3 Geneup生物技术有限公司产品目录-Cas9 V2 2015
phi29 DNA Polymerase产品说明书
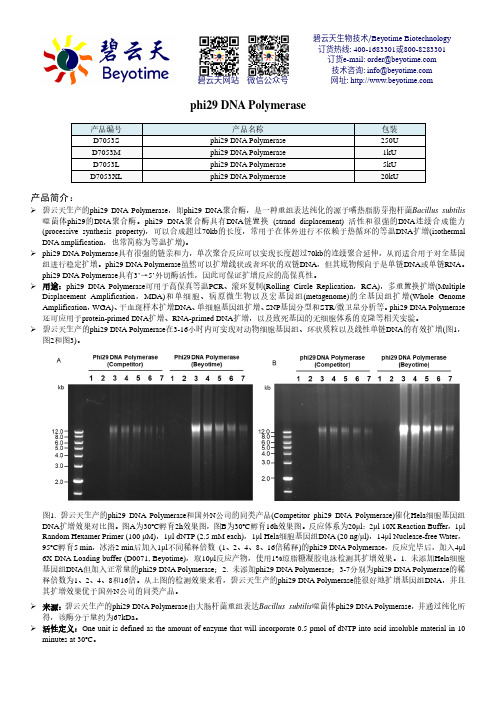
phi29 DNA Polymerase产品编号 产品名称包装 D7053S phi29 DNA Polymerase 250U D7053M phi29 DNA Polymerase 1kU D7053L phi29 DNA Polymerase 5kU D7053XLphi29 DNA Polymerase20kU产品简介:碧云天生产的phi29 DNA Polymerase ,即phi29 DNA 聚合酶,是一种重组表达纯化的源于嗜热脂肪芽孢杆菌Bacillus subtilis 噬菌体phi29的DNA 聚合酶。
phi29 DNA 聚合酶具有DNA 链置换 (strand displacement) 活性和很强的DNA 连续合成能力(processive synthesis property),可以合成超过70kb 的长度,常用于在体外进行不依赖于热循环的等温DNA 扩增(isothermal DNA amplification ,也常简称为等温扩增)。
phi29 DNA Polymerase 具有很强的链亲和力,单次聚合反应可以实现长度超过70kb 的连续聚合延伸,从而适合用于对全基因组进行稳定扩增。
phi29 DNA Polymerase 虽然可以扩增线状或者环状的双链DNA ,但其底物倾向于是单链DNA 或单链RNA 。
phi29 DNA Polymerase 具有3’→5’外切酶活性,因此可保证扩增反应的高保真性。
用途:phi29 DNA Polymerase 可用于高保真等温PCR 、滚环复制(Rolling Circle Replication ,RCA),多重置换扩增(Multiple Displacement Amplification ,MDA)和单细胞、病原微生物以及宏基因组(metagenome)的全基因组扩增(Whole Genome Amplification ,WGA)、干血斑样本扩增DNA 、单细胞基因组扩增、SNP 基因分型和STR/微卫星分析等。
诺禾致源2014产品手册

CONTENTS
建库测序
06 建库测序服务
基因组测序
08 动植物基因组测序 10 基因组特征评估 11 基因组de novo测序 14 泛基因组测序(pan-genome) 16 动植物重测序 17 变异检测(基于全基因组重测序) 19 变异检测(基于简化基因组测序) 21 单个性状定位 24 遗传图谱(基于全基因组重测序) 26 遗传图谱(基于简化基因组测序) 28 群体进化(基于全基因组重测序) 30 群体进化(基于简化基因组测序)
[2] Zhi X Y, Yao J C, Li H W, et al. Genome-wide identification, domain architectures and phylogenetic analysis provide new insights into the early evolution of shikimate pathway in prokaryotes[J]. Molecular phylogenetics and evolution, 2014, 75: 154-164.
[8] Xu X, Dong G X, Hu X S, et al. The genetic basis of white tigers[J]. Current Biology, 2013, 23(11): 1031-1035. [9] Jiang W, Liu Y, Xia E, et al. Prevalent role of gene features in determining evolutionary fates of whole-genome duplication duplicated genes in flowering plants[J]. Plant physiology, 2013, 161(4): 1844-1861. [10] Zhang G, Cowled C, Shi Z, et al. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity[J]. Science, 2013, 339(6118): 456-460. [11] Fan Y, Huang Z Y, Cao C C, et al. Genome of the Chinese tree shrew[J]. Nature communications, 2013, 4: 1426. [12] Wang M Y, Zhao P M, Cheng H Q, et al. The Cotton transcription factor TCP14 functions in auxin-mediated epidermal cell differentiation and elongation[J]. Plant physiology, 2013, 162(3): 1669-1680. [13] Lu S, Zong C, Fan W, et al. Probing meiotic recombination and aneuploidy of single sperm cells by wholegenome sequencing[J]. Science, 2012, 338(6114): 1627-1630. [14] Guo S, Zhang J, Sun H, et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions[J]. Nature genetics, 2013, 45(1): 51-58. [15] Li S, Li R, Li H, et al. SOAPindel: Efficient identification of indels from short paired reads[J]. Genome research, 2013, 23(1): 195-200. [16] Li M, Wu H, Luo Z, et al. An atlas of DNA methylomes in porcine adipose and muscle tissues[J]. Nature communications, 2012, 3: 850. [17] Fan W, Li R. Test driving genome assemblers[J]. Nature biotechnology, 2012, 30(4): 330. [18] Liu C M, Wong T, Wu E, et al. SOAP3: ultra-fast GPU-based parallel alignment tool for short reads[J]. Bioinformatics, 2012, 28(6): 878-879. [19] Hvilsom C, Qian Y, Bataillon T, et al. Extensive X-linked adaptive evolution in central chimpanzees[J]. Proceedings of the National Academy of Sciences, 2012, 109(6): 2054-2059. [20] Zhang G, Fang X, Guo X, et al. The oyster genome reveals stress adaptation and complexity of shell formation[J]. Nature, 2012, 490(7418): 49-54.
3-氧代酰基-[酰基载体蛋白]还原酶
![3-氧代酰基-[酰基载体蛋白]还原酶](https://img.taocdn.com/s3/m/59fd9a74a9956bec0975f46527d3240c8447a16a.png)
3-氧代酰基-[酰基载体蛋白]还原酶,英文名称为3-oxoacyl-[acyl carrier protein] reductase,简称为FabG,是一种参与细菌脂肪酸生物合成途径的酶之一。
该酶能够将3-氧代酰基-[酰基载体蛋白]还原为3-羟基酰基-[酰基载体蛋白],是脂肪酸合成途径中一个重要的还原酶。
在细菌中,这个途径是合成膜磷脂和细胞壁等生命必需物质的关键途径之一。
酰基载体蛋白(acyl carrier protein,ACP)是一个小分子质量为9 kDa的蛋白质,它是脂肪酸生物合成途径中的一个重要载体,能够在不同的酶之间传递脂肪酸中间体,参与脂肪酸的合成过程。
3-氧代酰基-[酰基载体蛋白]还原酶FabG在细菌中具有广泛的分布,是细菌脂肪酸生物合成途径中的一个重要酶。
该酶在抗生素和抗菌药物的研发中也具有重要的作用,是抗菌剂的重要靶点之一。
碧云天 L31600 pLenti-TLR2-sgRNA 产品说明书
pLenti-TLR2-sgRNA产品简介:pLenti-TLR2-sgRNA (TLR2基因敲除质粒)是一种在动物细胞中可以同时表达Cas9、目的基因的sgRNA 和puromycin 抗性基因的质粒。
用于在动物细胞中直接基于CRISPR/Cas9技术敲除目的基因,或者通过包装慢病毒后基于CRISPR/Cas9技术敲除目的基因。
本质粒中sgRNA 的有效性已经通过T7EI 法的验证。
本质粒在细菌中为Amp 抗性,全长约13,000bp 。
本质粒的关键图谱信息请参考图1。
本质粒可直接转染细胞用于目的基因的CRISPR/Cas9敲除,以及通过puromycin 筛选稳定细胞株;也可以与pMDLg 、Rev 及VSV-g 共转HEK293T 细胞进行重组慢病毒(lentivirus)的包装,然后再用于感染细胞或组织并进行目的基因的CRISPR/Cas9敲除。
图1. 表达sgRNA 、Cas9和puromycin 抗性的pLenti-sgRNA 质粒关键图谱信息。
本质粒中的sgRNA 基于碧云天研发的CRISPR/Cas9 sgRNA 快速筛选和验证体系获得,sgRNA 的有效性已经通过T7EI 法验证。
本质粒用于实验时,建议同时选购无任何靶向的对照质粒pLenti-Control-sgRNA (L00011)或靶向GFP 的对照质粒pLenti-GFP-sgRNA (L00013)。
碧云天同时提供基于CRISPR/Cas9技术的TLR2基因敲除的质粒(L31600 pLenti-TLR2-sgRNA)、慢病毒(L31601 TLR2 Knockout Lentivirus)、HEK293T 细胞(L31602 TLR2 Knockout HEK293T Cells)、HEK293T 敲除细胞的RIPA 裂解液(L31603 TLR2 Knockout HEK293T RIPA Lysate)、HEK293T 敲除细胞的Trizol 裂解液(L31604 TLR2 Knockout HEK293T Trizol Lysate)等产品,具体请在碧云天网站查询或在本产品网页点击相应产品。
申科sf9 hcp残留检测试剂盒 (一步酶联免疫吸附法)说明书

SHENTEK®Sf9HCP残留检测试剂盒(一步酶联免疫吸附法)说明书货号:1301310在实验前请完整阅读本说明书,务必重视注意点和常见问题!版本:A/0仅供研究用湖州申科生物技术股份有限公司⏹产品名称通用名:Sf9HCP残留检测试剂盒(一步酶联免疫吸附法)。
⏹包装规格96测试/盒。
⏹预期用途Sf9HCP残留检测试剂盒(一步酶联免疫吸附法)适用于昆虫细胞Sf9来源的宿主蛋白残留定量检测,如基于昆虫细胞-杆状病毒表达系统的生物制品,包括但不限于重组蛋白、疫苗,基因治疗AAV载体等。
该试剂盒仅供研究使用,不可用于临床。
⏹检测原理本试剂盒基于固相酶联免疫吸附法(Enzyme-linked Immunosorbent Assay,ELISA),采用双抗体夹心的方式对样品中残留Sf9HCPs进行定量检测。
该试剂盒内的多克隆抗体是通过裂解sf9细胞所得HCPs作为抗原,免疫绵羊获得血清,进而通过亲合纯化方法得到高质量抗体。
抗体通过目前主流的覆盖率分析法规方法评估其覆盖率水平。
该分析方法通过在预包被抗Sf9HCPs多克隆抗体的酶标板中加入校准品或待测样品、HRP标记的抗Sf9HCPs多克隆抗体进行共孵育;洗涤后,利用加入的TMB底物进行显色反应,最后使用终止液终止酶催化反应。
利用酶标仪在450nm波长下测读吸光度值,其吸光度与校准品和样品中的HCPs浓度成正相关,通过剂量-反应曲线可计算得出样品中Sf9HCPs的浓度。
本试剂盒对实际样品无需进行特殊处理,仅需通过合适的稀释比例进行适用性验证即可直接使用。
本试剂盒检测步骤少,快速,专一性强,性能稳定可靠。
图1检测原理示意图试剂盒组分表1.试剂盒组分组分产品号规格说明Sf9HCP 校准品PNB0112瓶冻干粉。
精确量取500μL复溶液,溶解,静置约5分钟,溶液应该澄清透明,无肉眼可见不溶物。
具体浓度见瓶身标注。
抗Sf9HCP 预包被酶标板PNA0128孔×12条已包被适量的绵羊抗Sf9HCPs多克隆抗体,铝箔袋密封包装,含干燥剂。
CRISPR-Cas9预制稳转株GFP荧光标记肿瘤细胞稳转株Luciferase

选择GeneCopoeia预制稳定细胞系:
CRISPR-Cas9 预制稳转株
稳定插入人源AAVS1或小鼠ROSA26的“Safe Harbor”安全位点,CRISPR-Cas9在表达过程中不影响细胞
的其它正常生物功能。
提供单克隆细胞现货。
Luciferase+GFP 双标签肿瘤细胞稳转株
持续表达高水平荧光素酶及荧光蛋白。
进行活体注射后,短时间即可检测荧光及荧光素报告基因,涵括17种
常用的人源癌细胞系的稳转株现货。
提供混合细胞株。
GFP 荧光标记肿瘤细胞稳转株
持续表达高水平荧光蛋白。
涵括18种常用的人源癌细胞系的稳转株现货。
提供混合细胞株。
细胞结构相关预制稳转株
可持续表达与细胞骨架构成相关的蛋白,可与RFP融合表达。
提供混合细胞株。
TRE 预制稳转株
可持续表达转录应答因子(Transcriptional Response Element )。
提供单克隆细胞现货。
• 出货前,细胞的存活率达到95%以上,活细胞比例高;
• 保证无支原体、细菌、真菌或其它影响细胞生长的毒素、污染;
• 报告基因表达稳定、均匀;
• Cas9预制稳转株利用Safe Harbor整合位点,保持Cas9稳定表达的同时无细胞毒副作用;
• Cas9预制稳转株为单克隆,保证稳定整合,在单一基因背景下持续高水平表达。
基于CRISPR

第38卷第2期2024年3月山东理工大学学报(自然科学版)Journal of Shandong University of Technology(Natural Science Edition)Vol.38No.2Mar.2024收稿日期:20230301基金项目:江苏省高校 青蓝工程 项目(2023);江苏省高职院校青年教师企业实践项目(2021QYSJ063);江苏省精准诊疗药物创制工程研究中心(苏州大学)开放课题(SDGC2239)第一作者:谢钰珍,女,295842442@;通信作者:覃鸿妮,女,qinhn@文章编号:1672-6197(2024)02-0067-06基于CRISPR /Cas9基因编辑技术构建PD -L1-GFP 报告基因HT29细胞株谢钰珍1,覃鸿妮1,2,吴凡1,孙曙光1,孟丽君3(1.苏州工业园区服务外包职业学院生物科技学院,江苏苏州215123;2.江苏省精准诊疗药物创制工程研究中心(苏州大学),江苏苏州215000;3.苏州东岭生物技术有限公司,江苏苏州215123)摘要:利用CRISPR /Cas9基因编辑技术和同源重组技术在PD -L1基因的特定位置敲入绿色荧光蛋白(GFP )序列,构建含有PD -L1-GFP 报告基因的结肠癌细胞(HT29)稳定细胞株㊂根据CRISPR -Cas9靶点设计原则,针对PD -L1基因终止密码子设计两对sgRNA ,退火形成双链后连接至Lenti -V2空载质粒中,转化培养后提取质粒并测序验证㊂对于正确的Lenti -V2-sgRNA 重组质粒,T7E1酶切验证其基因编辑效率㊂根据靶点位置设计左同源臂+GFP +右同源臂序列合成Donor 片段,双酶切后连接至pUC19,重组载体转化扩增后同样进行质粒提取和测序验证㊂将验证成功的Lenti -V2-sgRNA 和pUC19-donor -GFP 共转染HT29细胞,荧光显微镜检测GFP 表达情况,菌落PCR 及基因测序验证GFP 报告基因的靶向插入效果㊂经酶切和测序鉴定,靶向编辑PD -L1的Cas9载体Lenti -V2-gRNA 和含GFP 基因的Donor 质粒pUC19-donor -GFP 构建成功㊂两个重组质粒转染HT29细胞后,显微观察㊁多克隆验证㊁单克隆筛选及鉴定结果显示,GFP 成功转入HT29细胞并进行了表达,经有限稀释法筛选出的单克隆细胞荧光均一,且与对照组相比,阳性细胞克隆的基因组PCR 均检测到特异条带,表明在PD -L1终止密码子前成功插入了GFP 片段,细胞株构建成功㊂通过基因编辑技术成功构建了稳定表达PD -L1-GFP 报告基因的HT29结肠癌细胞株,建立了一个可以直观在细胞上观察PD -L1是否表达以及表达的强弱程度的系统,为后期体内外筛选调控PD -L1的上游新靶点及药物奠定了基础㊂关键词:PD -L1;CRISPR /Cas9;报告基因;GFP 中图分类号:TB532.1;TB553文献标志码:AConstruction of HT29cell line with PD -L1-GFPreport gene by CRISPR /Cas9systemXIE Yuzhen 1,QIN Hongni 1,2,WU Fan 1,SUN Shuguang 1,MENG Lijun 3(1.School of Biotechnology,Suzhou Industrial Park Institute of Services Outsourcing,Suzhou 215123,China;2.Jiangsu Province Engineering Research Center of Precision Diagnostics and Therapeutics Development,Soochow University,Suzhou 215000,China;3.Suzhou Dongling Biotechnology Company Limited,Suzhou 215123,China)Abstract :Using CRISPR /Cas9and homologous recombination technology to tap green fluorescent protein (GFP)sequence at specific position of PD -L1gene,we constructed human colon cancer cell (HT29)㊀stable cell line containing PD-L1-GFP reporter gene.According to the design principle of CRISPR-Cas9target,two pairs of sgRNAs were designed for the stop codon of PD-L1gene.After annealing,the sgRNAs were connected to the Lenti-V2plasmid.The plasmid was extracted and verified by sequencing after amplification of the receptor cells.For the correct Lenti-V2-gRNA recombinant plasmid,the effi-ciency of gene editing was verified by T7E1digestion.According to the target location,Donor fragment was synthesized from left homologous arm+GFP+right homologous arm sequence,which was ligated to pUC19after double enzyme digestion.Plasmid extraction and sequencing were also performed after re-combinant vector transformation and amplification.HT29cells were co-transfected with Lenti-V2-gRNA and pUC19-donor-GFP,and the expression of GFP protein was detected by fluorescence microscopy. Finally,the targeted insertion effect of GFP reporter gene was verified by colony PCR and gene sequen-cing.The Cas9vector Lenti-V2-gRNA and Donor plasmid pUC19-donor-GFP containing PD-L1were successfully constructed by enzyme digestion and sequencing.After the two recombinant plasmids were transfected into HT29cells,the results of microscopic observation,polyclonal verification,monoclonal screening and identification showed that GFP was successfully transferred into HT29cells and expressed, and the monoclonal cells screened by limited dilution method had uniform fluorescence.Moreover,com-pared with the control group,specific bands were detected in the genomic PCR of the clones of positive cells,indicating that the GFP fragment was successfully inserted before the PD-L1stop codon,and the cell line was successfully constructed.HT29colon cancer cell line with stable expression of PD-L1-GFP reporter gene was successfully constructed by gene editing technology,and a system was established to di-rectly observe whether PD-L1is expressed and the degree of expression,which laid a foundation for sub-sequent in vitro and in vivo screening of upstream new targets and drugs regulating PD-L1. Keywords:PD-L1;CRISPR/Cas9;reporter gene;GFP㊀㊀PD1(programmed death-1,程序性死亡受体-1)为PDCD1基因编码的Ⅰ型跨膜糖蛋白,主要表达于活化的免疫细胞表面,如T细胞㊁B细胞㊁NK细胞以及单核细胞等㊂PD1是维持自身耐受性的重要因子,在生理条件下可通过TCR识别抗原,调节外周组织中T细胞的功能,从而应对和清除外源病菌及内源异常细胞㊂PD-L1(程序性死亡配体-1)也称CD274,是PD1的配体之一,是由CD274基因编码的一种细胞表面糖蛋白,多过度表达于肿瘤细胞表面[1]㊂作为重要的负性免疫调节因子,当T细胞表面PD1受体与肿瘤细胞表面表达的PD-L1配体结合后,可向细胞内传递调控信号,抑制T细胞活化与增殖,从而帮助肿瘤细胞逃脱宿主的免疫监视,因此,抑制PD1/PD-L1通路或者通过特异性靶点抑制PD-L1蛋白的表达,可有效增强肿瘤治疗效果㊂近年来的临床研究结果显示,由PD1/PD-L1通路介导下的免疫抑制在非小细胞肺癌㊁胃癌㊁乳腺癌㊁大肠癌等多种恶性肿瘤的发生发展中均具有重要作用,PD-L1在以上各种癌组织的表达明显升高,且PD-L1的阳性表达与肿瘤大小㊁转移情况㊁分化程度及远期生存率存在密切关系[2-3];此外,从诱导肿瘤细胞表面高表达PD-L1的相关因素来看,目前已报道的肿瘤细胞中调控PD-L1表达的相关信号通路主要有Akt3信号通路和MAPK信号通路㊁IFN-γ信号通路和AR信号通路[4],而且这些通路的某些抑制剂已被证明可调节PD-L1㊂例如:TGFβR-1抑制剂LY364947可明显降低TGFβR-1诱导的PD-L1mRNA和蛋白表达[5];突变转录因子FOXP3在PD-L1启动子区的结合位点后,胰腺癌细胞PD-L1的表达明显受到抑制[6];但关于结肠癌肿瘤细胞PD-L1表达的具体调控机制仍需进一步研究㊂CRISPR/Cas9系统是一种新型基因编辑技术,通过sgRNA介导核酸酶Cas9对基因组特定位点进行识别㊁切割,从而实现基因编辑,操作相对简便,基因编辑效率高㊂本文利用CRISPR/Cas9技术,在PD-L1基因终止密码子之前插入绿色荧光蛋白GFP基因,通过构建稳定表达PD-L1-GFP报告基因的结肠癌细胞株HT29,旨在建立一个可直观观察细胞上PD-L1是否表达以及表达强弱的系统,为进一步研究结肠癌细胞中PD-L1表达的可能调控机制提供帮助,为后期体内外筛选调控PD-L1的上游新靶点及药物奠定基础㊂86山东理工大学学报(自然科学版)2024年㊀1㊀材料与方法1.1㊀材料HT29细胞和293T细胞(苏州东岭生物技术有限公司保存),Lenti CRISPR V2质粒载体和pUC19载体(苏州东岭生物技术有限公司),胶回收试剂盒(Thermo),Stbl3感受态大肠杆菌(TransGen Biotech),DMEM培养基和D10培养基(HYclone), T4连接酶(Takara公司),DNA聚合酶(NEB)㊂1.2㊀方法1.2.1㊀sgRNA引物的设计与合成根据NCBI查询的PD-L1基因序列,使用CRISP在线设计工具(/E-CRISP/),根据靶点设计原则,在PD-L1基因终止密码子处设计sgRNA,并在序列正义链与反义链的5 端添加Esp3I(BsmBI)酶切位点,合成的具体序列如下:sgRNA-1:GAGGAGACGTAATCCAGCAT gRNA-1-F:CACCGAGGAGACGTAATCCAGCA gRNA-1-R:AAACATGCTGGATTACGTCTCCTC sgRNA-2:GTCTCCTCCAAATGTGTATC gRNA-2-F:CACCGTCTCCTCCAAATGTGTATC gRNA-2-R:AAACGATACACATTTGGAGGAGA为检测突变是否成功,设置了一对检测引物,扩增产物为包含sgRNA靶点的基因组DNA片段,检测引物具体序列如下:Test-F:TGGGGGACAAGCCATCCCAA Test-R:ATGATTTGCTTGGAGGCTCC1.2.2㊀LentiV2-gRNA重组质粒的构建及鉴定根据所设计序列合成单链sgRNA,经梯度降温PCR退火形成双链sgRNA㊂退火结束后,将得到的双链gRNA连接到已用Esp3I酶切回收后的线性Lenti-V2空载质粒中,连接产物转化Stbl3感受态细胞,37ħ培养过夜后挑取阳性单克隆进行测序验证,测序引物:LKO1-5(GACTATCATATGCTTAC-CGT)㊂针对序列正确的单克隆,提取其对应菌液中的质粒DNA,即可得到正确的Lenti-V2-sgRNA重组质粒㊂1.2.3㊀pUC19-Donor-GFP重组质粒的构建及鉴定设计左同源臂+GFP+右同源臂序列,序列两端添加酶切位点XbaI/BamHI,送公司合成Donor片段,将合成的Donor片段㊁pUC19分别用XhaI和BamHI双酶切后连接㊂取10μL连接产物转化至50μL Stbl3感受态大肠杆菌中,37ħ培养1h 后,均匀涂在含有Amp的LB平板上,过夜培养㊂次日挑取6个克隆于4mL含有Amp的LB培养基中, 37ħ培养16h㊂16h后取1mL菌液抽提质粒,并对质粒进行菌落PCR,判断选取的单克隆中是否含有目的片段㊂将验证正确的质粒送至测序,测序引物:M13-R(CAGGAAACAGCTATGACC),测序正确后冻存菌种㊂1.2.4㊀细胞培养和细胞转染从-80ħ冰箱中取出冻存的HT29细胞,复苏结束后留2mol/L的细胞量在10cm培养皿中培养,隔天进行传代培养㊂培养至第4天时将HT29转移至24孔板中的2孔(一孔转染,一孔阴性对照),每孔0.5ˑ106细胞㊂转染前1~2h更换新鲜培养基(0.9mL/孔),按Lenti-V2-sgRNA(0.6μg)㊁Donor-GFP(0.4μg)㊁1mg/mL PEI(3μL)㊁DMEM(97μL)配制转染体系,室温孵育30min后,将混合液加入到1孔中,另一个孔无须加入,轻轻摇匀后放入培养箱培养(37ħ,5%CO2)㊂1.2.5㊀多克隆效果验证提取转染后的HT29细胞基因组DNA,利用在PD-L1基因组以及GFP上分别设计的上下游引物,对其进行PCR鉴定,以检测其中是否含有在PD-L1位点成功插入GFP片段的目的细胞㊂引物序列: PD-L1-F(TTCAAATTTATCATTTATCA),GFP-R (CCGGACACGCTGAACTTGTG),PCR体系:模板DNA10ng㊁PD-L1-F和GFP-R各1μL,2X PrimeSTAR HS DNA Polymerase MIX10μL,总体积20μL㊂PCR扩增程序:98ħ预变性20s,98ħ变性10s,55ħ退火5s,72ħ延伸35s,共30个循环,68ħ彻底延伸2min㊂扩增的PCR产物经1%琼脂糖凝胶电泳分析㊂1.2.6㊀单克隆细胞筛选转染72h后,观察细胞生长状况并计数,采用有限稀释法,用D10(10%的FBS+DMEM)按每200μL一个细胞稀释到96孔板中,培养24h 后,显微镜观察荧光及细胞状态,并做好标记㊂继续培养,当上一步标记的单克隆细胞密度达到80%后,消化并收集细胞,用基因组DNA抽提试剂盒提取基因组,作为检测引物(TestF㊁R)的扩增模板,最后将扩增产物送至公司测序,以检测PD-L1-GFP 报告基因是否插入成功㊂96第2期㊀㊀㊀㊀㊀谢钰珍,等:基于CRISPR/Cas9基因编辑技术构建PD-L1-GFP报告基因HT29细胞株2㊀结果与分析2.1㊀Lenti -V2-gRNA 重组质粒构建结果重组质粒基因测序结果显示,在酶切位点之间插入的片段位置㊁方向以及序列与预期一致(图1),含有本实验所需要的两条sgRNA 序列,证明LentiV2-gRNA 重组质粒构建成功㊂2.2㊀两条sgRNA 切割效果验证结果为了验证所设计的两条sgRNA 的切割效果,将两种Lenti -V2-gRNA 重组质粒转入293T 细胞后提取基因组DNA,并对其进行T7E1酶切鉴定,结果显示1和2两种sgRNA 都能切下相应大小的条带(图2),证明Lenti -V2-sgR1和Lenti -V2-sgR2都具有切割效果,本实验随机使用其中1条,采用sgRNA2,即Lenti -V2-sgR2㊂图1㊀Lentiv2-gRNA重组质粒测序结果图2㊀T7E1酶切图2.3㊀pUC19-Donor -GFP 重组质粒构建结果以挑选的6个单克隆为模板,经菌落PCR 均能扩增出目的片段(图3),说明菌落中含有目标质粒㊂将阳性质粒进一步送至金唯智测序,从图4可以看出,第一行的序列为测序结果,第二行的序列是所需的模板序列,序列比对完全正确,pUC19-Donor -GFP 质粒构建成功㊂2.4㊀Lenti -V2-sgR2和pUC19-donor -GFP 细胞转染结果㊀㊀将Lenti -V2-sgR2和pUC19-donor -GFP 转染至HT29细胞,72h 后荧光显微镜下观察细胞状态㊂由图5可知,培养72h后可观察到少量细胞表达绿注:1-6为随机挑取的6个单克隆,M 为DNA marker㊂图3㊀pUC19-Donor -GFP 重组质粒转化质粒菌落PCR 结果色荧光,证明GFP 成功转入HT29并进行了表达㊂2.5㊀PD -L1-GFP 报告基因工程细胞株阳性单克隆筛选结果2.5.1㊀多克隆效果验证结果图6为多克隆PCR 鉴定结果,可以发现实验组在200bp 位置有目的条带,而对照组没有,证明GFP 插入到指定位点㊂2.5.2㊀单克隆细胞筛选结果为将上述验证的插入正确GFP 序列位点的细胞挑选出来,将多克隆细胞进行单克隆筛选,图7为荧光显微镜观察结果,可以看到细胞形成了单克隆并且具有均一的荧光,可以进行后续的验证㊂7山东理工大学学报(自然科学版)2024年㊀图4㊀pUC19-Donor重组质粒测序结果(a)细胞图(b)荧光图图5㊀HT29细胞荧光蛋白表达为验证上述挑选的单克隆中所需的序列存在,对其基因组PCR 之后送至金唯智测序,图8显示与正常HT29细胞基因组相比,敲入的实验组所挑的单克隆在终止密码子前插入了GFP 片段,证明细胞系构建成功㊂3㊀讨论PD1表达于活化的免疫细胞表面,如B淋巴细图6㊀多克隆验证琼脂糖凝胶电泳图图7㊀单克隆细胞荧光状态图胞㊁CD4+T 细胞等㊂PD -L1作为PD1的配体之一,在癌组织上可见异常高表达,如肾癌㊁肝癌㊁肺癌㊁乳腺癌及卵巢癌等;但在肿瘤邻近的正常组织中低水平表达,提示它参与肿瘤发生发展,并在削弱抗肿瘤免疫反应中发挥重要作用㊂PD1/PD -L1是负性共刺激分子,当肿瘤细胞表面的PD -L1与免疫细胞表面的PD1结合时,可抑制免疫细胞的增殖与活性甚至诱导其凋亡,使癌细胞成功逃脱免疫杀伤㊂近年来,PD -L1及其受体PD1信号通路一直是肿瘤免疫领域的热门研究对象,针对阻断PD1/PD -L1通路的单克隆抗体已然成为了肿瘤免疫治疗的明星产品,目前FDA 已经批准了五种PD -L1抑制剂㊂作为非特异性免疫治疗产品,PD -L1抑制剂的抗癌效应具有广谱性,且在不同癌症疾病中显示出了很好的治疗[7-11],但由于治疗过程中的原发性和获得性耐药,相当大比例的患者无法从中受益㊂相较于单药疗法,近年来越来越多的研究正向着PD1/PD -L1耐药机制研究以及联合用药靶点的筛选上转移[12]㊂在一些研究报道中,针对不同类型免疫检查点的联合疗法已被证明对几种肿瘤有效㊂例如:采用抗PD -1抑制剂抗体㊁抗CD137激动剂抗体和疫苗治疗的三联疗法可以显著增强胰腺导管腺癌的治疗效果[13-14];联合anti -PD -L1和anti -TIGIT 在临床上对转移性NSCLC 患者非常有效[15];增强ITCH 活17第2期㊀㊀㊀㊀㊀谢钰珍,等:基于CRISPR /Cas9基因编辑技术构建PD -L1-GFP 报告基因HT29细胞株性可促进PD -L1泛素化降解从而降低肿瘤细胞PD -L1表达,化合物AK087与MAPK 抑制剂联用可明显增强MAPK 靶向治疗黑色素瘤的效果[16]等㊂但对于其他大部分肿瘤来说,不同疗法治疗过程中肿瘤表面PD -L1的表达量变化情况及产生治疗抗性的关键机制,仍需进一步研究㊂注:斜体为GFP 片段,下划线为PDL1终止密码子,WT 为正常HT29测序序列,Knock -in clone 为实验组测序序列㊂图8 对照组和实验组序列对比图㊀㊀为了探究结肠癌细胞表面PD -L1表达的更多可能调控机制,本研究利用CRISPR /Cas9系统和同源重组的原理,通过两次转染将GFP 荧光基团成功插入到PD -L1基因终止密码子之前,成功构建了PD -L1-GFP 报告基因HT29细胞株,通过荧光信号直观观察细胞上PD -L1是否表达以及表达的强弱程度,为进一步研究结肠癌细胞中PD -L1表达的可能调控机制提供了材料,并为后期体内外筛选调控PD -L1的上游新靶点及药物奠定了基础㊂参考文献:[1]谢丽叶,付杰军,卢奕,等.PD1/PD -L1激活促进癌症发生㊁发展和转移的研究进展[J].肿瘤防治研究,2017,44(6):423-427.[2]车章洪.PD -1/PD -L1通路在肿瘤的发生发展过程中对T 细胞的活化作用研究进展[J].中国新药杂志,2017,26(16):1913-1917.[3]方宇,王海娟,李征洋,等.PD -L1和A2aR 在大肠癌中的表达及意义[J].河北医药,2021,43(3):345-348,352.[4]丰江舟,杨梦梦,朱彬彬,等.人PD -L1基因启动子克隆及其荧光素酶报告基因载体的构建[J].皖南医学院学报,2016,35(6):524-526.[5]柯佳,李卓伟,李超,等.TGF -β1对结肠癌SW620细胞及其PD -L1表达的影响[C]//中国解剖学会.中国解剖学会2019年年会论文文摘汇编.昆明:解剖学杂志编辑部,2019:182.[6]王秀超.胰腺癌肿瘤细胞PD -L1表达调控机制及联合干预实验研究[D].天津:天津医科大学,2017.[7]李青丽,唐瑶,李富丽,等.抗PD -L1抗体抑制小鼠非小细胞肺癌移植瘤的生长及其机制[J].肿瘤,2020,40(8):531-540.[8]孙晨,镡云辉,耿波,等.PD -1/PD -L1抑制剂在肾癌中的研究进展[J].国际外科学杂志,2020,47(9):639-643.[9]张丽娜,杨艳芳,姜战胜.PD -1/PD -L1抑制剂治疗三阴性乳腺癌的研究进展[J].中国肿瘤生物治疗杂志,2021,28(8):844-849.[10]李一鑫,路丹.PD -1/PD -L1抑制剂治疗晚期卵巢癌的研究进展[J].现代肿瘤医学,2021,29(15):2741-2744.[11]陈茜,周建英,黎扬斯.抗PD -1/PD -L1治疗在晚期难治性肺鳞癌中的疗效和安全性[J].循证医学,2015,15(4):213-216.[12]YUAN Y,ADAM A,ZHAO C,et al.Recent advancements in themechanisms underlying resistance to PD -1/PD -L1blockade immu-notherapy[J].Cancers(BaseⅠ),2021,13(4):663.[13]韩丽炘,黄玉,温娟娟.PD -1/PD -L1抑制剂联合化疗对比免疫单药治疗晚期NSCLC 疗效及安全性的Meta 分析[J].山西大同大学学报(自然科学版),2022,38(4):84-90.[14]汝继轩,吴川林,张占田,等.PD -1/PD -L1免疫检查点抑制剂治疗胰腺癌研究进展[J].中华胰腺病杂志,2021,21(2):143-147.[15]陈丽丽.EGFR 联合PD -L1在可切除或临界可切除胰腺癌中的预后评价作用[C]//第二届中国临床分子诊断大会.第二届中国临床分子诊断大会论文集.成都:出版者不详,2019:73.[16]YANG Z T,WANG Y,LIU S X,et al.Enhancing PD -L1degra-dation by ITCH during MAPK inhibitor therapy suppresses acquiredresistance[J].Cancer Discovery,2022,12(8):1942-1959.(编辑:姚佳良)27山东理工大学学报(自然科学版)2024年㊀。
安捷伦产品目录

15
Real-Time PCR
16
Mx3000P QPCR System
17
Brilliant III Ultra-Fast SYBR Green QPCR and QRT-PCR Reagents
18
Brilliant III Ultra-Fast QPCR and QRT-PCR Reagents
Agilent / STRATAGENE
Agilent website: /genomics
Welgene | Agilent Stratagene
威健股份有限公司 | Stratagene 總代理
Table of Content
Table of Contents
/ XL1-Red Competent Cells SoloPack Gold Supercompetent Cells
/ TK Competent Cells Specialty Cells
/ Classic Cells / Fine Chemicals For Competent Cells
適用於 UNG 去汙染或 bisulphite
sequencing
適用於 TA Cloning
最高敏感性
取代傳統 Taq 的好選擇
-
2
威健股份有限公司 | Stratagene 總代理
PCR Enzyme & Instrument
Agilent SureCycler 8800
市場上領先的 cycling 速度和 sample 體積 10 ~ 100 μL 簡易快速可以選擇 96 well 和 384 well 操作盤 優秀的溫控設備讓各個 well 都能保持溫度的穩定 七吋的高解析度觸控螢幕讓操作上更為簡便 可以透過網路遠端操控儀器及監控儀器 Agilent 專業的技術支援可以幫助您應對各種 PCR 的問題
艾德KRAS试剂盒说明书

【产品名称】通用名称:人类KRAS基因7种突变检测试剂盒(荧光PCR法)英文名称:Human KRAS Gene 7 Mutations Fluorescence Polymerase ChainReaction (PCR) Diagnostic Kit【包装规格】12测试/盒【预期用途】KRAS基因是人体肿瘤中常见的致癌基因。
该基因的突变常见于多种恶性肿瘤,在肺癌患者中的突变率为15~30%,在结直肠癌患者中的突变率为20~50%。
导致KRAS处于激活状态的突变主要位于第12和13密码子上。
KRAS基因突变一般会使肺癌患者对EGFR酪氨酸激酶抑制剂产生耐药,使结直肠癌患者对抗EGFR抗体类药物产生耐药。
但是,2010年10月的最新研究发现第13密码子上的Gly13Asp(G13D)突变亦对抗EGFR抗体类药物有治疗反应性(参见:De Roock. W. JAMA. 2010;304(16):1812-1820)。
因此,KRAS基因突变检测能提高肿瘤临床治疗的针对性,降低治疗费用,节省宝贵的治疗时间。
大部分肿瘤的突变都是体细胞突变,突变细胞往往与野生型细胞混杂在一起,因此所提取的DNA常带有大量野生型DNA,所以对体细胞突变检测需要较高的特异性,而目前广泛使用的直接测序法检测能力有限,不能完全满足临床需要。
本试剂盒用于检测人类KRAS基因的12和13密码子上7种热点体细胞突变(见表1),试剂盒以DNA为检测样本,提供突变状态的定性评估。
辅助临床医生筛选出可受益于肿瘤靶向药物的大肠癌等癌症患者。
该产品用于组织中提取DNA的KRAS基因7种突变的检测,为临床医生对大肠癌或肺癌患者选择肿瘤靶向药物治疗提供参考。
表1 人类KRAS基因的12和13密码子上7种热点体细胞突变突变名称氨基酸变化碱基变化Cosmic ID 公司命名Gly12Asp 甘氨酸到天门冬氨酸GGT>GAT 521 12-2-A Gly12Ala 甘氨酸到丙氨酸GGT>GCT 522 12-2-C Gly12Val 甘氨酸到缬氨酸GGT>GTT 520 12-2-T Gly12Ser 甘氨酸到丝氨酸GGT>AGT 517 12-1-A Gly12Arg 甘氨酸到精氨酸GGT>CGT 518 12-1-C Gly12Cys 甘氨酸到胱氨酸GGT>TGT 516 12-1-T Gly13Asp甘氨酸到天门冬氨酸GGC>GAC 53213-2-A【检测原理】本试剂盒基于实时PCR平台结合了特异引物和双环探针两种技术,检测DNA样品中含有的突变基因。
法医DNA鉴定常用试剂盒

法医DNA鉴定常用试剂盒Applied Biosystems公司(美国):1、Identifiler PCR扩增试剂盒英文名称:AmpF/STR® Identifiler® PCR Amplification Kit产品概述AmpF STR Identifiler PCR扩增试剂盒可以在单次PCR反应中同时扩增15个STR基因座和Amelogenin性别基因。
Identifiler试剂盒中进行复合扩增的四核苷酸位点包括CODIS所必需的13个核心STR位点,即CSF1PO、D3S1358、D5S818、D7S820、D8S1179、D13S317、D16S539、D18S51、D21S11、FGA、TH01、TPOX、和vWA。
获得的数据可以满足ENFSI(欧洲法医学网)与Interpol(国际刑警组织)的要求。
另外两个四核苷酸位点(D2S1338与D19S433)与先前同FSS(英国法庭科学服务机构)联合开发的SGM Plus 扩增试剂盒相一致。
相比之前所有的AmpF STR试剂盒,包含了上述STR位点的Identifiler试剂盒是功能最强大的一个试剂盒。
6-FAM dye: CSF1PO, D7S820, D8S1179, D21S11VIC dye: D2S1338, D3S1358, D13S317, D16S539, TH01NED dye: D18S51, D19S433, TPOX, vWAPET dye: Amelogenin, D5S818, FGA特点:单管同时扩增16个位点,包括15个STR个体识别位点以及一个性别鉴定位点Amelogenin实验流程简单,一个反应管,一次进样,一个泳道分析采用与其他试剂盒完全相同的引物序列,不同试剂盒的结果具有可比性五色荧光技术,在维持扩增产物大小不变的条件下,有效提高了单个泳道分辨的片段数最大扩增产物小于350碱基,扩增容易,峰高均一提供完整的解决方案,由试剂、仪器、软件、技术支持等构成完整的体系2、Yfiler PCR扩增试剂盒英文名称:AmpF/STR® Yfiler® PCR Amplification Kit产品概述AmpF STR Yfiler PCR扩增试剂盒是一种能在单个PCR反应中复合扩增Y染色体上的17个STR 基因座的STR分析试剂盒。
Guide-it Recombinant Cas9 (3

Takara Bio USA, Inc. 1290 Terra Bella Avenue, Mountain View, CA 94043, USA U.S. Technical Support: *******************************United States/Canada Asia Pacific Europe Japan Page 1 of 9Takara Bio USA, Inc. Guide-it™ Recombinant Cas9 (3 µg/µl) User ManualCat. Nos. 632640, 632641 (021121)Table of ContentsI. Introduction (3)A. Summary (3)II. List of Components (3)III. Additional Materials Required (3)A. Electroporation Supplies (3)B. Mammalian Cell Culture Supplies (4)C. General Supplies (4)D. sgRNA Development and Production (4)E. Detection and Characterization of Gene Editing (4)IV. Protocol Overview (5)V. Electroporation Protocol for Neon Transfection System (5)A. Protocol: Preparation of Cells and Media (5)B. Protocol: Preparation of Cas9-sgRNA RNP Complex (6)C. Protocol: Electroporation (6)VI. Electroporation Protocol for 4D-Nucleofector System (7)A. Protocol: Preparation of Cells (7)B. Protocol: Preparation of Cas9-sgRNA RNP Complex (8)C. Protocol: Electroporation (8)VII. References (9)Table of FiguresFigure 1. Protocol overview for Guide-it Recombinant Cas9 (3 µg/µl). (5)I. IntroductionA. SummaryThe CRISPR/Cas9 system has emerged as a powerful tool for gene editing because of its high targetingspecificity, editing efficiency, and ease of use in virtually any organism. CRISPR/Cas9 technologyconsists of two key components that form a complex: Cas9 endonuclease and a single guide RNA(sgRNA) that directs Cas9 to cleave genomic DNA in a sequence-specific manner (Jinek et al. 2012).This RNA-programmable method exploits the error-prone nature of the non-homologous end joiningDNA repair pathway (NHEJ) to generate gene knockouts (via insertion/deletion). The method can also beused to generate knockins via the homology-directed repair (HDR) pathway.CRISPR/Cas9 system components have been delivered successfully into target cells through a variety ofapproaches, including vector-based expression systems, transfection of RNA, and more recently,introduction of Cas9-sgRNA ribonucleoprotein (RNP) complexes. Delivery of Cas9-sgRNA RNPsprovides a fast turnaround for gene-editing experiments while minimizing the likelihood of off-targeteffects compared to vector-based approaches (Sander and Joung 2014), and this approach has beenoptimized for various cell types using microinjection, electroporation, and lipid-mediated transfection(Liang et al. 2015).Guide-it Recombinant Cas9 (3 µg/µl) is a recombinant wild-type Streptococcus pyogenes Cas9 nucleaseexpressed with a C-terminal nuclear-localization signal (NLS)and purified from E. coli for use inCRISPR/Cas9-mediated gene editing experiments. The Cas9 protein solution has been verified to besterile and well-tolerated by mammalian cells when electroporated as a ribonucleoprotein complex (RNP)with a single guide RNA (sgRNA) for knockout experiments, or as an RNP with a donor repair templatefor knockin experiments.II. List of ComponentsGuide-it Recombinant Cas9 (3 µg/µl) (Cat. No. 632641)•100 µg Guide-it Recombinant Cas9 (3 µg/µl)Guide-it Recombinant Cas9 (3 µg/µl) (Cat. No. 632640)• 3 x 100 µg Guide-it Recombinant Cas9 (3 µg/µl)•Store Guide-it Recombinant Cas9 (3 µg/µl) at –70°C.•Avoid repeated freeze/thaw cycles. We recommend preparing aliquots upon initial thawing of Guide-it Recombinant Cas9 (3 µg/µl).III. Additional Materials RequiredThe following reagents/materials are required but not included.A. Electroporation SuppliesUse of this product requires an electroporator, electroporation chamber (typically cuvettes or tips), and anelectroporation buffer that is suitable for your target cells. Here we provide separate guidelines for theNeon Transfection System (Thermo Fisher Scientific, Cat. No. MPK5000) and the 4D-NucleofectorSystem (Lonza, Cat. No. AAF-1002B).B. Mammalian Cell Culture Supplies•Culture medium, supplies, and additives specific to your target cells•Cell culture plates•PBS without Ca2+ or Mg2+•Trypsin/EDTA or equivalent•Humidified incubator (set at 37º C, 5% CO2)C. General Supplies•Single-channel pipettes•Nuclease-free thin-wall PCR tubes or stripsD. sgRNA Development and ProductionCRISPR/Cas9 gene editing requires a custom sgRNA with a user-designed targeting sequence that ishomologous to the target gene or genomic region of interest. Selecting an appropriate DNA sequence at the target region is critical for maximizing the potential for efficient cleavage at the target site and forminimizing the likelihood of non-specific cleavage events. There are several freely available online tools that can be helpful for determining suitable sgRNA target sequences for a given organism and genomic target. For a list of these tools, please refer to:/US/Products/Genome_Editing/CRISPR_Cas9/Resources/Online_tools_for_gui de_RNA_design.NOTE: For many applications, it is advisable to design and test several variant sgRNAs against the same genomic target region.Candidate sgRNAs must ultimately be produced in sufficient quantity for the generation of functionalCas9-sgRNA RNPs. For development and production of user-designed sgRNAs, we recommend either of the following kits:•For constructing and purifying sgRNAs: Guide-it sgRNA In Vitro Transcription Kit (Takara Bio, Cat.No. 632635).•For constructing and purifying sgRNAs, and testing target cleavage efficiencies in vitro: Guide-it Complete sgRNA Screening System (Takara Bio, Cat. No. 632636).E. Detection and Characterization of Gene EditingThese items are recommended for determining the efficiency of gene editing and the nature of the edits:Cat. No. Product Size631443 Guide-it Mutation Detection Kit 100 rxns631448 Guide-it Mutation Detection Kit 25 rxns632611 Guide-it Genotype Confirmation Kit 100 rxns631444 Guide-it Indel Identification Kit 10 rxnsIV. Protocol OverviewPlease read each relevant protocol completely before starting. Successful results depend on understanding and performing the following steps correctly.Figure 1. Protocol overview for Guide-it Recombinant Cas9 (3 µg/µl).V. Electroporation Protocol for Neon Transfection SystemHere we provide protocols for performing knockout and knockin experiments in hiPS cells and CD34-positive stem cells using the Neon Transfection System. While these protocols may serve as a helpful starting point forelectroporation of other cell types as well, further optimization will be required. Please refer to the Neon Transfection System User Manual and manufacturer’s website for detailed operating instructions for the Neon Transfection System.A. Protocol: Preparation of Cells and MediaCultured target cells are harvested, washed, and resuspended in the appropriate buffer.1.Prepare a sufficient number of fresh cells for your experiment.NOTE: Each electroporation requires 1 x 105 cells. However, due to the potential variation of pipetteand tip volumes, we recommend preparing 1.5X the necessary volume of cell suspension (i.e., 1.5 x 105cells) for electroporation with a 10-µl Neon Tip to ensure that there is sufficient volume.2.For hiPS cells (adherent cells), continue to Step3. For CD34-positive stem cells (suspension cells),skip to Step 5.3.Aspirate the medium, wash the cell layer once with PBS (without Ca2+ and Mg2+), and dissociate thecells using TrypLE Select Enzyme (1X) (Thermo Fisher Scientific, Cat. No. 12563011).4.Harvest the cells in growth medium.5.Take an aliquot of the cell suspension and measure the cell density using your preferred method.6.Harvest the cells by centrifugation at 400g for 5 min in a 15-ml conical tube.7.Wash the cells once with PBS (without Ca2+ and Mg2+), and then resuspend hiPS cells in Buffer R andCD34-positive stem cells in Buffer T (included with Neon kits) at a concentration of 2 x 107 cells/ml(i.e., 1.5 x 105 cells in 7.5 µl).NOTE: Use Resuspension Buffer R for established adherent and suspension cells as well as primaryadherent cells, and use Resuspension Buffer T for primary blood-derived suspension cells.8.Keep the cell suspension on ice until use.B. Protocol: Preparation of Cas9-sgRNA RNP ComplexCas9 and sgRNA components are combined to form RNP complexes for electroporation.1.Thaw Guide-it Recombinant Cas9 (3 µg/µl) and sgRNA solutions at room temperature.NOTE: We recommend preparing aliquots upon initial thawing of Guide-it Recombinant Cas9(3 µg/µl) to avoid repeated freeze/thaw cycles.bine the following components in a 200-µl PCR tube to mix the Cas9 protein and sgRNA at a 5:1mass ratio. The molar ratio of Cas9 protein to sgRNA will be approximately 1:1 in this mixture, andthe total volume will be 7.5 µl. Be sure to use the same buffer that was used to resuspend the cells.NOTE: The reaction volume indicated below is 1.5X the required volume.Per reaction:0.45 μl* sgRNA (e.g., 1 µg/µl)0.75 μl Guide-it Recombinant Cas9 (3 µg/µl)6.3 μl* Resuspension Buffer R or T7.5 μl Total volume*The added volume of sgRNA will vary depending on sgRNA concentration, and the added volume ofResuspension Buffer should be adjusted such that the total reaction volume is 7.5 µl. The volumesindicated above are based on a sgRNA concentration of 1 µg/µl.NOTE: Make a master mix if you are performing multiple electroporations.NOTE: The optimal amount of RNP complex may vary for different cell types.NOTE: To maximize electroporation efficiency, the combined volume of the Cas9 and sgRNA solutionsshould be ≤20% of the total volume of the Cas9-sgRNA RNP complex reaction (e.g., for the 7.5-µlreaction specified above, the combined volume of the sgRNA and Cas9 solutions should be ≤1.5 µl).NOTE: If you plan to use donor DNA to induce HDR-mediated knockin, add the DNA after thesubsequent incubation step (Step 3). We recommend using ≤1 µg of DNA for knockin experiments.Adjust the volume of Resuspension Buffer R or T included in the reaction such that the final volumeupon addition of donor DNA is 7.5 µl.3.Mix the reaction well by gently pipetting up and down. Incubate using a thermal cycler preheated to37°C with the following program:37°C 5 min4°C hold4.OPTIONAL: Add donor DNA and keep on ice until use.C. Protocol: ElectroporationCas9-sgRNA RNPs are electroporated into target cells.1.Fill the Neon Tube with 3 ml of Buffer E (included with Neon kits) and insert the Neon Tube into theNeon Pipette Station.ing the touchscreen on the Neon system, set up the electroporation parameters as follows:Pulse voltage / Pulse width / Pulse number = 1100 v / 20 ms / 2 pulsesNOTE: We have used these parameters successfully for hiPS cells and CD34-positive stem cellsusing the Neon Transfection System. Optimization of electroporation parameters will be required fordifferent target cell types. Suggested parameters for different cell types are included in thesupplementary material for (Liang et al. 2015).3.Gently resuspend the cells by tapping, and transfer 7.5 µl of the cell suspension into the PCR tubecontaining the 7.5 µl of Cas9-sgRNA RNP complex solution.4.Mix well by gently pipetting up and down.5.Insert the Neon Pipette into the Neon Tip and confirm that the pipette and tip are tightly connected.ing the Neon Pipette, aspirate the mixture slowly into the Neon Tip.NOTE: Avoid any air bubbles in the tip. If you notice air bubbles, place the sample back into thePCR tube and aspirate again into the tip without any air bubbles.7.Insert the Neon Pipette into the Neon Tube placed in the Neon Pipette Station and run the program.8.Remove the pipette very carefully and transfer the cells into a cell culture plate with pre-warmedmedium.NOTE: Use an appropriate well plate for your target-cell type. We had success using 24 and 48-wellplates for CD34-positive stem cells and hiPS cells, respectively. hiPS cells typically require greaterconfluence than regular adherent cells.9.Shake the plate appropriately to disperse the cells and incubate at 37°C in a humidified incubator with5% CO2 until the next necessary procedure.VI. Electroporation Protocol for 4D-Nucleofector SystemHere we provide protocols for performing knockout and knockin experiments in Jurkat and CD34-positive stem cells using the 4D-Nucleofector System with 16-well Nucleocuvette Strips. While these protocols may serve as a helpful starting point for electroporation of other cell types as well, further optimization will be required. Please refer to the 4D-Nucleofector System User Manual and manufacturer’s website for more detailed information.A. Protocol: Preparation of CellsCultured target cells are harvested, washed, and resuspended in the appropriate solution.1.Prepare a sufficient number of fresh cells for your experiment.NOTE: Each electroporation requires 2 x 105 cells.2.Take an aliquot of the cell suspension and measure the cell density using your preferred method.3.Harvest the cells by centrifugation at 400g for 5 min in a 15-ml conical tube.4.Wash once with PBS (without Ca2+ and Mg2+), and then resuspend Jurkat cells in SE NucleofectorSolution (with supplement) and CD34-positive stem cells in P3 Nucleofector Solution (withsupplement) at a concentration of 1 x 107 cells/ml (i.e., 2 x 105 cells in 20 µl).NOTE: Please refer to the 4D-Nucleofector System User Manual and manufacturer’s website formore information about working with other cell types.NOTE: 20 µl of cell suspension will be needed per well of the Nucleocuvette Strip.5.Keep the cell suspension on ice until use.B. Protocol: Preparation of Cas9-sgRNA RNP ComplexCas9 and sgRNA components are combined to form RNP complexes for electroporation.1.Thaw Guide-it Recombinant Cas9 (3 µg/µl) and sgRNA solutions at room temperature.NOTE: We recommend preparing aliquots upon initial thawing of Guide-it Recombinant Cas9(3 µg/µl) to avoid repeated freeze/thaw cycles.bine the following components in a 200-µl PCR tube to mix the Cas9 protein and sgRNA at a 5:1mass ratio:Per reaction (e.g. 5 µl):2μl* sgRNA (e.g., 1 µg/µl)3.3 μl Guide-it Recombinant Cas9 (3 µg/µl)5.3 μl Total volume*The added volume of sgRNA will vary depending on sgRNA concentration. The volume indicated aboveis based on an sgRNA concentration of 1 µg/µl.NOTE: Make a master mix if you are performing multiple electroporations.NOTE: The RNP volume required for efficient transfection needs to be optimized for different celltypes. Usually ≤10 µl of the RNP solution will be tested for electroporation in each well of the 16-well Nucleocuvette Strip.3.Mix the reaction well by gently pipetting up and down. Incubate using a thermal cycler preheated to37°C with the following program:37°C 5 min4°C HoldC. Protocol: ElectroporationCas9-sgRNA RNPs are electroporated into target cells.bel wells of Nucleocuvette Strips to be used for electroporation.bine 20 µl of cell suspension with 5 µl Cas9-sgRNA RNP complex solution in each well of theNucleocuvette Strip, and mix well by gently pipetting up and down.NOTE: If you plan to use donor DNA to induce HDR-mediated knockin, add the donor DNA at thisstep.3.Select the program CL-120 for Jurkat cells or the program D0-100 for CD34-positive stem cells.4.Insert the Nucleocuvette Strip into the Nucleofector machine and run the program.5.Add 80 µl of pre-warmed medium to each cuvette and allow electroporated cells to recover for 12min post-transfection at room temperature.6.Gently collect the cells along with the media from each well and transfer to individual wells of a 48-well plate containing pre-warmed medium.7.Shake the plate appropriately to disperse the cells and incubate at 37°C in a humidified incubator with5% CO2 until the next necessary procedure.VII. References1.Jinek, M., Chylinsky, K., Fonfara, I., Hauer, M., Doudna, J.A., & Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337(6096), 816–21 (2012).2.Liang, X. et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J.Biotechnol.208, 44–53 (2015).3.Sander, J.D. & Joung, J.K. CRISPR-Cas9 systems for genomic editing, regulation and targeting. Nat.Biotechnol. 32, 347-55 (2014).Contact UsCustomer Service/Ordering Technical Supporttel: 800.662.2566 (toll-free) tel: 800.662.2566 (toll-free)fax: 800.424.1350 (toll-free) fax: 800.424.1350 (toll-free)web: web: e-mail: **********************e-mail: ********************Notice to PurchaserOur products are to be used for Research Use Only. They may not be used for any other purpose, including, but not limited to, use in humans, therapeutic or diagnostic use, or commercial use of any kind. Our products may not be transferred to third parties, resold, modified for resale, or used to manufacture commercial products or to provide a service to third parties without our prior written approval.Your use of this product is also subject to compliance with any applicable licensing requirements described on the product’s web page at . It is your responsibility to review, understand and adhere to any restrictions imposed by such statements© 2021 Takara Bio Inc. All Rights Reserved.All trademarks are the property of Takara Bio Inc. or its affiliate(s) in the U.S. and/or other countries or their respective owners. Certain trademarks may not be registered in all jurisdictions. Additional product, intellectual property, and restricted use information is available at .This document has been reviewed and approved by the Quality Department.。
凯杰digene HC2 HPV检测系统产品应用及售后服务

凯杰digene HC2 HPV DNA检测系统产品用途及售后服务旋转混合器DML3000基因杂交信号放大仪自动恒温加热器digene® HC2 HPV DNA 检测试剂盒凯杰企业管理(上海)有限公司1产品用途1.1产品原理基于杂交捕获二代技术(Hybrid Capture® 2,简称HC2),采用专利全长8,000 个碱基对的RNA混合鸡尾酒探针,结合基因杂交、抗体捕获和化学发光信号放大方法,定性且定量地检测世界卫生组织(WHO)国际癌症研究机构(IARC)最新(迄今)公布的13 种高危型HPV:16、18、31、33、35、39、45、51、52、56、58、59 和68 型。
*HC2检测是目前临床HPV DNA检测领域的金标准,也是子宫颈癌筛查的首选方法。
1.1.1HC2技术原理A.样本DNA双链被释放并分解为DNA单链;B.HC2专利全长8000碱基对的RNA探针与DNA单链结合为RNA-DNA杂交体;C.固定在微孔壁的特异性抗体(一抗)捕获RNA-DNA杂交体;D.携带碱性磷酸酶的特异性抗体(二抗)与RNA-DNA杂交体结合;E.基因信号放大仪检测由碱性磷酸酶催化底物释放的化学发光信号。
1.2临床应用1.2.1子宫颈癌筛查A.高度的阴性预测值,安全地将子宫颈癌筛查间隔延长至3 年,由此降低的检查成本远远高于因采用HPV 检测所增加的费用;B.发现高危型HPV持续阳性而细胞学涂片未见异常的妇女,因这一人群中有20–25%会在四年内发展为CIN III或浸润癌;C.找出高危型阳性的妇女,浓缩高危人群,便于进行有效监控,早期发现宫颈病变。
1.2.2ASCUS 的分流与管理A.把潜在患子宫颈癌风险妇女从低风险妇女中分离出来,合理指导阴道镜的使用,提高诊断率,降低漏诊风险;B.相比复查细胞学,HC2 HPV检测提供了更快捷的结果,准确有效地把HPV 阴性的低风险妇女重新放回常规筛查人群中,减少过度诊断和过度治疗;C.作为细胞病理学实验室质量控制的有效手段,通过HC2 HPV 辅助检测,从病因学角度进行分子水平检测来监测细胞学形态结果。
CRISPR 产品介绍

CRISPR/Cas9相关产品介绍简介CRISPR-Cas9系统是基于原核生物适应性免疫机制改造的基因编辑技术。
其包含两个模块,一是识别特异DNA序列的gRNA及其骨架,二是结合和编辑DNA的蛋白质Cas9。
自被发明以来,该技术快速发展,目前CRISPR-Cas9及其变体已经被广泛应用于活细胞内核酸的编辑、检测、标记、成像等各个领域。
由于其在不同物种中所展示出的高效易用的特点,该技术已经革新了基础生命科学和生物医学的研究方式。
该技术的发明人也被授予了2020年诺贝尔化学奖。
如今CRISPR-Cas系统仍在蓬勃发展,无论在基础科学研究领域还是在基因治疗、细胞治疗等临床应用领域都具有广泛的前景和深厚的潜力。
产品及服务pSpCas9(BB)-2A-EGFP/PURO 瞬时转染,低转染效率Lentivirual vector-mediated spCas9 system 整合基因组,有脱靶潜在风险Adenoviral vector-mediated spCas9 system 非整合,高效细胞KO体系AAV-saCas9 在体基因编辑利器Knockout / Knockin plasmid and virus construction单克隆敲除细胞株(编码基因、非编码基因)组织特异性表达Cas9的各种病毒应用实例汉恒合作发表的基因编辑论文:AAV-SaCas9在肌肉组织中在体编辑目的基因。
中国药科大学客户使用汉恒构建的pHBcas9/ gRNA puro vector成功建立了SCAP基因敲除的HL-7702细胞株。
温州医科大学附属第二医院客户使用汉恒生物生产的CRISPR-Cas9慢病毒成功建立了NSUN2敲除的BGC-823细胞株。
武汉华中农业大学生物医学中心兽医学院客户使用汉恒生产的CRISPR-Cas9腺病毒通过尾静脉注射感染小鼠肝脏,成功对目的基因进行了编辑。
汉恒CRISPR-Cas9产品客户发表文献节选Anterior cingulate cortex dysfunction underlies social deficits in Shank3 mutant mice杂志:Nature NeuroscienceIF:21.126 单位:西安第四军医大学基础医学院神经生物学系SLE non-coding genetic risk variant determines the epigenetic dysfunction of an immune cell specific enhancer that controls disease-critical microRNA expression杂志:Nature communicationsIF:14.919 单位:上海交通大学医学院附属仁济医院上海风湿研究所Discovery of a potent SCAP degrader that ameliorates HFD-induced obesity, hyperlipidemia and insulin resistance via an autophagyindependent lysosomal pathway 杂志:AutophagyIF:11.059 单位:中国药科大学Prevention of Muscle Wasting by CRISPR/Cas9-mediated Disruption of Myostatin In Vivo杂志:MOLECULAR THERAPYIF:8.986 单位:中科院营养所/汉恒研究中心NSUN2 modified by SUMO-2/3 promotes gastric cancer progression and regulates mRNA m5C methylation杂志:Cell death & diseaseIF:8.469单位:温州医科大学附属第二医院Deletion of miR-126a Promotes Hepatic Aging and Inflammation in a Mouse Model of Cholestasis杂志:MOLECULAR THERAPY-NUCLEIC ACIDSIF:5.66 武汉华中农业大学生物医学中心兽医学院。
CRISPR-cas9 试剂盒说明书

V2.0
北京英茂盛业生物科技有限公司 北京市昌平区沙河镇青年创业大厦 B‐916 Tel:010‐62495135 Emai:order@ Web site:
产品内容
Cas9 基因敲除试剂盒 货号 CR0001 内容 pCas9/gRNA1 载体 pTYNE 验证载体 pCas9‐P 阳性对照载体 pCas9‐N 阴性对照载体
靶点 2
AATGGGGAGTGTAAAGAGGCACTAGCAAAGTCCGAGATGAATGTGAATATGAAGTATCAGCTTCCCAACTTCACCGCGGA
AACACCCATCCAGAATGTCATTCTACATGAGCATCACATTTTCCTTGGTGCCACTAACTACATTTATGTTTTAAATGAGG
4)Cas9Nicknase 需要挑选成对的靶点。我们一般在正义链和反义链上分别挑选相距 20‐30bp 的靶点配对。 多对靶点的敲除效率常有较大差异。由于基因敲除实验时间长,在正式对目的细胞进行敲除前对靶点进行 验证和挑选非常必要。
1.2 插入片段设计
干扰靶点序列通过 EcoRV 位点插入 pCas9/gRNA1 或 pCas9/gRNA3 载体中。
2 北京英茂盛业生物科技有限公司 Tel: 010‐62495135 Email: order@
用 pCas9/gRNA1 或 pCas9/gRNA3 载体进行基因敲除流程
产品说明书
设计基因敲除靶点
本
试
构建pCas9/gRNA载体
剂
盒
中
包
含
5 北京英茂盛业生物科技有限公司 Tel: 010‐62495135 Email: order@
产品说明书
1 基因敲除靶点和寡核苷酸设计
CRISPR-dCas9系统简介

CRISPR-dCas9系统简介dCas9蛋⽩是Cas9蛋⽩的突变体,即Cas9内切酶的RuvC1和 HNH两个核酸酶活性区域同时发⽣突变。
因此,dCas9蛋⽩的内切酶活性全部消失,只保留由gRNA引导进⼊基因组的能⼒。
CRISPR-dCas9系统提供了⼀个研究定点转录调控的平台。
在这个平台中,dCas9主要是与其他效应蛋⽩融合(如GFP、转录因⼦、组蛋⽩修饰等),进⾏基因调控,基因组成像,染⾊质或DNA修饰以及染⾊质免疫沉淀等。
Figure 8 :The Cas9 null mutant has lostboth of its DNA cleavage domains while still retaining its ability to targetgenomic loci through gRNA:DNA interactions. By fusing effector domains to theCas9 null mutant, it is possible to activate gene expression (i.e. V964 orNF-kB), repress gene expression (i.e. KRBA domain), visualize genomic loci(i.e. EGFP), perform chromatin remodelling (i.e. TET proteins), and simplifychromatin immunopercipitation (through an antibody epitope tag)⼀、转录调控系统2013年,齐等⼈将来⾃化脓性链球菌的Cas9的核酸酶结构域(在HNH结构域中产⽣H840A突变和在RuvC结构域中产⽣D10A突变)进⾏突变,以产⽣核酸酶缺陷的“dCas9”。
这种“钝化”和“死亡”Cas9失去切割DNA的功能,但在gRNA的指导下仍能以相同的精确度靶向和结合DNA。
CRISPR Cas9技术说明书

What is CRISPR/Cas9?Melody Redman,1Andrew King,2Caroline Watson,3David King41HYMS Centre for Education Development(CED),Hull York Medical School,University of York,York,UK2Molecular Haematology Unit, MRC Weatherall Institute of Molecular Medicine,University of Oxford,John Radcliffe Hospital,Oxford,UK3Department of Haematology, Oxford University NHS Foundation Trust,Churchill Hospital,Oxford,UK4Academic Unit of Child Health, Sheffield Children’s Hospital, Sheffield,UK Correspondence toDr David King,Academic Unit of Child Health,Sheffield Children’s Hospital,Western Bank, Sheffield S102TH,UK;*********************.uk Received5January2016 Revised18February2016 Accepted19February2016INTRODUCTIONClustered regularly interspaced palin-dromic repeats(CRISPR)/Cas9is agene-editing technology causing a majorupheaval in biomedical research.It makesit possible to correct errors in the genomeand turn on or off genes in cells and organ-isms quickly,cheaply and with relativeease.It has a number of laboratory applica-tions including rapid generation of cellularand animal models,functional genomicscreens and live imaging of the cellulargenome.1It has already been demonstratedthat it can be used to repair defective DNAin mice curing them of genetic disorders,2and it has been reported that humanembryos can be similarly modified.3Otherpotential clinical applications include genetherapy,treating infectious diseases such asHIV and engineering autologous patientmaterial to treat cancer and other diseases.In this review we will give an overview ofCRISPR/Cas9with an emphasis on how itmay impact on the specialty of paediatrics.Although it is likely to have a significanteffect on paediatrics through its impact inthe laboratory,here we will concentrate onits potential clinical applications.W e willalso describe some of the difficulties andethical controversies associated with thisnovel technology.OVERVIEW OF CRISPR/CAS9CRISPR/Cas9is a gene-editing technologywhich involves two essential components:a guide RNA to match a desired targetgene,and Cas9(CRISPR-associatedprotein9)—an endonuclease which causesa double-stranded DNA break,allowingmodifications to the genome(seefigure1).POTENTIAL CLINICAL USESCorrection of genetic disordersOne of the most exciting applications ofCRISPR/Cas9is its potential use to treatgenetic disorders caused by single genemutations.Examples of such diseasesinclude cystic fibrosis(CF),Duchenne’smuscular dystrophy(DMD)and haemo-globinopathies.The approach so far hascurrently only been validated in preclin-ical models,but there is hope it can soonbe translated to clinical practice.Schwank et al used CRISPR/Cas9toinvestigate the treatment of ingadult intestinal stem cells obtained fromtwo patients with CF,they successfullycorrected the most common mutationcausing CF in intestinal organoids.Theydemonstrated that once the mutation hadbeen corrected,the function of the CFtransmembrane conductor receptor(CFTR)was restored.4Another disease in which CRISPR/Cas9has been investigated is DMD.T abebordbaret al recently used adeno-associated virus(AA V)delivery of CRISPR/Cas9endonu-cleases to recover dystrophin expression ina mouse model of DMD,by deletion of theexon containing the original mutation.Thisproduces a truncated,but still functionalprotein.T reated mice were shown to par-tially recover muscle functional deficien-cies.5Significantly,it was demonstrated thatthe dystrophin gene was edited in musclestem cells which replenish mature muscletissue.This is important to ensure anytherapeutic effects of CRISPR/Cas9do notfade over time.T wo similar studies havedescribed using the CRISPR/Cas9system invivo to increase expression of the dys-trophin gene and improve muscle functionin mouse models of DMD.67Other studieshave used CRISPR/Cas9to target duplica-tion of exons in the human dystrophin genein vitro and have shown that this approachcan lead to production of full-length dys-trophin in the myotubules of an individualwith DMD.8CRISPR/Cas9could also be used totreat haemoglobinopathies.Canver et al9recently showed BCL11A enhancer dis-ruption by CRISPR/Cas9could inducefetal haemoglobin in both mice andprimary human erythroblast cells.In thefuture such an approach could allow fetalhaemoglobin to be expressed in patientswith abnormal adult haemoglobin.Thiswould represent a novel therapeutic strat-egy in patients with diseases such assickle cell disease orthalassaemias.RESEARCH IN PRACTICE Redman M,et al.Arch Dis Child Educ Pract Ed2016;0:1–3.doi:10.1136/archdischild-2016-3104591 Education & Practice Online First, published on April 8, 2016 as 10.1136/archdischild-2016-310459 Copyright Article author (or their employer) 2016. Produced by BMJ Publishing Group Ltd under licence. copyright. on December 25, 2023 by guest. Protected by / Arch Dis Child Educ Pract Ed: first published as 10.1136/archdischild-2016-310459 on 8 April 2016. Downloaded fromKnock-in of a fully functional β-globin gene is much more challenging,which is the reason for this some-what unusual approach.Treatment of HIVAnother potential clinical application of CRISPR/Cas9is to treat infectious diseases,such as HIV .Although antiretroviral therapy provides an effective treatment for HIV ,no cure currently exists due to permanent integration of the virus into the host genome.Hu et al showed the CRISPR/Cas9system could be used to target HIV-1genome activity .This inactivated HIV gene expression and replication in a variety of cells which can be latently infected with HIV ,without any toxic effects.Furthermore,cells could also be immu-nised against HIV-1infection.This is a potential thera-peutic advance in overcoming the current problem of how to eliminate HIV from infected individuals.After further refinement,the authors suggest their findings may enable gene therapies or transplantation of genet-ically altered bone marrow stem cells or inducible pluripotent stem cells to eradicate HIV infection.10Engineering somatic cells ex vivo to treat malignancy or other diseasesThere has been increasing interest in the possibility of using CRISPR/Cas9to modify patient-derived T-cells and stem/progenitor cells which can then be reintro-duced into patients to treat disease.This approach may overcome some of the issues associated with how to efficiently deliver gene editing to the right cells.T-cell genome engineering has already shown success in treating haematological malignancies and has the potential to treat solid cancers,primary immune deficiencies and autoimmune diseases.Genetic manipulation of T-cells has previously been inefficient.However,Schumann et al recently reported a more effective approach in human CD4+T-cells based on the CRISPR/Cas9system.Their tech-nique allowed experimental and therapeutic knock-out and knock-in editing of the genome in primary human T-cells.They demonstrated T-cells could be manipulated to prevent expression of the protein PD-1,which other work has shown may allow T-cells to target solid cancers.11There is also interest in using CRISPR/Cas9-mediated genome editing in pluripotent stem cells or primary somatic stem cells to treat disease.For example Xie et al 12showed the mutation causing β-thalassaemia could be corrected in human induced pluripotent stem cells ex vivo.They suggest that in the future such an approach could provide a source of cells for bone marrow transplantation to treat β-thalassaemia and other similar monogenic diseases.LIMITATIONSA number of challenges remain before the potential of CRISPR/Cas9can be translated to effective treatments at the bedside.A particular issue is how to deliver gene editing to the right cells,especially if the treat-ment is to be delivered in vivo.T o safely deliver Cas9-nuclease encoding genes and guide RNAs in vivo without any associated toxicity ,a suitable vector is needed.AA V has previously been a favoured option for gene delivery .1However,this delivery system may be too small to allow efficient transduction of the Cas9gene.1A smaller Cas9gene could be used,but this has additional implications on efficacy .1A number of other non-viral delivery systems are under investi-gation and this process requires further optimisation.Another significant concern is the possibility of off-target effects on parts of the genome separate from the region being targeted.Unintentional edits of the genome could have profound long-term complications for patients,including malignancy .The concentration of the Cas9nuclease enzyme and the length of time Cas9is expressed are both important when limiting off-target activity .1Although recent modifications in the nuclease have increased specificity ,further work is required to minimise off-target effects and to establish the long-term safety of any treatment.The therapeutic applications of CRISPR/Cas9con-sidered in this article have predominantly been direc-ted at somatic cells.A particularly controversial issue surrounding CRISPR/Cas9is that of gene editinginFigure 1The CRISPR/Cas9system.1Clustered regularlyinterspaced palindromic repeats (CRISPR)refers to sequences in the bacterial genome.They afford protection against invading viruses,when combined with a series of CRISPR-associated (Cas)proteins.Cas9,one of the associated proteins,is anendonuclease that cuts both strands of DNA.Cas9is directed to its target by a section of RNA.This can be synthesised as a single strand called a synthetic single guide RNA (sgRNA);the section of RNA which binds to the genomic DNA is 18–20nucleotides.In order to cut,a specific sequence of DNA of between 2and 5nucleotides (the exact sequence depends upon the bacteria which produces the Cas9)must lie at the 3’end of the guide RNA:this is called the protospacer adjacent motif (PAM).Repair after the DNA cut may occur via twopathways:non-homologous end joining,typically leading to a random insertion/deletion of DNA,or homology directed repair where a homologous piece of DNA is used as a repair template.It is the latter which allows precise genome editing:thehomologous section of DNA with the required sequence change may be delivered with the Cas9nuclease and sgRNA,theoretically allowing changes as precise as a single base-pair.Research in practice2Redman M,et al .Arch Dis Child Educ Pract Ed 2016;0:1–3.doi:10.1136/archdischild-2016-310459copyright.on December 25, 2023 by guest. Protected by /Arch Dis Child Educ Pract Ed: first published as 10.1136/archdischild-2016-310459 on 8 April 2016. Downloaded fromembryos.It has already been shown that CRISPR/ Cas9technology can alter the genome of human embryos3which theoretically could prove useful in the preimplantation treatment of genetic diseases. However,any genetic modification of the germline would be permanent and the long-term consequences are unclear.Many oppose germline modification under any circumstances,reasoning that an eventual consequence could be non-therapeutic genetic enhancement.13It is clear that the ethical boundaries, within which CRISPR/Cas9can be used,remain to be fully determined.▸CRISPR/Cas9technology has the potential to revolu-tionise the treatment of many paediatric conditions.▸A number of practical and ethical challenges must be overcome before this potential can be realised at thebedside.Contributors DK conceived the idea for this article.All authors were involved in writing and reviewing the final manuscript. Funding AK is supported by a W ellcome T rust Fellowship (108785/Z/15/Z).Competing interests None declared.Provenance and peer review Not commissioned;externally peer reviewed.Open Access This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution(CC BY4.0)license,which permits others to distribute,remix,adapt and build upon this work,for commercial use,provided the original work is properly cited. See:/licenses/by/4.0/REFERENCES1Hsu PD,Lander ES,Zhang F.Development and Applications of CRISPR-Cas9for Genome Engineering.Cell2014;157:1262–78.2Yin H,Xue W,Chen S,et al.Genome editing with Cas9in adult mice corrects a disease mutation and phenotype.NatBiotech2014;32:551–3.3Liang P,Xu Y,Zhang X,et al.CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes.Protein Cell2015;6:363–72.4Schwank G,Koo BK,Sasselli V,et al.Functional repair of CFTR by CRISPR/Cas9in intestinal stem cell organoids ofcystic fibrosis patients.Cell Stem Cell2013;13:653–8.5T abebordbar M,Zhu K,Cheng JK,et al.In vivo gene editing in dystrophic mouse muscle and muscle stem cells.Science2016;351:407–11.6Nelson CE,Hakim CH,Ousterout DG,et al.In vivo genome editing improves muscle function in a mouse model ofDuchenne muscular dystrophy.Science2016;351:403–7.7Long C,McAnally JR,Shelton JM,et al.Prevention of muscular dystrophy in mice by CRISPR/Cas9–mediated editing of germline DNA.Science2014;345:1184–8.8W ojtal D,Kemaladewi Dwi U,Malam Z,et al.Spell checking nature:versatility of CRISPR/Cas9for developing treatmentsfor inherited disorders.Am J Hum Genet2016;98:90–101.9Canver MC,Smith EC,Sher F,et al.BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis.Nature2015;527:192–7.10Hu W,Kaminski R,Y ang F,et al.RNA-directed gene editing specifically eradicates latent and prevents new HIV-1infection.Proc Natl Acad Sci USA2014;111:11461–6.11Schumann K,Lin S,Boyer E,et al.Generation of knock-in primary human T cells using Cas9ribonucleoproteins.ProcNatl Acad Sci USA2015;112:10437–42.12Xie F,Y e L,Chang JC,et al.Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs usingCRISPR/Cas9and piggyBac.Genome Res2014;24:1526–33. 13Lanphier E,Urnov F,Haecker SE,et al.Don’t edit the human germ line.Nature2015;519:410–1.Research in practiceRedman M,et al.Arch Dis Child Educ Pract Ed2016;0:1–3.doi:10.1136/archdischild-2016-3104593copyright. on December 25, 2023 by guest. Protected by / Arch Dis Child Educ Pract Ed: first published as 10.1136/archdischild-2016-310459 on 8 April 2016. Downloaded from。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
2. pLVc-505 (pLVc/U6.sgRNA-Bla-P2A-EGFP)
载体特点: � 使用人 U6 启动子表达 sgRNA 序列; � 用 BsmB1 酶切载体,与 crRNA 插入片段连接; � 含有 EGFP 标记基因,可以实时跟踪转染效率; � 含有杀稻瘟菌素(Blasticidin)抗性筛选基因,可筛选稳定表达细胞系; � 既可用瞬时转染,也可包装成慢病毒后感染细胞; � 设计 crRNA 插入片段的引物结构为 上游引物: 5`-ACCG NNNN NNNNN NNNNN NNNNN-3’ 下游引物: 3`- NNNN NNNNN NNNNN NNNNN CAAA-5’ (三)sgRNA/Cas9 共表达载体 1. 常规质粒载体 (pCas9 V1.0)
Geneup 生物技术有限公司产品目录
第三类 CRISPR/Cas9 系列
一、CRISPR/Cas9 技术简介 CRISPR/Cas9(Clustered Regularly Interspaced Short Palindromic Repeats )是 来自细菌及古细菌的一套特异性免疫系统。 在该系统中, crRNA (CRISPR-derived RNA)与 tracrRNA(trans-activating RNA)结合形成的复合物能特异性识别靶基 因序列,并引导 Cas9 核酸内切酶在靶定位点剪切双链 DNA,从而达到对目的基 因组 DNA 进行特异性编辑。自 2013 年 1 月首次报道利用 CRISPR/Cas9 系统可 以对哺乳动物细胞进行基因组改造以来, CRISPR/Cas9 系统高效的基因组编辑功 能已被应用于多种模式生物,包括人、小鼠、大鼠、斑马鱼、秀丽隐杆线虫、 植 物 及 细 菌 。 研 究 表 明 , 与 锌 指 核 酸 酶 ( ZFNs ) 和 转 录 激 活 样 效 应 核 酸 酶 (Transcription activator-like effector nucleases, TALEN)相比较,CRISPR-Cas 系 统介导的基因组靶向实验在细胞或动物中具有相似甚至更高的效率。
载体特点:
� � 使用人 U6 启动子表达 sgRNA 序列; 用 Bbs1 酶切载体,与 crRNA 插入片段连接。
2. 基于 EBNA1 和 oriP 的载体 (pEP-KO) 基于 EBNA1 和 oriP 元件的非整合型附着体载体(Episomal vector)导入真 核细胞后,则以游离体方式转运入核,载体上携带的目的基因能够获得较高水平 表达,并随着细胞分裂保持较长时间,但最终会将逐渐去除。
� 既可用瞬时转染,也可包装成慢病毒后感染细胞; � 与第四代慢病毒包装质粒匹配,可制备高滴度慢病毒颗粒。 载体图谱:
为什么要选择诱导型 Cas9 表达体系? 利用 Cas9 技术可以进行高通量筛选,现有的做法是在 Cas9 稳转细胞系上进行操作。 研 究表明,Cas9 的持续表达对细胞会产生一定的毒性,因此我们开发了诱导型 Cas9 过表达慢 病毒载体,可以方便地构建稳转细胞系。当对细胞转染过表达 sgRNA 的载体或者体外合成 的 sgRNA 时, 加 Dox 便可诱导 Cas9 表达并启动对靶 DNA 的编辑, 一旦剪切筛选任务结束, 更换为不含 Dox 的培养液,便可有效避免由于 Cas9 持续表达对细胞造成的各种毒副作用。
图 1:CRISPR/Cas9 技术编辑基因原理示意图
二、利用 CRISPR/Cas9 技术进行基因敲除的优点 1. 可实现多个靶基因位点及多个基因的同时敲除; 2. RNA 导向的基因组 DNA 识别,无需考虑 DNA 甲基化; 3. 无种属限制,适用于各种细胞系的靶向敲除; 4. 系统灵活简单,敲除效率更高。 三、Geneup 公司研发的 Cas9 相关产品及服务 (一)诱导型 Cas9/Cas9n 过表达慢病毒载体 (pLVi-503,pLVi-504) 载体特点: � 使用改良的四环素反应元件 (TRE3G) 和反义四环素转录激活子 (TETON3G) 并反向克隆到同一慢病毒载体,显著提高了诱导效果,并降低了本底,在非 诱导条件下几乎检测不到“泄漏”的人源化 Cas9 蛋白; � 加入药物 Dox 时,TETON3G 与其结合后发生构象改变并结合到 TRE3G 位 点,从而促使 miniCMV 启动子控制下的 Cas9 或者 Cas9n 的表达; � 含有嘌呤霉素(puromycin)抗性筛选基因,可筛选稳定表达细胞系;
载体特点:
� � � 使用人 U6 启动子表达 sgRNA 序列; 含有嘌呤霉素(puromycin)抗性筛选 基因,可筛选转染后的细胞; 用 Sap1 酶切载体, 与 crRNA 片段连接。
服务内容 1. 靶向目标序列 sgRNA 载体构建 2. 诱导型 Cas9/Cas9n 稳转细胞系的构建 服务价格
(二)sgRNA 过表达慢病毒载体 1. pLVc-502 (pLVc/U6.sgRNA-copGFP-Puro)
载体特点: � 使用人 U6 启动子表达 sgRNA 序列; � 用 BsmB1 酶切载体,与 crRNA 插入片段连接; � 含有 copGFP 标记基因,可以实时跟踪转染效率; � 含有嘌呤霉素(puromycin)抗性筛选基因,可筛选稳定表达细胞系; � 既可用于瞬时转染,也可包装成慢病毒后感染细胞; � 设计 crRNA 插入片段的引物结构为 上游引物: 5`-ACCG NNNN NNNNN NNNNN NNNNN-3’ 下游引物: 3`- NNNN NNNNN NNNNN NNNNN CAAA-5’
服务项目 内容 sgRNA 克隆服务 每个靶基因设计 3 条 sgRN