
审评
信息公开
工作程序1——药学研究
工作程序2——BE试验
伦理审查
BE试验合同
BE试验备案
信息登记
开展BE试验
719家药物 临床试验 机构
工作程序3-受理
2016年第105号公告
2017年第100号公告
17
《关于阿莫西林胶囊等7个品种规格
通过仿制药质量和疗效一致性评价的 7
公告(第三批)》(2018年第6号)
《关于蒙脱石散等16个品种通过仿制
药质量和疗效一致性评价的公告(第 五批)》(2018年第49号)
16
《关于瑞舒伐他汀钙片等5个品种通过仿制
5 药质量和疗效一致性评价的公告(第二
批)》(2018年第20号)
关于仿制药质量和疗效一 致性评价工作有关事项的 公告(2017年第100号)
关于规范使用“通过一致 性评价”标识的通知
关于阿托伐他汀钙片等12 个品种规格通过仿制药质 量和疗效一致性评价的公 告(第四批)(2018年第 24号) 附件2关于“通过一致性评 价”标识使用有关事宜的 说明
工作成效——推动供给侧结构性改革
国家食品药品监管总局对《关于仿制药质量和疗效一致性评价工作有关事项的公告》的政策解读

国家食品药品监管总局对《关于仿制药质量和疗效一致性评价工作有关事项的公告》的政策解读一.《关于仿制药质量和疗效一致性评价工作有关事项的公告》(2017年第100号,以下简称《公告》)出台的背景和意义?自《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》(国办发〔2016〕8号)发布以来,仿制药质量和疗效一致性评价(以下简称一致性评价)工作扎实推进,有的企业已经完成了部分品种研究工作,进入申报审评阶段。
为进一步加强对企业的指导,提高工作效率,我局对前期工作进行了总结和分析,研究制定了本《公告》,对一致性评价工作各环节进行了优化调整,旨在保障受理、检查、检验和审评等环节顺畅衔接,保障评价标准统一。
二.针对参比制剂确定和获得,《公告》中提出了哪些优化措施?为了便于企业开展研究工作,总局目前已发布8批610个品种规格的参比制剂,包括《关于落实﹤国务院办公厅关于开展仿制药质量和疗效一致性评价的意见﹥有关事项的公告》(2016年第106号)中公布的《2018年底前须完成仿制药一致性评价品种目录》(以下简称《289品种目录》)中的163个品种(219个品规)。
该目录中另约有90左右品种为改规格、改剂型、改盐基的品种,按照《仿制药质量与疗效一致性评价工作中改规格药品(口服固体制剂)评价一般考虑》《仿制药质量与疗效一致性评价工作中改剂型药品(口服固体制剂)评价一般考虑》《仿制药质量与疗效一致性评价工作中改盐基药品评价一般考虑》等技术指南,上述改规格、改剂型、改盐基的约90左右品种的参比制剂选择依据也已明确。
至此,已经对《289品种目录》中大多数品种的参比制剂选择给出指导。
《公告》一方面对参比制剂选择顺序进一步明确,另一方面明确我局将继续对企业备案的参比制剂进行遴选和确认,符合参比制剂要求的发布参比制剂目录。
关于参比制剂获得事宜,企业可以通过申报一次性进口申请及进口备案、通关等程序来获得参比制剂,除此之外,《公告》明确企业还可以通过其他方式获得参比制剂,在提交一致性评价资料时,仅需在资料中提供购买凭证、产品包装及说明书等材料,或以其他适当方法证明参比制剂真实性即可。
2020年药审中心审评通过的境外生产原研药

67
乙磺酸尼达尼布软胶囊#
系统性硬化病相关间质性肺疾病;具有进行性表型的其他慢性纤维化性间质性肺疾病
68
红斑狼疮5岁及以上患者
69
阿达木单抗注射液#
4岁及以上儿童与青少年重度慢性斑块状银屑病;非感染性葡萄膜炎
70
恩曲他滨替诺福韦片#
HIV-1暴露前预防
法布雷病
5
阿替利珠单抗注射液
联合含铂化疗用于初治广泛期小细胞肺癌
6
艾地骨化醇软胶囊
绝经后女性骨质疏松症
7
艾度硫酸酯酶β注射液
黏多糖贮积症Ⅱ型
8
艾托格列净片
配合饮食和运动改善成人2型糖尿病患者的血糖控制。
9
达依泊汀α注射液
需血液透析的慢性肾病患者的贫血
10
地舒单抗注射液
骨折风险增高的绝经后妇女的骨质疏松症
71
乌司奴单抗注射液/
乌司奴单抗注射液(静脉输注)#
成人中重度活动性克罗恩病患者
72
左乙拉西坦注射用浓溶液#
4岁以上儿童及成人癫痫患者部分性发作
73
富马酸卢帕他定片*
过敏性鼻炎和荨麻疹
74
盐酸二甲双胍片*
2型糖尿病
注:1.“#”为新增适应症品种。
2.“*”是国内已有仿制品种上市的境外生产原研药,不纳入2020年统计范围内。
11
甘精胰岛素注射液
需胰岛素治疗的成人2型糖尿病
12
格隆溴铵福莫特罗吸入气雾剂
慢性阻塞性肺疾病
13
氯化镭[223Ra]注射液
伴骨转移且无已知内脏转移的去势抵抗性前列腺癌
14
马来酸奈拉替尼片
HER2阳性早期乳腺癌的强化辅助治疗。
武汉市肺科医院2018年药物临床试验质量管理规范(GCP)考试

武汉市肺科医院2018年药物临床试验质量管理规范(GCP)考试1、现行的《药物临床试验质量管理规范》是从何时开始实行的? ( ) [单选题] *A.2003年9月1日(正确答案)B.2004年2月19日C.2007年10月1日D.2015年4月24日2、对于首次人体研究(FIH),以下哪项不是需要考虑的因素? ( ) [单选题] *A.试验设计B.受试者C.评估指标(正确答案)D.伴随用药3、样本均数的95%可信区间可以解释为: ( ) [单选题] *A.有95%的把握认为总体均数在这个围绕样本均数的区间内(正确答案)B.有95%个体值在该范围内C.95%可信区间是单侧范围D.可信区间估计法与假设检验结论不可能相同4、在当前数据管理模式中,以下哪个环节临床医生无需参与? ( ) [单选题] *A.数据采集B.数据核查与产生疑问(正确答案)C.数据修正D.回答数据疑问5、临床试验中,哪种数据不是原始数据? ( ) [单选题] *A.医生撰写的病历资料B.实验室检查报告C.填写的病例报告表数据(正确答案)D.影像学图像资料6、下列哪项內容不属于监查员的职责? ( ) [单选题] *A.确认临床研究机构的人员和条件符合GCP要求B.确认CRF表内容和原始资料一致C.核实CRF表内容,如有错误直接进行修改(正确答案)D.核实试验用药的供应、分发储藏和回收符合规定并记录7、叙述试验的背景、理论基础和目的、试验设计、方法和组织,包括统计学考虑、试验执行和完成条件的临床试验的主要文件,称为: ( ) [单选题] *A.知情同意书B.研究者手册C.病例报告表D.试验方案(正确答案)8、有关一种试验用药品在进行人体研究时已有的临床与非临床数据汇编,称为:( ) [单选题] *A.原始资料B.知情同意书C.试验方案D.研究者手册(正确答案)9、由申办者任命具备相关知识的人员对试验的数据进行核实,并将试验进展报告给申办者,督促临床试验的进行,以保证临床试验按方案和法规执行,称为: ( ) [单选题] *A.稽查B.监查(正确答案)C.视察D.质量保证10、为了有效地实施和完成临床试验而针对每一工作环节、每一步骤或每一具体操作而制定的标准而详细的书面规程是? ( ) [单选题] *A.管理制度B.标准操作规程(SOP)(正确答案)C.制定SOP的SOPD.试验方案11、病人或临床试验受试者接受一种药品后出现的不良医学事件,但怀疑可能与试验药物存在一定的因果关系,称为:( ) [单选题] *A.不良事件B.药物不良反应(正确答案)C.严重不良事件D.重大不良事件12、按照多中心临床试验协作审查程序,组长单位的伦理委员会审查内容不包括以下哪一条: ( ) [单选题] *A.试验方案的科学性和伦理合理性B.该项试验在所有临床试验单位实施的可行性(正确答案)C.临床试验实施过程中的跟踪审查D.本试验单位发生的严重不良事件13、下列哪项不是知情同意书必需的内容? ( ) [单选题] *A.试验目的B.试验可能的受益和可能发生的风险C.研究者的专业资格及经验(正确答案)D.说明可能被分配到不同组别14、以下哪一项不是受试者中止研究的标准? ( ) [单选题] *A.受试者出现不能继续治疗的不良事件或伴发情况B.受试者不愿继续治疗C.受试者在研究期间服用其他非试验药物(正确答案)D.受试者妊娠15、试验用药品的使用由谁负责? ( ) [单选题] *A.受试者B.研究者(正确答案)C.申办者D.医疗机构16、下列哪项不属于研究者的职责? ( ) [单选题] *A.做出相关医疗决定,保证受试者安全B.保证试验结果通过新药审评(正确答案)C.填写病历报告表D.报告不良事件17、以下不属于《药物临床试验质量管理规范》目的的是? ( ) *A.保证药品临床试验的过程规范,结果科学可靠,保护受试者的权益及保障其安全B.药品临床试验在科学上具有先进性(正确答案)C.保证临床试验对受试者无风险(正确答案)D.保证药品临床试验的过程按计划完成(正确答案)18、Ⅲ期临床试验主要目的是什么? ( ) *A.治疗作用的确证阶段,验证试验药物安全有效(正确答案)B.确定药物对目标适应症,评价利益与风险关系(正确答案)C.为药物注册获得批准提供充分的临床试验依据(正确答案)D.为完成药品使用说明书提供所需要的信息(正确答案)19、临床试验开始前,研究者应要求申办者提供哪些资料? ( ) *A.CFDA批文(正确答案)B.药检报告(正确答案)C.研究者手册(正确答案)D.临床试验方案(正确答案)20、发生严重不良事件时,研究者需要报告的部门或组织: ( ) *A.药品监督管理部门(正确答案)B.申办者(正确答案)C.本机构伦理委员会(正确答案)D.其他机构伦理委员会21、以下哪些是药物临床试验伦理审查的宗旨: ( ) *A.提高药物临床试验的质量管理水平B.保证受试者尊严、安全和权益(正确答案)C.促进药物临床试验科学、健康的发展(正确答案)D.增强公众对药物临床试验的信任和支持(正确答案)22、试验设计中假设检验的类型有哪些? ( ) *A.优效性检验(正确答案)B.等效性检验(正确答案)C.非等效性检验D.非劣效性检验(正确答案)23、伦理委员会委员组成的要求: ( ) *A.至少5人(正确答案)B.多学科背景(正确答案)C.男女性别均衡(正确答案)D.至少一名委员独立于研究/试验单位(正确答案)24、以下不属于伦理审查批准范围的是: ( ) *A.研究实施条件B.其他伦理委员会的审查意见(正确答案)C.试验实施过程中的临床监查和稽查计划(正确答案)D.研究者手册25、研究实施过程中的跟踪审查包括以下哪些审查类别: ( ) *A.修正案审查(正确答案)B.年度/定期跟踪审查(正确答案)C.严重不良事件审查(正确答案)D.不依从/违背方案审查(正确答案)1、研究者必须详细阅读和了解试验方案的内容,与申办者一同签署临床试验方案,并严格按照方案和GCP的规定进行临床试验。
2018年医药行业政策梳理回顾,你看了吗?

2018年医药行业政策梳理回顾,你看了吗?2018年是“十三五”承上启下的关键一年,在这一年里,国务院大部制机构改革,政策发布之密集、力度之大、落地速度之快远超过往。
医保局、卫健委、药监局三大医疗机构崛起;全面破除以药补医,深化公立医院综合改革文件发布;仿制药一致性评价进入关键阶段;进口抗癌药实施零关税;九部委联合发文严打医药商业贿赂;17种抗癌药纳入医保报销目录;新版《国家基本药物目录》发布;国家4+7带量采购方案公布……2018年医药行业政策仍以三医联动为核心,医药端支持创新、保障药品质量;医疗端综合推进医药分开与分级诊疗,限制辅助用药品种;医保端保障用药,控制费用合理支出,本文将从医药、医疗器械、医疗、医保四方面对2018年行业政策进行梳理回顾。
医药深化审评审批政策,鼓励药品创新2018年,在推动创新药物研发方面,备受关注的药品试验数据保护制度、接受药品境外临床试验数据的技术指导原则的通告、临床试验申请默认制等政策陆续出台。
4月26日,国家药品监督管理局公布《药品试验数据保护实施办法(暂行)(征求意见稿)》,办法对于创新药、罕见病和儿童专用药给予一定期限的数据保护期,突破性地拓展了创新药和专用药的数据保护时间和范围。
作为与药品专利保护完全不同的知识产权保护体系,这一鼓励创新的后继保护措施对于支持医药研发和技术转化具有十分重要的意义。
7月10日,国家药品监督管理局发布了《关于发布接受药品境外临床试验数据的技术指导原则的通告(2018年第52号)》(以下简称为《通告》),明确境外临床试验数据可用于在中国的药品注册申报,国外新药进入中国的速度将越来越快。
7月27日,国家药品监督管理局发布《关于调整药物临床试验审评审批程序的公告》,标志着我国临床试验由“批准制”改为“默认制”。
在“批准制”情况下,我国药品临床试验的平均启动时间约为14-20个月,“默认制”的实施则意味着我国临床试验申请自申请受理并缴费之日起60日内,申请人未收到国家食品药品监督管理总局药品审评中心(CDE)否定或质疑意见的,即可开展临床试验,此举将大大提升国内创新药物临床开发进程。
【独家】从创新到创举,这8款重磅药品打开了2018国内抗感染领域新格局!

【独家】从创新到创举,这8款重磅药品打开了2018国内抗感染领域新格局!如果您喜欢药智网推送的这篇文章欢迎点赞和转发哦~每年很多疾病都与感染有关,从轻微的真菌病毒感染,到比较严重的复发性感染,再到致命的危害性感染。
我们似乎时刻都需准备着与之为战。
因此在我国的医药市场中,抗感染药物一直占有很大的比重,每年市场耗用量都很大,虽然国家在此领域有所布局希望能够减少一些不适当、不必要的使用,但随着病毒、病菌的不断变异,抗感染药物还是有其刚性的需求,也因此不得不在这种矛盾之中继续发展。
我国抗感染领域药品现状据药智上市药物筛选系统数据库统计,在我国市场上的抗感染类药品共计31458个(以批准文号计,下同),而这些药品被列入国家医保的共计20187个,有效地减轻了病患的负担;另外国产药物为30987个,可见我国的抗感染药物主要是靠国家本土企业,国内这片市场很大。
而依据我国基本的药物研发生产情况,抗感染系统的药物是大类,很多的国产优质药品已经冲击到国际市场。
据药智数据统计,截止2017年12月15日(下同),CDE承办的药品中,系统用抗感染药所占比重最多,为17.22%。
这是于市场需求相契合的。
而在抗感染药品之中,系统用抗菌药所占比例最大,为74.19%。
图1 CDE承办的抗感染领域药品在所有药品中的占比情况2017年8款新药面世,创新与创举齐发近几年,随着国家相继出台政策规范抗菌药物的临床使用,导致抗感染类药物增长速度放缓,而依据政策,合理使用抗感染药物成为主流。
因此对于抗感染药物开启了重新的定位,我们需求更多的疗效佳、耐药性强的药品,而各企业也在向这一目标调整。
2017年截止12月15日,我们迎来了8款抗感染领域的新药,均是可圈可点的重磅好药,这是在新形势、新环境下踏出的良好一步。
下面我们分别来看看这8款药品。
图2 2017年(截止12月15日)CDE批准上市的抗感染领域新药1、乙肝一线药品富马酸替诺福韦二吡呋酯目前,全球乙肝患者较多,国内患者对乙肝药品需求量很大,但又碍于其价格比较昂贵,让国内患者压力较大。
化学仿制药参比制剂目录(第六十七批)
株式会社
未进口原研药品
67-22
普瑞巴林缓释片
Pregabalin Extended release Tablets/Lyrica Cr
165mg
Pf Prism CV/UPJOHN US 2 LLC
未进口原研药品
美国橙皮书
67-23
普瑞巴林缓释片
Pregabalin Extended release Tablets/Lyrica Cr
国际公认的同种药品
美国橙皮书
67-34
头孢地尼干混悬剂
Cefdinir for Oral Suspension
125mg/5mL
Aurobindo Pharma Limited
美国橙皮书
67-35
蔗糖铁注射液
Iron Sucrose Injection/Venofer
5ml:100mg铁和1.6g蔗糖
0.25%
(10ml:25mg)
Hospira Inc
未进口原研药品
美国橙皮书
67-27
左卡尼汀口服溶液
Levocarnitine Oral Solution/CARNITOR SF
10ml:1g
Leadiant Biosciences, Inc.
未进口原研药品
美国橙皮书
67-28
碳酸氢钠血滤置换液(钾4mmol/L)
Hemofiltration Replacement Fluid of Sodium Bicarbonate
(4mmol/L Potassium Calcium free)/PrismaSol
5000ml(250ml/4750ml)
BAXTER HEALTHCARE CORP
【独家】列入优先审评药品简析:正大天晴、恒瑞、华海、拜耳、诺华...

【独家】列入优先审评药品简析:正大天晴、恒瑞、华海、拜耳、诺华...看点:1. 承办趋势,2015-2016关键年;2. 报产批准率:新药25.78%,仿制药26.84%,进口药65.82%;3. 报临床批准率:新药78.69%,仿制药50%,进口药55.24%;4. 1类新药:临床批准率92.31%;23个报产制剂值得期待,百济神州、恒瑞等重磅产品在列;5. 3类仿制药:5品种批产,33品种审评中,恒瑞、人福、正大天晴大比拼;6. 优先审评受理数量三甲企业:新药正大天晴、恒瑞、豪森,仿制药华海、东阳光、齐鲁,进口药拜耳、诺华、勃林格殷格翰、辉瑞。
1.优先审评全览截止2019年2月23日,纳入优先审评的药品受理号达791个。
其中仿制药申报占比最多,为39.70%,其次为新药,约有三分之一的占比。
在这791个受理号申请中,其承办时间如下所示,其中近3年承办的药品,纳入优先审评数量最多。
且新药纳入数量在8年间直线上升,2018年超过进口申请;可见在政策及创新驱动下,越来越多国研新药进入收获期。
而仿制药在2015-2016间纳入优先申评数量少于进口及新药,这与当时处于仿制药变革期有一定的关系,渡过风口之后的仿制药在2017、2018年数量大增,热度再次回归。
以下从申报类型新药、仿制药、进口药三个维度具体分析。
2.申报类型简析2.1新药2.1.1申报批准情况新药列入优先审评主要理由有具有明显临床价值的创新药、重大专项、儿童用药、罕见病新药、具有明显治疗优势的新药等。
目前列入优先审评的新药受理号总计达189个,其主要申请为申报生产,数量达128个,占比67.72%,已经批准33个受理号,批准率为25.78%。
而申报临床数量虽不及报产,但批准率较高,达78.69%。
2.1.2申报企业申报受理号(包括原料药)数量上,正大天晴受理数量最多,达17个,其次为恒瑞医药及豪森药业。
作为国内新药产出较多的企业,3企业纳入优先审评的药品数量情况,一定程度也能窥见其创新力所受认可度。
BE审评工作-20181211-来源药审中心

Guideline on the investigation of bioequivalence (2010)
EMA
使申请人的研究有章可循 使BE试验技术审评有据可依 申请人对结果具有可预见性
10
BE审评参考——国外指导原则
仿制药BE指导原则(2013年, draft) 新药的BE指导原则(2014年,draft)
FDA Bioanalytical Method Validation Guidance for Industry(May 2018)
Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System(December 2017)
0
5
10
15
20
一致性评价(前5批)
方法验证及 其他:23% 样品检
测:23%
样品检测&其他:54%
方法验证及样品检测
样品检测&其他
其他 12
BE审评的总体考虑
•PK-BE?PD-BE? •交叉?平行?重复? •采血点设计? •空腹?餐后?
• ……
•剔除数据? •数据集? •随机化? •敏感性分析? •……
适用范围
• 无全身吸收的局部作用 药物 • 生物样本中活性成分浓 度过低,不能可靠测定 • 活性成分的浓度与药品 的有效性和安全性无关
FDA_胃肠道局部作用复杂仿制药的等效性评价

药事管理㊀作者简介:魏赫ꎬ女ꎬ硕士ꎬ主管药师ꎬ研究方向:药品技术审评ꎬE-mail:weih@cde.org.cn通信作者:李雪梅ꎬ女ꎬ博士ꎬ主任药师ꎬ研究方向:药品技术审评ꎬTel:010-85242663ꎬE-mail:lixm@cde.org.cnFDA胃肠道局部作用复杂仿制药的等效性评价魏赫ꎬ李晓锋ꎬ李雪梅(国家药品监督管理局药品审评中心ꎬ北京100022)摘要:复杂仿制药具有广泛的临床应用价值ꎬ其研发和评价难度均较大ꎮ本文介绍和讨论了部分美国食品药品监督管理局(FDA)胃肠道局部作用复杂仿制药的生物等效性指南ꎬ该类药物具有复杂的活性成分ꎬ体内基本无吸收ꎬ可在证明仿制药活性成分与参比药物相同的基础上ꎬ采用多种体外试验进行生物等效性研究ꎬ从而评价治疗等效性ꎮ建议我国制药行业和药品监管机构关注复杂仿制药的研发ꎬ实现国产仿制药高质量发展ꎮ关键词:复杂仿制药ꎻ活性成分相同性ꎻ生物等效性ꎻ体外研究ꎻ胃肠道中图分类号:R95㊀文献标志码:A㊀文章编号:2095-5375(2023)09-0734-007doi:10.13506/j.cnki.jpr.2023.09.017FDAᶄsequivalenceevaluationforlocallyactinggastrointestinalcomplexgenericdrugsWEIHeꎬLIXiaofengꎬLIXuemei(CenterforDrugEvaluationꎬNationalMedicalProductsAdministrationꎬBeijing100022ꎬChina)Abstract:Complexgenericdrugshavewideclinicalapplicationsꎬwhiletheirdevelopmentandevaluationarebothdiffi ̄cult.ThispaperintroducesanddiscussessomeoftheFDAᶄsspecificguidanceoflocallyactinggastrointestinalcomplexge ̄nericdrugsꎬwhichhavecomplexactiveingredientsandarenotabsorbedinvivo.Onthebasisofprovingthattheactivein ̄gredientsofgenericdrugsarethesameasthereferencelisteddrugsꎬbioequivalencestudiescanbeconductedbyusingava ̄rietyofinvitroteststoevaluatethetherapeuticequivalence.Itissuggestedthatthepharmaceuticalindustryanddrugregula ̄torsinChinapayattentiontotheresearchanddevelopmentofcomplexgenericdrugsꎬinordertoachievehigh-qualityde ̄velopmentofdomesticgenericdrugs.Keywords:ComplexgenericdrugsꎻAPIsamenessꎻBioequivalenceꎻInvitrostudyꎻGastrointestinalgeneric㊀㊀仿制药(genericdrugs)是指具有与参比药物(referencelisteddrugsꎬRLD)相同的活性成分㊁剂型㊁规格㊁适应证㊁给药途径和用法用量ꎬ并证明质量和疗效与RLD相同的药品[1-2]ꎮ仿制药需要进行药学等效性评价(pharmaceuticalequivalenceꎬPE)和生物等效性(bioequivalenceꎬBE)评价ꎬ用以证明与参比药物的治疗等效性(therapeuticequivalenceꎬTE)ꎮ复杂仿制药一般包括具有复杂活性成分的产品ꎬ具有复杂剂型或给药途径的产品ꎬ复杂的药械组合产品等[3-4]ꎮ一般来说ꎬ结构相对简单的小分子药物比较容易通过药学质量研究和人体生物等效性试验进行PE和BE评价ꎬ而复杂仿制药的研发和评价难度较大ꎮ对于具有复杂活性成分的一类仿制药ꎬ如合成肽㊁复杂混合物等ꎬ由于其分子结构的复杂性需要进行充分的研究来证明仿制药与RLD活性成分(activepharmaceuticalingredientꎬAPI)的相同性ꎬ从而确证PE[5]ꎮBE是评价药物是否具有可替换性的重要指标ꎮBE研究方法包括药代动力学研究㊁药效动力学研究㊁临床研究和体外研究ꎮ以药代动力学参数为终点的BE研究最为常用ꎬ但部分胃肠道局部作用的药品ꎬ未进入血液循环ꎬ无法通过药代动力学终点指标进行BE评价ꎬ而以药效学参数为终点的BE研究和以临床疗效为终点的BE研究则具有较大的变异和较低的灵敏度ꎮ近年来随着监管理念的提升和药物评价的发展ꎬ对于胃肠道局部作用药物ꎬ可根据产品特点㊁作用机制㊁体内外相关性等以体外方法来评价BE[6]ꎮ国家卫生健康委联合国家药监局和国家知识产权局等部门制定了两批鼓励仿制药品目录清单ꎬ2019年发布的第一批目录中包括格拉替雷㊁盐酸考来维仑复杂仿制药ꎬ但截至目前尚无批准ꎮ表1汇总了部分复杂仿制药中美获批情况对比ꎬ可以看出我国此类药品的获批情况与美国有一定的差距ꎬ上市时间较早的硫糖铝则出现了过度仿制的情况[7]ꎮ表1㊀中美部分复杂仿制药获批情况对比剂型RLD持有人及首次获批时间美国批准的仿制药数量中国批准的仿制药数量中国在审的仿制药数量硫糖铝咀嚼片㊁片剂㊁口服混悬剂ABBVIEINCꎻ1982年772(过度仿制)2盐酸考来维仑片剂㊁口服混悬剂COSETTEPHARMACEUTICALSINCꎻ2000年1302碳酸司维拉姆片剂㊁口服混悬剂混悬剂:GENZYMECORP片剂:SANOFIGENZYMEꎻ2007年15414环硅酸锆钠口服混悬剂ASTRAZENECAPHARMACEUTICALSLPꎻ2018年000醋酸格拉替雷皮下注射液TEVAPHARMACEUTICALSUSAꎻ2002年402(进口仿制)氯替泼诺混悬滴眼液BAUSCHANDLOMBINCꎻALCONLAB ̄ORATORIESINCꎻ1998年302㊀注:以上数据来源于国家药品监督管理局药品审评中心官方网站ꎬ查询日期截至2023年6月20日ꎮ㊀㊀本文主要以胃肠道局部作用的具有复杂活性成分的仿制药为例ꎬ汇总了近年美国食品药品监督管理局(FDA)发布的具体产品指南ꎬ以期为该类药物的研发和等效性评价提供参考ꎮ1㊀硫糖铝硫糖铝为抗酸药(结构示意图见图1)ꎬ用于治疗胃㊁十二指肠溃疡及胃炎ꎮ在酸性环境下ꎬ解离出硫酸蔗糖复合离子ꎬ复合离子聚合成不溶性的带负电荷的胶体ꎬ能与溃疡面带正电荷的蛋白质渗出物相结合ꎬ形成一层保护膜覆盖于溃疡面ꎬ促进溃疡愈合ꎮ还具有吸附胃蛋白酶和胆汁酸的作用ꎮFDA硫糖铝口服混悬剂生物等效性研究指南首次发布于2014年ꎬ目前现行有效的版本为2023年2月发布[8]ꎮ推荐的研究内容包括1项API相同性研究ꎬ1项理化特性对比研究和4项生物学测定研究ꎮ受试制剂和RLD的Q1和Q2应相同(具有相同的非活性成分ꎬ且用量差异在ʃ5%以内)ꎬBE应建立在API相同性评估基础上ꎬ并且受试制剂和RLD体外研究具有可比性ꎮ1.1㊀API相同性研究㊀硫糖铝结构式和分子式如图1所示ꎮ仿制药申请人应该对受试制剂的API进行表征ꎬ并且要证明其组成和分子式与RLD标签中的结构信息是一致的ꎮ结构表征至少采用3批受试制剂ꎬ推荐的结构表征包括但不限于:①API的组成:辛硫酸蔗糖和铝的含量ꎻ②元素分析测定C㊁H㊁S㊁Al的数据ꎬ以及C/S比和C/Al比ꎻ③酸中和能力ꎻ④光谱学表征ꎬ例如傅立叶变换红外光谱(FT-IR)㊁紫外光谱(UV)㊁固态27Al核磁共振㊁差式扫描量热法(DSC)㊁热重量分析法(TGA)㊁粉末X射线衍射法(PXRD)ꎮAl8(OH)16(C12H14O35S8)[Al(OH)3]x[H2O]y㊀x=8to10ꎬy=22to31图1㊀硫糖铝结构示意图1.2㊀受试制剂和RLD的理化特性对比㊀理化特性对比包括以下内容:pHꎬ比重ꎬ未处理制剂的黏度分布ꎬ表观黏度随酸度的变化情况ꎬ再分散性(再分散的时间㊁沉降时间和体积)ꎬ酸中和能力ꎬ铝在pH1.2介质中的释放情况ꎮ1.3㊀受试制剂和RLD的生物学测定㊀在进行生物学测定之前受试制剂和RLD均用酸预处理ꎬ如果可能ꎬ在与体内生理条件相关的条件下进行试验ꎮ应提供生物学测定的方法开发和方法学验证资料ꎮ1.3.1㊀体外胆盐平衡结合研究㊀该项研究是评估生物等效性的关键性研究ꎮ应在至少8种不同浓度的胆盐中进行孵育ꎬ浓度应分布合理直到达到最大结合ꎮ每项结合研究应至少重复12次ꎮ此外ꎬ研究数据应该表明选择的孵育时间长度可以产生最大的结合ꎮ生物等效性评价指标为Langmuir结合常数k2的90%置信区间ꎮ1.3.2㊀体外胆盐动力学结合研究㊀该项研究用以支持关键性的平衡结合研究ꎮ应在2种不同浓度的胆盐溶液中孵育至少8个不同的时间长度ꎬ时间长度应分布合理直到达到最大结合ꎮ每项结合研究应至少重复12次ꎮ生物等效性评价指标是受试制剂和RLD硫糖铝与胆盐结合率的比较ꎮ1.3.3㊀体外人血白蛋白(HSA)或牛血清白蛋白(BSA)平衡结合研究㊀该项研究试验设计和数据分析可参考体外胆盐平衡研究ꎮ1.3.4㊀体外酶(胃蛋白酶)活性研究㊀该项研究应采用至少5种不同浓度的受试制剂和RLD进行ꎮ生物等效性评价指标是受试制剂和RLD关于胃蛋白酶活性降低百分比的比较ꎮ1.4㊀讨论㊀表2为FDA对硫糖铝产品指南的修订情况ꎬ可以看出近年来FDA对该药的BE评价观念逐渐转变ꎬ由体外评价方法替代以临床终点为指标的评价方法ꎬ更加体现科学性ꎬ同时也降低了仿制药企业的研发成本和时间成本ꎮ根据硫糖铝作用机制ꎬ设计4种体外研究方法ꎬ随着监管能力的提升ꎬ对体外BE研究的要求也越来越趋于完善ꎮ表2㊀FDA硫糖铝产品指南修订情况剂型发布/修订时间推荐的等效性研究内容片剂2014年首次发布1项以临床终点为指标的BE研究2015年撤销/2019年修订1项API相同性研究ꎬ1项理化特性对比研究ꎬ4项体外生物学测定研究2023年修订/现行版本1项API相同性研究ꎬ1项理化特性对比研究(增加了崩解时限要求)ꎬ4项体外生物学测定研究(细化了结合试验浓度㊁时间㊁重复次数等要求)口服混悬剂2014年首次发布1项以临床终点为指标的BE研究2015年撤销/2017年修订1项API相同性研究ꎬ1项理化特性对比研究ꎬ4项体外生物学测定研究2023年修订/现行版本1项API相同性研究ꎬ1项理化特性对比研究ꎬ4项体外生物学测定研究(细化了结合试验浓度㊁时间㊁重复次数等要求)2㊀碳酸司维拉姆碳酸司维拉姆(结构示意图见图2)是丙烯胺化合物的交联聚合体ꎬ用于治疗控制正在接受透析治疗的慢性肾病成人患者的高磷血症ꎮ口服后在胃肠道内水合膨胀成数倍于原体积的凝胶ꎬ胺根以质子化的形式存在ꎬ通过离子键和氢键与磷酸分子相互作用结合消化道中的磷酸根ꎬ降低血清中的磷酸根浓度ꎬ被聚合物结合的磷酸盐不能吸收入体内ꎬ以粪便方式排出[9]ꎮFDA碳酸司维拉姆片生物等效性研究指南首次发布于2008年ꎬ目前现行有效的版本为2015年9月发布[10]ꎮ推荐的研究内容包括1项API相同性研究ꎬ2项体外研究ꎮ2.1㊀API相同性研究㊀碳酸司维拉姆结构如图2所示ꎮAPI相同性研究通过合成路线以及理化性质对比研究建立ꎮFDA鼓励申请人采用与参比API相同的合成路线ꎬ即聚丙烯胺盐酸盐与环氧氯丙烷交联来生产碳酸司维拉姆ꎬ如果采用不同的合成路线ꎬ则需要联系仿制药办公室取得同意ꎬ并且可能需要指南以外的特性研究来证明API的相同性ꎮ申请人应该向监管机构提供生产工艺和工艺过程控制信息ꎮ应将至少3批仿制API与至少3批从RLD中提取的API进行背对背比较研究ꎬ用以评价API的相同性和生产工艺的稳定性ꎮRLD中API的提取方法应该在申报资料中递交ꎮ推荐的理化性质对比研究内容包括:①交联度(即交联氨基与总氨基的比例):建议采用C13固态核磁共振光谱(13CSS ̄NMR)ꎬ通过定量峰面积分析API的交联度ꎻ②质子化程度(碳酸盐含量):建议采用TGAꎻ③总可滴定胺:建议采用滴定法ꎻ④粒径分布ꎻ⑤元素分析:结果应该包括C㊁H㊁N元素ꎻ⑥溶胀指数ꎻ⑦鼓励申请人进行更多的理化性质表征研究ꎬ包括但不限于FT-IR㊁拉曼光谱㊁PXRD和DSC等ꎮ2.2㊀体外研究2.2.1㊀体外平衡结合研究㊀该项研究是评估生物等效性的关键性研究ꎮ应将受试制剂和RLD整片样品ꎬ在pH4和pH7条件下采用酸预处理或不处理的方式ꎬ于至少8种不同浓度的磷酸盐溶液中进行孵育ꎬ每种磷酸盐溶液中应包含80mmol L-1的氯化钠和100mmol L-1的NꎬN-双(羟乙基)-2-氨基乙磺酸(BES)ꎮ磷酸盐浓度应分布合理直到达到最大结合ꎮ试验应该在37ħ条件下进行ꎮ每一项结合试验应重复至少12次ꎮ此外应提供数据ꎬ表明选择的孵育时间长度可使磷酸盐产生最大结合ꎮ2.2.2㊀体外动力学结合研究㊀该项研究用以支持关键性的平衡结合研究ꎮ应将受试制剂和RLD整㊀aꎬb-伯胺基团数㊀a+b=9ꎻc-交联基团数㊀c=1ꎻm-很大的数目ꎬ以表示延伸的聚合物网状结构ꎮ图2㊀碳酸司维拉姆结构示意图片样品ꎬ在pH4和pH7条件下采用酸预处理或不处理的方式ꎬ于2种不同浓度的磷酸盐溶液中孵育至少8个不同的时间长度ꎮ磷酸盐的浓度应选择相应平衡结合研究中的最高和最低浓度ꎮ时间长度应分布合理直到达到最大结合ꎮ试验应该在37ħ条件下进行ꎮ每一项结合试验应重复至少12次ꎮ2.2.3㊀数据分析㊀检测滤液中未结合的磷酸盐ꎬ间接计算出与碳酸司维拉姆结合的磷酸盐量ꎮ在体外平衡结合研究中ꎬ计算Langmuir结合常数k1和k2ꎬ受试制剂与RLDk1比值㊁k2的90%置信区间ꎮ体外动力学结合研究中ꎬ比较不同时间受试制剂与RLD的磷酸盐结合率ꎬ但可不计算90%置信区间ꎮ生物等效性的评价指标为平衡结合研究中k2的90%置信区间应在80%~120%范围内ꎮ2.3㊀讨论㊀仿制药与RLD具有相同的活性成分是研发的第一步ꎬ也是能够发挥相同治疗作用的重要前提ꎮ碳酸司维拉姆是高度交联的聚合物ꎬ不是单一分子ꎬ证明仿制药与RLDAPI的相同性较为复杂ꎬ不仅需要进行API理化特性对比ꎬ更需要从合成路线㊁工艺控制等方面入手ꎬ利用质量源于设计的理念使API的质量从源头得到保证ꎮ体外研究方面ꎬ根据随餐服用的特点ꎬ建议在pH4和pH7条件下进行磷结合试验ꎬ能够反映体内生理条件ꎮ对于选用的磷酸盐浓度ꎬ孵育时间长度等应进行合理优化ꎬ提供确定依据ꎮ采用的检测方法应具有较高的专属性和灵敏度ꎬ并开展完整的方法学验证[11]ꎮ3㊀盐酸考来维仑盐酸考来维仑(结构示意图见图3)是一种胆汁酸螯合剂ꎬ用于治疗原发性高脂血症ꎮ非吸收性的聚合物盐酸考来维仑在肠道中能与胆汁酸结合ꎬ阻滞胆汁酸的重吸收ꎬ从而加速肝内胆固醇经7-α-羟化酶向胆汁酸的转化ꎮ肝细胞对胆固醇的需求增加ꎬ使胆固醇生物合成酶羟甲基戊二酰辅酶A还原酶(HMG-CoA)的转录和活性增加ꎬ肝脏低密度脂蛋白受体数量增加ꎮ这些代偿作用导致血液中的低密度脂蛋白胆固醇(LDL-C)清除率增加ꎬ从而降低血清LDL-C水平[12]ꎮFDA盐酸考来维仑口服混悬剂生物等效性研究指南首次发布于2010年ꎬ目前现行有效的版本为2021年9月发布[13]ꎮ推荐的研究内容包括1项API相同性研究ꎬ4项体外研究ꎮ3.1㊀API相同性研究㊀盐酸考来维仑结构如图3所示ꎮAPI相同性研究可以通过基本反应方案的相同性㊁化学结构和组成的相同性㊁理化性质相同性3方面进行确证ꎮ应将至少3批仿制API与至少3批从RLD中提取的API进行背对背比较研究ꎬ用以评价API的相同性和生产工艺的稳定性ꎮRLD中API的提取方法应该在申报资料中递交ꎮ㊀a+aᶄ=2ꎻb=1ꎻc+cᶄ=7ꎻd+dᶄ=6ꎻm=聚合物网状结构的数量ꎻ基本聚合物单元的分子式为(C3H8NCl)2(C9H20N2OCl2)1(C13H28NCl)7(C12H28N2Cl2)6图3㊀盐酸考来维仑结构示意图3.1.1㊀基本反应方案相同性研究㊀FDA建议仿制药申请人根据RLD标签信息ꎬ采用聚丙烯胺盐酸盐与环氧氯丙烷交联ꎬ与1-溴癸烷和(6-溴己基)-三甲基溴化铵烷基化的工艺生产盐酸考来维仑ꎮ如果采用替代合成工艺ꎬ应该联系FDA取得同意ꎬ并且可能需要指南以外的表征来证明API的相同性ꎮ3.1.2㊀化学结构和组成相同性研究㊀仿制药申请人应定义API的结构ꎬ如a+aᶄꎬbꎬc+cᶄꎬd+dᶄ的数值等ꎬ受试API和参比API应具有相同的化学结构和组成ꎮ仅从理化性质表征可能不足以确证API的化学组成ꎬ因此建议仿制药申请人对交联中间体进行表征ꎬ推荐的研究包括13CSSNMR和玻璃化转变温度(Tg)ꎬ具体可参照司维拉姆产品指南ꎻ此外ꎬ应对烷基化试剂随反应进行的消耗程度进行研究ꎮ3.1.3㊀理化性质相同性研究㊀API理化性质相同性研究包括以下内容:①13CSSNMRꎻ②元素分析测定C㊁H㊁N㊁Cl和Br含量ꎻ③滴定法测定氯含量ꎬ确定质子化程度ꎻ④滴定法测定总可滴定胺ꎻ⑤滴定法测定溴含量ꎻ⑥粒径分布ꎻ⑦溶胀指数ꎻ⑧FT-IR和拉曼光谱ꎻ⑨DSC法测定玻璃化转变温度ꎻ⑩TGAꎮ3.2㊀体外研究3.2.1㊀体外平衡结合研究㊀该项研究是评估生物等效性的关键性研究ꎮ应将受试制剂和RLD在pH6.8条件下采用酸预处理或不处理的方式ꎬ于至少8种不同浓度的总胆盐溶液中进行孵育ꎬ孵育介质总胆盐中应含有甘氨胆酸(GCA)㊁甘氨鹅去氧胆酸(GCDA)和牛磺脱氧胆酸(TDCA)ꎮ总胆盐浓度应分布合理直到达到最大结合ꎮ试验应该在37ħ条件下进行ꎮ每一项结合试验应重复至少12次ꎮ此外应提供数据表明选择的孵育时间长度可使总胆盐产生最大结合ꎮ3.2.2㊀体外动力学结合研究㊀该项研究用以支持关键性的平衡结合研究ꎮ应将不经酸预处理的受试制剂和RLDꎬ于2种不同浓度(0.3mmol L-1和3.0mmol L-1)的总胆盐溶液中孵育至少8个不同的时间长度ꎮ时间长度应分布合理直到达到最大结合ꎮ试验应该在37ħ条件下进行ꎮ每一项结合试验应重复至少12次ꎮ3.2.3㊀水和饮料条件下的体外平衡结合研究㊀在37ħ条件下将样品采用水㊁3种无糖软饮㊁3种弱酸性或中性果汁(葡萄柚汁除外)处理后ꎬ离心弃去上清液ꎬ然后对样品酸预处理或不处理ꎬ进行平衡结合研究ꎬ试验设计与 3.2.1 项下一致ꎮ3.2.4㊀水和饮料条件下的体外动力学结合研究㊀在37ħ条件下将样品采用水㊁3种无糖软饮㊁3种弱酸性或中性果汁(葡萄柚汁除外)处理后ꎬ离心弃去上清液ꎬ进行动力学结合研究ꎬ试验设计与 3.2.2 项下一致ꎮ3.2.5㊀数据分析㊀检测滤液中未结合的胆盐ꎬ间接计算出与盐酸考来维仑结合的胆盐量ꎮ体外平衡结合研究及体外动力学结合研究需要进行数据分析的内容ꎬ以及生物等效性评价指标与碳酸司维拉姆相同(见 2.2.3 项下)ꎮ3.3㊀讨论㊀FDA盐酸考来维仑产品指南在2016年以后增加了API相同性研究要求ꎬ此前仅有体外结合研究要求ꎮ与碳酸司维拉姆类似ꎬ需要与从RLD中提取出来的API进行理化特性对比研究ꎬ除此之外还建议对交联中间体进行研究ꎬ从工艺过程控制方面对API相同性研究提出了更高的要求ꎬ同时也更能保证物质基础的一致性ꎮ盐酸考来维仑混悬剂可以水㊁果汁或无糖软饮送服ꎬ因此体外研究较片剂增加了水㊁3种无糖软饮㊁3种弱酸性或中性果汁(葡萄柚汁除外)前处理条件下的体外平衡结合研究和体外动力学研究ꎮ模拟临床应用条件ꎬ使得体外研究更加充分ꎮ4㊀环硅酸锆钠环硅酸锆钠(结构示意图见图4)用于治疗成人高钾血症ꎮ环硅酸锆钠是一种不可吸收的非聚合物无机粉末ꎬ具有均匀的微孔结构ꎬ优先捕获钾ꎬ置换出氢和钠ꎮ体外试验中ꎬ即便存在钙和镁等其他阳离子ꎬ环硅酸锆钠仍对钾离子具有高度选择性ꎮ环硅酸锆钠可在整个胃肠道中捕获钾离子ꎬ并降低胃肠腔中游离钾的浓度ꎬ从而降低血清钾浓度并促进粪便钾排泄以缓解高钾血症[14]ꎮFDA环硅酸锆钠口服混悬剂生物等效性指南首次发布于2020年ꎬ目前现行有效的版本为2021年8月发布[15]ꎮ推荐的研究内容包括一项API相同性研究ꎬ两项体外研究ꎮ4.1㊀API相同性研究㊀环硅酸锆钠的结构如图4所示ꎮAPI表征应包括范不限于化学组成㊁晶体结构㊁孔径㊁粒径分布㊁FT-IR㊁密度㊁TGA㊁DSC㊁钾交换能力ꎮ仿制药申请人应采用至少3批受试制剂与3批对照药进行背对背比较研究ꎮ分子式:Na~1.5H~0.5ZrSi3O9 2-3H2O图4㊀环硅酸锆钠结构示意图4.2㊀体外研究4.2.1㊀体外平衡结合研究㊀该项研究是评估生物等效性的关键性研究ꎮ应将受试制剂和RLD在pH1.2㊁pH4.5㊁pH6.8条件下ꎬ于至少8种不同浓度的钾盐溶液中进行孵育ꎮ每个研究中应选择合适的钾盐浓度ꎬ以描绘吸附曲线ꎬ包括最大结合(即达到吸附平台)ꎮ试验应该在37ħ条件下进行ꎮ每一项结合试验应重复至少12次ꎮ此外应提供数据表明选择的孵育时间长度能够满足吸附平衡条件ꎮ4.2.2㊀体外动力学结合研究㊀该项研究用以支持关键性的平衡结合研究ꎮ应将受试制剂和RLD在pH1.2㊁pH4.5㊁pH6.8条件下ꎬ于至少3种浓度的钾盐溶液中孵育ꎬ包括平衡结合研究中的最高浓度㊁最低浓度㊁中间浓度(约50%最高浓度)ꎬ每种条件下应该选择8个时间点直至24hꎬ包括达到结合平衡的时间ꎬ以充分描绘结合率曲线ꎮ试验应该在37ħ条件下进行ꎬ并持续轻微振摇ꎮ每一项结合试验应重复至少12次ꎮ4.2.3㊀数据分析㊀检测滤液中未结合的钾盐ꎬ间接计算出与环硅酸锆钠结合的钾盐量ꎮ体外平衡结合研究及体外动力学结合研究需要进行数据分析的内容ꎬ以及生物等效性评价指标与碳酸司维拉姆相同(见 2.2.3 项下)ꎮ4.3㊀讨论㊀环硅酸锆钠通过与胃肠道中的钾结合而局部发挥其作用ꎬ不被全身吸收ꎬ从而使全身毒性的风险最小化或消除ꎮ采用离子阱技术ꎬ与现有高钾血症治疗方案相比ꎬ提高了捕获过量钾离子的能力㊁选择性和速度[16]ꎮ具有较明显的临床优势ꎮ环硅酸锆钠口服混悬剂为2018年FDA批准的新药ꎬ虽然上市时间较短ꎬ但推荐的体外BE研究方法已在多种药物指南中应用ꎬFDA对于此类研究的评价具有较为丰富的经验ꎮ由于数据保护问题ꎬ国内外尚无仿制药上市ꎮ5㊀总结不同于一般小分子化合物ꎬ复杂仿制药需要进行API相同性研究ꎬ只有在满足API一致的前提下进行的BE研究才有意义ꎮFDA硫糖铝产品指南在2019年修订时增加了API相同性要求ꎬ盐酸考来维仑产品指南在2016年修订时增加了API相同性要求ꎬ2022年发布了仿制药API相同性评估指南ꎬFDA对API相同性研究的要求越来越明确ꎮ国内在此类药品研发和评价时也应给予足够重视ꎮ不同复杂仿制药对于API相同性评估的要求不同ꎬ根据复杂程度和结构特点等ꎬ可能需要从工艺路线和过程控制入手ꎬ并采用不同的结构确证分析手段与RLD进行多批次对比研究ꎬ使API的相同性得到保证ꎮ对于胃肠道局部作用的药物ꎬ例如表3列举的11种药物ꎬ与磷酸盐㊁钾盐㊁胆盐等结合形成不溶性复合物随粪便排出以产生治疗效果ꎬ体外结合研究可以反映作用机制ꎬ已成为FDA比较成熟的体外BE研究方法ꎮ本文提及的胃肠道局部作用制剂硫糖铝ꎬ国内仿制药批准时间均约在2005年以前ꎬ推测未采用体外BE研究获得批准ꎮ2020年以后ꎬ国内已有采用体外BE试验获批的碳酸司维拉姆片ꎬ目前仍有多家仿制药在审ꎮ尽管如此ꎬ国内制药企业和监管机构对于此类体外BE研究和评价的经验仍有限ꎬ需要进行更深入的探讨ꎬ具体案例具体分析ꎬ科学地进行更多的探索和实践ꎮ表3㊀FDA推荐采用体外结合研究的胃肠道局部作用药物[17]类别药物名称胃肠道磷酸盐结合剂醋酸钙ꎬ碳酸镧ꎬ枸橼酸铁ꎬ氢氧化铁ꎬ碳酸司维拉姆ꎬ盐酸司维拉姆胃肠道胆盐结合剂考来烯胺ꎬ盐酸考来维仑ꎬ盐酸考来替泊胃肠道钾盐结合剂环硅酸锆钠胃肠道胆盐和蛋白结合剂硫糖铝㊀㊀复杂仿制药的研发面临较大的挑战ꎬ一方面由于仿制药企业缺乏相关经验ꎬ另一方面我国药品监管机构对复杂仿制药的研发缺少激励政策ꎬ且相关的技术要求和法规文件等发布滞后ꎬ使得国产复杂仿制药的可及性处于比较被动的状态ꎮ建议相关部门及时发布具有较大临床价值或专利即将到期等复杂仿制药清单ꎬ并制定和发布与之相应的特定产品指南ꎬ加强行业指导ꎬ体现科学监管理念ꎻ完善注册申请人与药品监管机构的沟通交流机制ꎮ另外ꎬ仿制药企业应结合市场需求制定合理的开发策略ꎬ及时跟进国内外监管机构发布的指南和法规文件ꎬ高质量研发ꎬ满足广大人民群众的用药需求ꎮ参考文献:[1]㊀国家药品监督管理局.国家药监局关于发布化学药品注册分类及申报资料要求的通告(2020年第44号)[EB/OL].(2020-06-29)[2023-05-19].https://www.nmpa.gov.cn/yaopin/ypggtg/ypqtgg/20200630180301525.html.(下转第744页)[2]李大林.重庆市RC区农村药品安全监管存在的问题及对策研究[D].重庆:重庆大学ꎬ2021.[3]施京京.落实 四个最严 保障药品安全 新版«中华人民共和国药品管理法»解读[J].中国质量技术监督ꎬ2019(9):21-22.[4]孙亚平ꎬ张中胜.基层药品安全监管现状及对策[J].首都食品与医药ꎬ2017ꎬ24(8):23.[5]常宝泉.基层医疗机构药品监管现状及建议[J].商业文化ꎬ2021(16):134-135.[6]常远.L市药品安全监管问题研究[D].郑州:郑州大学ꎬ2017.[7]霍增辉.药品管理法修订背景下医疗机构制剂监管创新与完善[J].中国药事ꎬ2020ꎬ34(5):514-519. [8]薛媛ꎬ吴威威ꎬ李翠华ꎬ等.基层门诊部药品储存管理的体会和思考[J].继续医学教育ꎬ2021ꎬ35(8):148-150. [9]沈瑞麟ꎬ邵敏ꎬ范珊珊.从执法实践看新修订«药品管理法»中存在的若干问题[N].中国市场监管报ꎬ2020-07-07(004).[10]黄聪.以非药品冒充药品或以他种药品冒充此种药品ꎬ如何定性处罚[N].中国医药报ꎬ2021-9-6(003). [11]王业.基层药品监管问题及对策研究 以T区药品监管为例[D].长春:吉林大学ꎬ2022.[12]肖珏ꎬ林银银.安徽省桐城市基层医疗机构药事管理质量控制现状分析[J].安徽医药ꎬ2022ꎬ26(9):1892-1896. [13]郭静ꎬ唐媛ꎬ杨志强. 两法 实施在基层药品监管中的实践与思考[J].中国食品药品监管ꎬ2021(8):83-90. [14]顿彬ꎬ补世明ꎬ宋阔魁.药品智慧监管推动政府治理能力现代化的实践探索与思考[J].中国医药导刊ꎬ2022ꎬ24(1):89-93.[15]李旭.盘锦市医疗机构药品使用监管问题研究[D].大连:大连理工大学ꎬ2017.[16]熊井柱.基层医疗机构药品监管现状及对策[J].世界最新医学信息文摘ꎬ2016ꎬ16(51):167.(收稿日期:2022-11-07)(上接第739页)[2]㊀FDA.Section505(j)oftheFederalFoodꎬDrugꎬandCosmeticAct[EB/OL].(2018-03-29)[2023-05-19].https://www.fda.gov/regulatory-information/laws-enforced-fda/federal-food-drug-and-cosmetic-act-fdc-act.[3]李宛潞ꎬ杨悦ꎬ邢花.美国复杂仿制药研发激励政策研究[J].中国新药杂志ꎬ2022ꎬ31(13):1241-1247. [4]李斯文ꎬ杨悦.质量源于设计在美国复杂仿制药监管中的应用[J].中国医药工业杂志ꎬ2022ꎬ52(6):846-854. [5]FDA.SamenessEvaluationsinanANDA ActiveIngre ̄dientsGuidanceforIndustry[EB/OL].(2022-11-08)[2023-04-15].https://www.fda.gov/regulatory-infor ̄mation/search-fda-guidance-documents/sameness-eval ̄uations-anda-active-ingredients.[6]EMA.Equivalencestudiesforthedemonstrationoftherapeuticequivalenceforlocallyappliedꎬlocallyactingproductsinthegastrointestinaltract[EB/OL].(2019-04-30)[2023-04-15].https://www.ema.europa.eu/en/equiva ̄lence-studies-demonstration-therapeutic-equivalence-locally-applied-locally-acting-products.[7]国家食品药品监督管理总局.总局关于发布过度重复药品提示信息的公告(2016年第153号)[EB/OL].(2016-09-14)[2023-06-01].https://www.nmpa.gov.cn/xxgk/ggtg/qtggtg/20160914165601324.html. [8]FDA.DraftGuidanceonSucralfate[EB/OL].(2023-02-16)[2023-05-15].https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_019183.pdf.[9]司延斌ꎬ徐蓓.新型磷结合剂碳酸司维拉姆临床应用[J].药品评价ꎬ2013ꎬ10(24):36-38.[10]FDA.DraftGuidanceonSevelamerCarbonate[EB/OL].(2015-09-18)[2023-05-15].https://www.accessdata.fda.gov/drugsatfda_docs/psg/Sevelamer%20carbonate_oral%20tablet_022127_RV09-15.pdf.[11]刘淑洁ꎬ张丹ꎬ马婧怡ꎬ等.碳酸司维拉姆片体外磷结合生物等效性研究技术要点[J].中国医药导刊ꎬ2021ꎬ23(11):877-880.[12]FDA.LabelsforNDA022362[EB/OL].(2021-10-20) [2023-05-19].https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/022362s029ꎬ021176s049lbl.pdf.[13]FDA.DraftGuidanceonColesevelamHydrochloride[EB/OL].(2021-11-8)[2023-05-20].https://www.ac ̄cessdata.fda.gov/drugsatfda_docs/psg/PSG_022362.pdf. [14]国家药品监督管理局药品审评中心.环硅酸锆钠散说明书[EB/OL].(2021-06-07)[2023-06-01].ttps://www.cde.org.cn/main/xxgk/postmarketpage?acceptidhCODE=b8ac10e8017696af88366d025463197e.[15]FDA.DraftGuidanceonSodiumZirconiumCyclosilicate[EB/OL].(2021-08-20)[2023-05-15].https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_207078.pdf.[16]国家药品监督管理局药品审评中心.环硅酸锆钠散申请上市技术审评报告[EB/OL].(2021-06-07)[2023-06-01].https://www.cde.org.cn/main/xxgk/postmarketpage?acceptidCODE=b8ac10e8017696af88366d025463197e. [17]FDA.Product-SpecificGuidancesforGenericDrugDevel ̄opment[EB/OL].[2023-06-07].https://www.accessdata.fda.gov/scripts/cder/psg/index.cfm.(收稿日期:2023-07-04)。
药品一致性评价审评流程
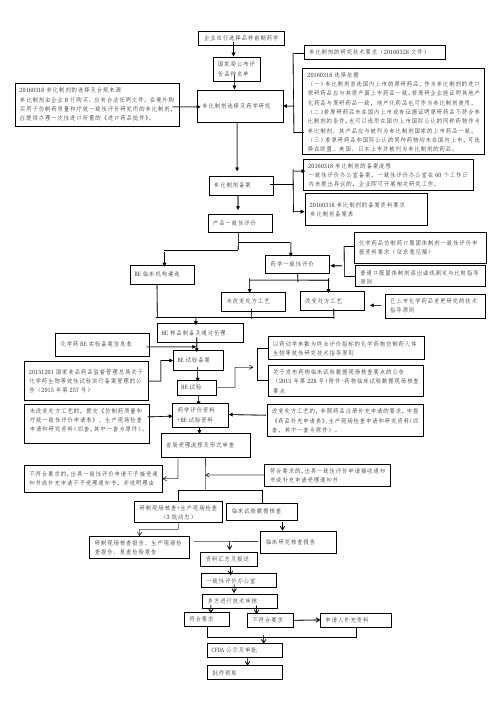
2018年11月,中央全面深化改革委员会第五次会议审议通过了《国家组织药品集中采购试点方案》,同意开展国家组织药品集中采购和使用试点工作。
根据规定,在药品一致性评价的基础上,仿制药才能进入国家组织药品集中采购。
而通过一致性评价的仿制药品种将优先纳入《国家基本药物目录》,未通过一致性评价的仿制药品种将逐步调出目录。
但到底什么是药品一致性评价?通过一致性评价的药品和进口药品的区别在哪里?在此给大家介绍一下。
一、原研进口药原研药是指境内外首个获准上市,且具有完整和充分的安全性、有效性数据作为上市依据的药品。
按照通俗的说法,原研药作为新药上市前,需要进行药物临床前研究和临床试验。
临床前研究包括药物的合成工艺、提取方法、理化性质及纯度、剂型选择、处方筛选、制备工艺、检验方法、质量指标、稳定性、药理、毒理、动物药代动力学研究等。
药物临床试验分为Ⅰ期临床试验、Ⅱ期临床试验、Ⅲ期临床试验、Ⅳ期临床试验以及生物等效性试验。
根据药物特点和研究目的,研究内容包括临床药理学研究、探索性临床试验、确证性临床试验和上市后研究。
原研药在上市之前需要进行大量的研究,上市后还需要定期对药品不良反应的监测数据、临床研究、文献等资料进行评价。
只有提供了充分可靠的研究数据,证明了药品的安全性、有效性和质量可控性,才可上市,并须在上市后持续进行临床验证。
二、国内仿制药仿制药是指与被仿制药具有相同的活性成分、给药途径、剂型、规格和相同治疗作用的药品。
顾名思义,也就是仿原研药品或者国际公认的同品种的药品。
三、国内仿制药一致性评价仿制药一致性评价全称是仿制药质量和疗效一致性评价,既包括质量,也包括疗效,在药品的原辅料、生产工艺、质量检测和疗效等方面均有严格标准。
仿制药应当与参比制剂(原研药品或者国际公认的同种药品)质量和疗效一致,即仿制药须在质量与药效上达到与原研药一致的水平。
换句话说,仿制药必须达到和原研药“管理一致性、中间过程一致性、质量标准一致性等全过程一致”的高标准要求。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
雷珠单抗注射液
脉络膜新生血管导致的视力损害
63
阿柏西普眼内注射溶液
新生血管性年龄相关性黄斑变性
64
注射用A型肉毒毒素
暂时性改善成人中度至重度鱼尾纹
65
盐酸莫西沙星片
轻至中度盆腔炎性疾病
66
多种维生素片(6)
用于维生素B1、维生素B6、维生素B12需求量增加,通过饮食摄入不能满足需求的患者
67
10%脂肪乳(OO)/5.5%氨基酸(15)/葡萄糖(20%)注射液
2018年药审中心审评通过的进口原研药
序号
药品名称
获批时的适应症小结
(具体详见药品说明书)
1
注射用重组人凝血因子Ⅷ
用于成人和儿童A型血友病患者出血事件的控制和预防
2
口服五价重配轮状病毒减毒活疫苗(Vero细胞)
预防6周至32周龄婴儿疫苗相关型别的轮状病毒胃肠炎
3Hale Waihona Puke 九价人乳头瘤病毒疫苗(酿酒酵母)
预防16~26岁女性疫苗相关型别的宫颈癌、癌前病变或不典型病变、及持续性感染。
早期特发性帕金森病
40
利那洛肽胶囊
成人便秘型肠易激综合征
41
拉莫三嗪分散片
癫痫
42
拉考沙胺片
癫痫
43
地诺孕素片
子宫内膜异位症
44
艾考恩丙替片
HIV-1感染
45
依达赛珠单抗注射液
快速逆转达比加群酯(泰毕全®)抗凝效果
46
左炔诺孕酮宫内节育系统(III)
避孕
47
小儿法罗培南钠颗粒
敏感细菌所致的儿童皮肤及皮肤组织感染、淋巴管炎、肺炎等感染性疾病。
4
棕榈帕利哌酮酯注射液
精神分裂症
5
达芦那韦考比司他片
人类免疫缺陷病毒(HIV)感染
6
注射用盐酸美法仑
多发性骨髓瘤
7
注射用盐酸苯达莫司汀
复发难治的惰性B细胞非霍奇金淋巴瘤
8
注射用全氟丁烷微球
超声造影剂,检查肝脏病变。
9
注射用拉布立海
控制儿童白血病、淋巴瘤患者的尿酸水平
10
注射用醋酸地加瑞克
前列腺癌
11
HIV-1感染
54
德拉马尼片
成人耐多药肺结核
55
泊沙康唑肠溶片
预防侵袭性曲霉菌和念珠菌感染
56
艾尔巴韦格拉瑞韦片
成人慢性丙型肝炎
57
司来帕格片
肺动脉高压(PAH,WHO第1组)
58
帕妥珠单抗注射液
乳腺癌
59
甲磺酸仑伐替尼胶囊
肝细胞癌
60
醋酸阿比特龙片
前列腺癌
61
注射用英夫利西单抗
成人中重度活动性溃疡性结肠炎
胃肠道外营养
68
盐酸帕洛诺司琼注射液*
预防化疗引起的恶心、呕吐
69
非布司他片*
痛风患者高尿酸血症的长期治疗
注:“*”是指国内已有仿制品种上市的进口原研药,如盐酸帕洛诺司琼注射液等,不纳入此次的67个进口原研药统计范围内。
依那西普注射液
类风湿关节炎,强直性脊柱炎
12
依库珠单抗注射液
阵发性睡眠性血红蛋白尿症
非典型溶血尿毒症综合征
13
盐酸托莫西汀口服溶液
注意缺陷/多动障碍
14
盐酸度洛西汀肠溶胶囊
治疗抑郁症,焦虑障碍,慢性肌肉骨骼疼痛。
15
盐酸阿来替尼胶囊
晚期非小细胞肺癌
16
乌美溴铵维兰特罗吸入粉雾剂
慢性阻塞性肺病
17
噻托溴铵奥达特罗吸入喷雾剂
慢性阻塞性肺病
18
塞瑞替尼胶囊
晚期非小细胞肺癌
19
普乐沙福注射液
非霍奇金淋巴瘤造血干细胞动员
20
哌柏西利胶囊
晚期乳腺癌
21
帕博利珠单抗注射液
晚期黑色素瘤
22
纳武利尤单抗注射液
晚期非小细胞肺癌
23
来那度胺胶囊
多发性骨髓瘤
24
糠酸氟替卡松维兰特罗吸入粉雾剂
哮喘和慢性阻塞性肺病
25
枸橼酸伊沙佐米胶囊
多发性骨髓瘤
33
依托孕烯炔雌醇阴道环
女性避孕
34
盐酸莫西沙星滴眼液
敏感微生物引起的细菌性结膜炎
35
特立氟胺片
复发型多发性硬化
36
培哚普利氨氯地平片(Ⅲ)
单药治疗不能充分控制的高血压
37
培哚普利氨氯地平片(Ⅱ)
单药治疗不能充分控制的高血压
38
培哚普利氨氯地平片(I)
培哚普利和氨氯地平联合降压治疗的替代
39
罗替高汀贴片
26
格拉司琼透皮贴片
预防化疗引起的恶心和呕吐
27
富马酸喹硫平缓释片
治疗精神分裂症和双相情感障碍的抑郁发作。
28
丁丙诺啡纳洛酮舌下片
用于戒毒
29
奥拉帕利片
复发性上皮性卵巢癌
30
奥达特罗吸入喷雾剂
慢性阻塞性肺病
31
艾美赛珠单抗注射液
存在抑制物的A型血友病的常规预防性治疗
32
依洛尤单抗注射液
纯合子型家族性高胆固醇血症
48
索磷布韦维帕他韦片
成人慢性丙型肝炎
49
丙酚替诺福韦片
慢性乙型肝炎
50
来迪派韦索磷布韦片
慢性丙型肝炎病毒感染
51
拉替拉韦钾干混悬剂
大于4周龄且体重3~20公斤婴幼儿中的HIV-1感染
52
甲苯磺酸艾多沙班片
预防高风险人群卒中和体循环栓塞。治疗深静脉血栓和肺栓塞。预防深静脉血栓和肺栓塞复发。
53
恩曲他滨丙酚替诺福韦片(I)