不同药品包装材料微生物限度检测方法适用性验证
药包材微生物限度检查操作规程

药包材微生物限度检查操作规程药包材微生物限度检查操作规程一、目的与范围为保障药包材的卫生质量,规范药包材微生物限度检查操作,本操作规程适用于药包材微生物限度检查。
二、术语与定义1. 药包材:指用于药品包装的材料,如玻璃瓶、铝箔、袋子等。
2. 微生物限度:指药包材中的微生物污染的数量不能超过一定的限度。
3. 检查样品:指从药包材中取样检查的样品。
三、操作流程1. 准备工作1.1 检查人员应在工作前进行手部消毒,并佩戴洁净工作服、帽子和口罩。
1.2 检查仪器设备应进行清洁和消毒,确保无污染。
1.3 首次使用检查样品前,应进行无菌处理。
2. 取样2.1 根据药包材的特点,选择合适的取样方法,如剪刀剪取、刮取等。
2.2 取样时应注意避免污染,避免手部和工具的直接接触。
2.3 取样应充分代表药包材的不同部位和不同批次。
3. 样品处理3.1 将取样的药包材放入无菌容器中,避免接触外界环境。
3.2 添加合适的培养基,保证微生物生长。
3.3 药包材中的微生物落菌数较多时,可对样品进行稀释处理。
4. 培养和观察4.1 将培养基接种完毕后,密封容器,确保无菌。
4.2 将培养基置于适宜的温度和湿度条件下培养。
4.3 观察培养基是否出现细菌、真菌等微生物的生长。
5. 计数和判断5.1 培养基培养一定时间后,根据培养基上的菌落形态、颜色等特征进行初步判断。
5.2 将培养基进行放大培养,使用显微镜观察细菌、真菌的形态和结构,进行进一步鉴定。
5.3 使用计数板对菌落进行计数,记录每种微生物的数量。
5.4 根据国家规定的微生物限度标准,判断样品是否合格。
6. 结果记录和分析6.1 将检查结果记录在相应的记录表中,包括药包材名称、样品编号、微生物种类和数量等信息。
6.2 对合格的样品进行合格认证,对不合格的样品进行处理和记录。
6.3 对不合格的样品进行原因分析,查明污染源,并采取相应的措施进行处理和纠正。
四、操作注意事项1. 所有操作应在洁净室中进行,避免外界空气和微生物的污染。
药品的微生物限度检查

24~48h
紫红胆盐葡 萄糖平板
10-1
无菌落
18~24h
有菌落
报告未检出
10g/10ml/ 100cm2供试品
3.耐胆盐革兰阴性菌定量检查操作示意图
以TSB为稀释液制供试液
一定量 肠道增菌肉汤
20~25℃, 2h预增菌
1ml
24~48h
紫红胆盐 葡萄糖平板
报告结果
10-1
18~24h
(六)梭菌的检查
梭菌 增菌 培养 基
供试液的制备
书写检 验记录
哥伦比亚琼脂培养基分离
1.梭菌检查程序
无菌落生长
过氧化氢 酶试验
配培养基和稀释液
最终结果判断
有 菌 落
确证 试验
10g/10ml/ 100cm2供试品
2.供试品梭菌检查操作示意图
各10ml
分别接种
一份80℃保温10min后迅速冷却
10-2
10-31ml10-110-210-3
各管加1ml
查可能菌数表
9ml 稀 释 液
有/无菌落
(二)大肠埃希菌检查
大肠埃希菌作为检验供试品是否受粪便污染的指示菌。
2015年版《中国药典》规定经口及呼吸道服给药的制剂,每1g、1ml或10cm2不得检出大肠埃希菌。
TSB 增菌 培养
30℃~35℃
3~5d
1.平皿法
(1)基本程序:
数据 处理
结果观察与计数
书写检 验记录
平板 接种
需氧菌 的培养
供试液 的制备
配培养基和稀释液
倒置 培养 3~5d
30~35℃
TSA
不少于 0.1ml
微生物限度里需氧菌指的是什么?

微生物限度里需氧菌指的是什么?微生物限度是指药品、原料、辅料等在正常使用条件下所允许的微生物污染程度的标准。
微生物限度检查是一种检测非无菌产品的微生物安全性的方法,包括需氧菌总数、霉菌和酵母菌总数以及控制菌检查三个项目。
需氧菌是指在有氧条件下能够生长繁殖的微生物,包括细菌、放线菌、真菌等。
需氧菌总数是指在特定培养条件下能够形成可见菌落的需氧菌的数量,通常用胰酪大豆胨培养基进行计数。
需氧菌总数反映了产品中微生物的总体水平,与产品的质量和稳定性有关。
不同类型的产品有不同的需氧菌总数限度,根据药典或内控标准进行检查和判断。
本文介绍了微生物限度检查的目的、原理、方法、标准和结果判断,以及需氧菌总数检查的注意事项和常见问题。
本文旨在帮助读者了解微生物限度里需氧菌指的是什么,以及如何正确地进行需氧菌总数检查。
一、微生物限度检查概述1.1 微生物限度检查的目的微生物限度检查是一种评价非无菌产品(如口服固体制剂、外用制剂、原料药、辅料等)受微生物污染程度的方法。
微生物限度检查不仅可以反映产品中微生物的总体水平,还可以检测是否存在特定的有害微生物(如大肠埃希菌、沙门菌等),从而保证产品的安全性。
微生物限度检查的目的是:确保产品符合药典或内控标准规定的微生物限度要求,保障产品质量和安全性。
监测产品在贮存期内的微生物变化情况,评估产品的稳定性。
评价原料、辅料、包装材料等对产品微生物质量的影响。
评价生产过程中各环节对产品微生物质量的控制效果。
1.2 微生物限度检查的原理微生物限度检查主要包括三个项目:需氧菌总数、霉菌和酵母菌总数以及控制菌检查。
需氧菌总数:指在特定培养条件下能够形成可见菌落的需氧菌的数量,通常用胰酪大豆胨培养基进行计数。
需氧菌总数反映了产品中微生物的总体水平,与产品的质量和稳定性有关。
霉菌和酵母菌总数:指在特定培养条件下能够形成可见菌落的霉菌和酵母菌的数量,通常用沙氏葡萄糖琼脂培养基进行计数。
霉菌和酵母菌总数反映了产品中真菌的水平,与产品的质量和稳定性有关。
微生物限度检查方法适用性验证方案

目录1.概述 (2)1.1验证目的 (2)1.2验证频次 (2)1.3适用范围 (2)1.4参照标准 (2)1.5验证小组人员及职责 (2)2.试验前准备 (3)2.1验证所需的文件资料 (3)2.2验证条件 (3)2.3回收试验 (5)2.4结果判断 (7)1.概述1.1验证目的进行产品的微生物计数检查时,应进行方法适用性试验(即微生物回收试验),以确认所采用的方法适合于微生物限度检查。
本次验证的目的就是确认本法适合生产原料、初包装材料、一次性妇用(壳聚糖)生物高分子止血吸附栓的微生物限度检查。
1.2验证频次1.2.1建立生产原料、初包装材料一次性妇用(壳聚糖)生物高分子止血吸附栓的微生物限度检查法时。
1.2.2检验方法更改时。
1.2.3原料、初包装材料、产品发生变化或检验程序发生改变时。
1.3适用范围生产原料、初包装材料、一次性妇用(壳聚糖)生物高分子止血吸附栓的微生物限度检查。
1.4参照标准2015版《中国药典》第四部通则。
2.验证前准备2.1验证所需的文件资料2.2验证条件2.2.1环境要求微生物计数检查在万级洁净区内的百级单向流空气区域内进行,其全过程应严格遵守无菌操作,防止微生物污染。
单向流空气区与工作台面必须进行洁净度验证。
2.2.3验证用培养基所有培养基均采用即用型培养基,临用时按使用说明使用。
2.2.4验证用菌株:菌种来源()2.2.5菌液制备-表3-接种金黄色葡萄球菌、铜绿假单胞菌、枯草芽孢杆菌的新鲜培养物至胰酪大豆胨液体培养基中或胰酪大豆胨琼脂培养基上,接种生孢梭菌的新鲜培养物至硫乙醇酸盐流体培养基中,30〜35℃培养18〜24小时;接种白色念珠菌的新鲜培养物至沙氏葡萄糖液体培养基中或沙氏葡萄糖琼脂培养基上,20〜25℃培养24〜48小时,上述培养物用PH7.0无菌氣化钠-蛋白胨缓冲液或0.9%无菌氣化钠溶液制成每lml含菌数小于l00cfu(菌落形成单位)的菌悬液。
接种黑曲霉的新鲜培养物至沙氏葡萄糖琼脂斜面培养基上,20〜25℃培养5〜7天,加入3〜5ml含0.05% (ml/ml)聚山梨酯80的pH7无菌氣化钠-蛋白胨缓冲液或0.9% 无菌氣化钠溶液,将孢子洗脱。
微生物限度检查法汇总

微生物限度检查法1范围本标准适用于非规定灭菌制剂及其原料、辅料检查项目包括细菌数、霉菌及酵母菌数、控制菌检查及活螨的检查。
2依据标准《中华人民共和国药典》《药品微生物学检验手册》YBB00132002药品包装用复合膜、袋通则3人员资质及培训3.1从事药品微生物检验工作的负责人应具备微生物学或相近专业知识的教育背景。
3.2检验人员应依据所在岗位和职责接受相应的培训,在确认可以承担某一试验前,不能独立从事该项微生物试验。
应保证所有人员在上岗前接受胜任工作所必需的设备操作、微生物检验技术和实验室生物安全等方面的培训,经考核合格后方可上岗,同时,实验室应制定所有级别实验人员的继续教育计划。
3.3检验人员必须熟悉相关检验方法、程序、检测目的和结果评价。
3.4实验室应通过参加内部质量控制、能力验证或使用标准菌株等方法客观评价检验人员的能力,必要时对其进行再培训并重新评估。
当使用一种非经常使用的方法或技术时,有必要在检测前确认微生物检测人员的操作技能。
4培养基4.1培养基的制备4.1.1培养基可按处方配制。
也可使用按处方生产的符合规定的脱水培养基。
4.1.2在制备培养基时,应选择质量符合要求的脱水培养基或单独配方组分进行配制。
脱水培养基应附有处方和使用说明,配制时应按使用说明上的要求操作以确保培养基的质量符合要求,不得使用结块或颜色发生改变的脱水培养基。
4.1.3脱水培养基或单独配方组分应在适当的条件下贮藏,如低温、干燥和避光,所有的容器应密闭,尤其是盛放脱水培养基的容器。
商品化的成品培养基除了应附有处方和使用说明外,还应注明有效期、贮藏条件、适用性检查试验的质控菌和用途。
4.1.4为保证培养基质量的稳定可靠,各脱水培养基或各配方组分应准确称量,并要求有一定的精确度。
配制培养基最常用的溶剂是纯化水,特殊情况下,可能需要去离子水和蒸馏水。
应记录各称量物的重量和水的使用量。
4.1.5配制培养基所用容器和配套器具应洁净,可用纯化水冲洗玻璃器皿以消除清洁剂和外来物质的残留。
无菌、微生物限度检查及方法验证

01
02
03
直接接种法
将样品接种在培养基上, 观察是否有微生物生长。
薄膜过滤法
将样品通过薄膜过滤,收 集滤膜上的微生物,再进 行培养。
离心沉淀法
将样品离心,收集沉淀物 中的微生物,再进行培养 。
无菌检查的注意事项
确保环境洁净
无菌检查需要在洁净的环境中进行,以避免 外界微生物的污染。
避免样品中防腐剂的影响
方法验证
方法验证的定义与目的
定义
方法验证是对检测方法的可靠性、准确性和可重复性的评估过程,以确保该方 法能够满足预期的检测要求。
目的
方法验证的目的是确保所采用的无菌、微生物限度检查方法具有足够的灵敏度 、特异性、重现性和可操作性,以保证检测结果的准确性和可靠性。
方法验证的流程
准备验证计划
制定详细的验证计划,包括验 证实验的设计、实验步骤、数 据收集和分析等内容。
进出口检验
进出口药品需要进行严格的微生物限度检查,以确保药品符合进口 国或地区的质量标准,保障公众健康。
方法验证在药品质量控制中的应用
验证无菌、微生物限度检查方法的可靠性
通过方法验证可以确保无菌、微生物限度检查方法的准确性和可靠性,提高药品质量控制 水平。
评估检测方法的性能指标
方法验证过程中需要对检测方法的性能指标进行评估,如灵敏度、特异性、重现性等,以 确保检测结果的准确性和可靠性。
如果样品中含有防腐剂,可能会抑制微生物 的生长,因此需要进行相应的处理。
正确选择培养基
根据待测样品的特性,选择适合的培养基, 以确保微生物能够正常生长。
定期进行方法验证
无菌检查方法需要定期进行验证,以确保其 可靠性。
0义与目的
药包材微生物检验

已配制好的培养基必须立即灭菌 湿热灭菌必须严格控制灭菌温度和时间(121度,15-30分钟,20分钟)
培养基制备过程中的注意事项
培养基的保温
培养基灭菌后应立即取出,不得储藏在高压灭菌器中,避免造成过度 灭菌,影响培养基的营养成分或选择性效果。建议用45℃—55℃水浴 锅保温不得超过8小时(60℃的培养箱,温度不同,长时间高温影响 培养基质量)。
培养基水化使用的容器应是玻璃器皿或搪瓷器皿如三角烧瓶。蒸馏水洗 净,烘干后方可使用。不得用铜锅或铁锅,以防有离子混入培养基中,影 响微生物的生长;
配制培养基最常用的溶剂是纯化水 ,不能含有任何可能抑制或影响微生 物生长的物质;
培养基的溶解
在培养基分装灭菌前, 应溶解均匀, 使用电炉等设备煮沸培养基时,应用 小火加热,并边加热边搅拌,避免培养基焦化或爆沸溢出。
• 实验器材、消耗品的准备
a.无菌衣、手套、口罩、试管、瓶子、培养皿、消毒剂 配制缸、脱脂棉花球
b.一次性薄膜过滤滤器、微生物检查薄膜过滤仪 c.消毒剂、酒精灯
培养基的制备
培养基是微生物试验的基础,直接影响微生 物试验结果。培养基的制备是检验的重要一 环。
培养基制备过程中的注意事项
培养基的水化
培养基的贮藏
配制好的培养基应保存在避光的环境。一般在三周内使用(琼脂培养 基不得在0℃或0℃以下存放,防止冷冻破坏凝胶特性)。
固体培养基灭菌后的再融化应在加热的水浴中或采用流通蒸汽进行, 只允许一次,不得再灭菌。
人流、物流程序
➢物流
将样品、灭菌后的物品、集菌过滤器等转移入传递窗内,打开传递窗 内的紫外灯,照射不少于0.5小时。
实验准备
洁净室的启用
微生物限度检查应在环境洁净度10000级下的局部洁净 度100级的单向流空气区域内进行。在检验开始前,应 确保: a.洁净室房间、台面等已清洁消毒 b.洁净室已臭氧灭菌不少于半小时 c.洁净室净化空调系统持续运转不少于1小时,紫外灯已 开启 d.洁净室中有压差要求的房间压差达到规定
微生物限度检查及效价检查技术

五、 供试品检查:检验量、供试品检查
菌数报告规则 如各稀释级的平皿均无菌落生长,或仅最低稀释级的平 板有菌落生长,但平均菌落数小于1 时,以﹤1 乘以最低稀 释倍数的值报告菌数。
五、 供试品检查:检验量、供试品检查
供试品检查 ② 薄膜过滤法 取相当于1g、1ml 或10 cm² 供试品的供试液,若供试 品所含的菌数较多时,可取适宜稀释级的供试液,照方法适 用性试验确认的方法加至适量稀释液中,立即过滤,冲洗, 冲洗后取出滤膜,菌面朝上贴于胰酪大豆胨琼脂培养基或沙 氏葡萄糖琼脂培养基上培养。 培养和计数 培养条件和计数方法同平皿计数法,每张滤 膜上的菌落数应不超过100cfu。
⑵药品染菌反映药品生产工艺的科学性、合理性及质量 管理水平。 微生物限度检查的各项数据,是对药品生产工艺、生 产环境、质量管理及人员素质的综合评价依据之一。
只有优良的GMP企业,才能提供优质的医药产品,凡 工艺条件、生产管理水平、生产人员素质差,不注意文明生 产的单位和企业,其生产的药品染菌必然严重,不合格率高。
五、 供试品检查:检验量、供试品检查
供试品检查 ① 平皿法 包括倾注法和涂布法。 每稀释级每种培养基至少制备2个平皿。胰酪大豆胨琼 脂培养基平板在30~35℃培养3天,沙氏葡萄糖琼脂培养基 平板在20~25℃培养5 天, 观察菌落生长情况,点计平板上 生长的所有菌落数。菌落蔓延生长成片的平皿不宜计数。点 计菌落数后,计算各稀释级供试液的平均菌落数,按菌数报 告规则报告菌数。
五、 供试品检查:检验量、供试品检查
⑶ 油脂类供试品 取供试品,加入无菌十四烷酸异丙 酯使溶解,或与最少量并能使供试品乳化的无菌聚山梨酯 80或其他无抑菌性的无菌表面活性剂充分混匀。表面活性 剂的温度一般不超过 40℃(特殊情况下,最多不超过 45℃),小心混合,若需要可在水浴中进行,然后加入预 热的稀释液使成 1∶10供试液,保温,混合,并在最短时 间内形成乳状液。必要时,用稀释液或含上述表面活性剂 的稀释液进一步 10倍系列稀释。
无菌药品GMP检查指南

无菌药品GMP检查指南2015年10月目录一、目的3二、适用范围及检查依据 (3)三、无菌药品生产工艺概述 (3)四、检查要点6(一)质量管理系统6(二)厂房、设施及设备系统9(三)物料系统15(四)生产系统18(五)包装和贴签系统24(六)实验室控制系统26五、参考文献29一、目的本指南的主要目的是为检查员在实施无菌药品生产企业检查时提供指导.检查组应参照本指南的要求检查无菌药品生产质量管理情况,评价企业无菌保证的能力,以确定企业是否符合《药品生产质量管理规范(2010年修订)》(以下简称GMP)的要求。
二、适用范围及检查依据本指南适用于无菌药品的GMP检查,包括无菌制剂生产全过程和无菌原料药的灭菌和无菌生产过程。
无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,通常包括大容量注射剂、小容量注射剂、粉针剂、冻干粉针剂、眼用制剂、耳用制剂、埋植剂、供雾化器用的液体制剂、冲洗剂、外用制剂、无菌原料药等。
无菌药品按生产工艺可分为两类:采用最终灭菌工艺的为最终灭菌产品;部分或全部工序采用无菌生产工艺的为非最终灭菌产品。
本指南适用于对上述不同生产工艺及不同类型的无菌药品的检查。
检查过程中,检查员应依据《药品生产质量管理规范(2010年修订)》及其附录来确定检查缺陷所涉及的条款.三、无菌药品生产工艺概述无菌药品按生产工艺可分为最终灭菌产品和非最终灭菌产品两类,部分或全部工序采用无菌生产工艺的为非最终灭菌产品。
无菌药品、直接接触药品的包装材料应尽可能采用热力灭菌方式进行最终灭菌。
采用湿热灭菌方法进行最终灭菌的,通常标准灭菌时间F0值应当大于8分钟,流通蒸汽处理不属于最终灭菌。
最终灭菌产品中的微生物存活概率(即无菌保证水平,SAL)不得高于10—6。
(一)最终灭菌工艺产品的无菌保证水平不能仅依赖最终灭菌.灭菌工艺必须与产品注册批准要求相一致,且应当经过验证。
最终灭菌工艺通常将产品在洁净度级别较高洁净区(不得低于C级)进行灌装和密封,以降低产品的微生物和微粒污染。
WHO发布《使用不同方法—包括HBEL—建立清洁验证限度指南》!

WHO发布《使⽤不同⽅法—包括HBEL—建⽴清洁验证限度指南》!Thanks for your attention近⽇,WHO发布了新的清洁验证指南——《不同⽅法——包括HBEL——建⽴清洁验证的残留限度以确定共⽤设施⽣产污染风险的考量》,该指南解读如下:传统的⽅法是进⾏清洁验证并基于GMP⽂件中建议的接受标准来判断清洁程序的适⽤性。
这种做法可能不再是可以接受的和合理的,因为没有考虑到HBEL。
鉴于污染和交叉污染的风险,应毫不拖延地实施下⽂所述的新⽅法。
新的清洁验证⽅法包括:清洁能⼒研究;风险评估及风险控制;技术和组织控制;设定HBELs分析程序;及清洁确认,并通过统计学评价证实清洁能⼒。
设备图纸应保持更新,准确和可⽤。
在计算设备表⾯积时应使⽤。
应有这些计算的源数据。
计算的数值应应⽤于清洁验证的计算。
难以清洁的设备和部件,如筛⼦、筛⽹和袋⼦,也应包括在清洁验证和计算中。
关于取样,应使⽤⾄少两种或三种取样⽅法的组合。
包括擦拭取样、冲洗取样和⽬视检查的组合。
应当选择适当的取样程序和技术来收集样品。
应在程序和规程中加以明确说明。
收集样品的地点(擦拭取样)及⽅式应清楚说明,并具科学理据⽀持。
淋洗⽔取样应有详细的描述。
规程应清洗明确。
收集的样品进⾏分析的⽅式应适当,并详细说明。
在验证清洁程序并⽇常使⽤之前,应进⾏清洁能⼒研究,以确定物料、产品残留、清洁剂和微⽣物的清除程序是否合适。
关于清洁能⼒研究,对于不同结构材料上的不同物料、中间体和产品,应确定通过清洁程序能够除去的物质的最低浓度。
该浓度可⽤mg/m2表⽰。
清洁能⼒研究应在批准的⽂件中进⾏描述,例如规程和程序。
该⽅法应具有科学性,可包括对不同结构材料进⾏涂布。
可以使⽤所谓的烧杯法或其他适当的⽅法。
应制定和执⾏程序,说明如何获取关于HBEL的科学数据和毒理学信息。
关于HBEL报告,数据和信息应收集并呈现在报告中。
这些数据应没有偏差。
如此服务外包,应采取适当措施,以确保所获得的数据可靠。
药用包装材料质量标准

(一)聚酯/铝/聚乙烯药品包装用复合膜、袋来源国家药品监督管理局YBB00172002本品系指聚酯(PET)与铝箔(Al)及聚乙烯(PE)通过黏合剂复合而成的膜。
本品的袋系将上述膜通过热合的方法制成。
本标准适用于固体药品包装用的复合膜、袋。
[外观] 取本品适量,照药品包装用复合膜、袋通则(试行)(YBB00132002)外观项下的方法检查,应符合规定。
[鉴别]红外光谱取本品适量,采用内表面反射方法,照分光光度法(中华人民共和国药典2000年版附录ⅣC)测定,PET及PE层应分别与对照图谱基本一致。
[阻隔性能] 水蒸气透过量照塑料薄膜和片材透水蒸气性试验方法杯式法(GBl037-88)的规定进行。
试验时PE层向湿度低的一侧,试验温度(38±2)℃,相对湿度(90±5)%,不得过0.5(g/m2·24h)。
氧气透过量照塑料薄膜和薄片气体透过性试验方法压差法(GB/T 1038-2000)的规定进行。
试验时PE层向氧气低压侧,试验温度为(23±2)℃,不得过0.5cm3/(m2·24h·0.1MPa)。
[机械性能] PE层与A1层剥离强度照药品包装用复合膜、袋通则(试行)(YBB00132002)内层与次内层剥离强度项下的方法检查,纵、横向剥高强度平均值均不得低于2.5N/15mm。
[热合强度] 膜除另有规定外,裁取100mm×100mm试片四片,将任意两个试片PE面叠合,置热封仪上进行热合,热合温度150℃~170℃,压力0.2~0.3MPa,时间1秒。
从热合的中间部位各裁取3条15mm宽的试样,进行试验。
试样应在温度23℃±2℃,相对湿度50%±5%的环境中,放置4小时以上,并在上述条件下进行试验。
以热合部位为中心线,打开呈180度,把试样的两端夹在试验机的两个夹具上,试样轴线与上下夹具中心线相重合,并松紧适宜,夹具间距离为50mm,试验速度为(300±30)mm/min,读取试样断裂时的最大载荷,平均值不得低于12N/15mm。
微生物限度检查操作规程(中国药典2015版四部通则)
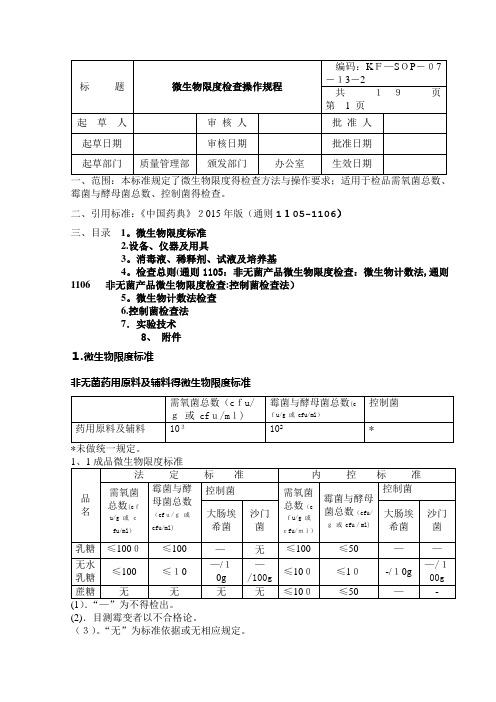
霉菌与酵母菌总数、控制菌得检查。
二、引用标准:《中国药典》2015年版(通则1105-1106)三、目录1。
微生物限度标准2.设备、仪器及用具3。
消毒液、稀释剂、试液及培养基4。
检查总则(通则1105:非无菌产品微生物限度检查:微生物计数法,通则1106非无菌产品微生物限度检查:控制菌检查法)5。
微生物计数法检查6.控制菌检查法7.实验技术8、附件1.微生物限度标准非无菌药用原料及辅料得微生物限度标准(2).目测霉变者以不合格论。
(3)。
“无”为标准依据或无相应规定。
准依据或无相应规定.2.设施、仪器及用具2、1、设施:2、1、1.微生物限度检查室及相关设施:微生物计数试验环境应符合微生物限度检查得要求。
检验全过程必须严格遵守无菌操作,防止再污染,防止污染得措施不得影响供试品中微生物得检出。
单向流空气区域、工作台面及环境应定期进行监测。
2、1、2.其她设备:高压蒸汽灭菌器;细菌培养箱(30~35℃);霉菌培养箱(25~28℃);电炉(或其她适宜得加热装置);恒温水浴;电热干燥箱(250~300℃);电冰箱。
生化试剂储存箱。
2、2仪器及器皿2、2、1。
菌落计数器;显微镜(1500X);电子天平或药物天平(感量0、1g);pH 系列比色计。
2、2、2.玻璃器皿:锥形瓶(250~300ml,内装玻璃珠若干)、研钵(玻璃或陶瓷制,∮10~12cm)、培养皿(∮9 cm)、量筒(100ml)、试管(18×180mm)及塞、吸管(1ml分度0、01,10ml分度0、1)、载玻片、盖玻片、玻璃消毒缸(带盖)。
2、2、3新购得玻璃器皿得清洁:先用流水冲洗,浸泡于1%~2%盐酸(工业用)液中约2~6小时,除去游离碱质,再用流水冲洗.用于化学分析得玻璃仪器,需用重铬酸钾清洁液浸泡数分钟后,再用流水冲洗,最后以纯化水涮洗2~3次,晾干备用。
2、3用过得玻璃器皿:2、3、1未被病原微生物污染得器皿:可随时洗涤.用清水冲洗(或浸泡),除容量仪器外,可用毛刷与肥皂粉,内外刷洗,再用清水涮洗干净,晾干备用.容量仪器宜用清洁液浸泡或涮洗,再用流水冲洗,最后以纯化水涮洗2~3次.试管及培养皿:先正放或直立于高压蒸汽灭菌器内,经121℃灭菌30 分钟.趁热倾出培养物,再以清水或用毛刷及肥皂粉刷洗,最后以流水涮净。
药品包装检测的几类项目

药品包装检测的几类项目医药包装检测机构对药品包装检测,通常是根据包装形式的不同而采取不同的检测方法。
那么,你知道药品包装检测形式都有哪些吗?药品行业包装形式主要有泡罩包装、条形包装、袋包装、双铝包装、水针剂塑料包装、软质瓶包装等等。
汇总我国及国际相关标准规范,对药品包装检测及材料检测与控制的指标主要有:阻隔性能、机械性能、滑爽性、厚度、溶剂残留、密封性能、瓶盖扭力、顶空气体分析、印刷质量等。
药品包装检测主要指通过检测仪器进行药包材阻隔性能检测(气体透过量测试与水蒸气透过量测试)等。
大家在吃药的时候难免会抠下这层类似“锡纸”的材质甚至觉得十分解压感到舒适那你们有没有想过这层“锡纸”是什么材质呢?为什么要用这种材质呢?这种药品包装叫泡罩包装泡罩包装的主要材料是药用PTP铝箔、塑料硬片(聚氯乙烯PVC),及粘合剂小敏今天先带大家了解一下药用铝箔。
一、外观在自然光明亮处,正视目测检查样品是否有破损、裂变等。
小敏聊检测:铝箔表面应该清洁、平整、涂层均匀;文字、图案印刷应正确、清晰、牢固。
二、针孔度铝箔生产过程中采用轧制工艺,不可避免会出现缺陷,针孔就是其中最主要的一种。
小敏聊检测:铝箔针孔的大小和数量对铝箔及其复合材料的防潮性、阻气性和遮光性有着决定性的影响。
铝箔不应该有密集的、连续性的、周期性的针孔;每一平方米中,直径大于0.3mm的针孔不允许有,直径为0.1~0.3mm的针孔数不得过1个。
三、阻隔性能以包装材料的氧气透过量和水蒸气透过量衡量包装材料的阻隔性能。
小敏聊检测:铝箔的阻隔性能是药品选择适宜包材产品的重要考察指标。
阻隔性能优良的包装容器可有效保护药品质量、延长药品的有效期。
四、粘合层热合强度泡罩与铝箔热粘合强度的高低与热封温度、热封压力、热封时间等热封参数有直接关系。
小敏聊检测:保护层的粘合性较差,在热合或者其他受热过程中发生脱落时,则对油墨的保护作用减弱,故油墨层易出现被磨花、脱落等问题。
五、保护层耐热性保护层的耐热性较差,在热封过程等受热环节易出现脱落现象。
药品微生物限度检查用培养基的配制、灭菌和贮存效期验证的

药品微生物限度检查用培养基的配制、灭菌和贮存效期验证的研究张颖安秀华王建平曹凤兰(天士力制药集团股份有限公司,天津300410)摘要目的:本试验通过对药品微生物限度检查用5种培养基的配制方法、灭菌程序以及配制后贮存条件和贮存效期进行验证,确保每次配制的培养基质量均满足2010年版《中国药典》要求。
方法:3次独立试验中确定了培养基的配制方法、灭菌参数(液体程序,121℃,15min)、贮存条件(洁净度为C级的环境)及贮存时间(45天),并对贮存0天、15天、30天和45天的培养基进行适用性检查(包括促生长能力、抑制能力、指示能力和无菌性)。
结果:培养基在贮存0天、15天、30天和45天后的适用性检查结果均满足2010年版《中国药典》对培养基质量控制的要求。
结论:通过该验证试验,确立了培养基的配制方法、灭菌参数、贮存条件及30天的贮存效期。
关键词:微生物限度检查,培养基适用性检查,配制方法,灭菌参数,贮存效期Study on verification of the preparation, sterilization and the storage life of the media usedin drug microbial enumeration TestsZHANG Ying, AN Xiu-hua, WANG jian-ping, CAO feng-lan(Tasly Pharmaceutical group CO., LTD, Tianjin 300410,China)Abstract Objective: In order to ensure the quality of every batch of prepared media which meet Chinese Pharmacopoeia requirement, the media preparation methods, sterilization procedures and the shelf life of prepared media are validated. Methods: The preparation procedure, sterilization parameters( Liquid procedure, 121℃,15min), storage condition(cleanliness environment for class C) and storage time(45 days) of the culture media for each test is determined, the performance of the media after sterilization stored 0 days, 15 days, 30 days and 45 days were checked(included sterilization, growth promotion indicative and inhibitory properties of the media.)Results: All media prepared in three tests which stored 0 days, 15 days, 30 days and 45 days in the cleanliness environment for class C met Chinese Pharmacopoeia requirement. Conclution:The preparation methods, sterilization procedures and the storage life of the 30 days of prepared media is confirmed through the verification test.Key Words: microbiological examination,applicability inspection of the culture medium, media preparation methods, sterilization parameters , storage life of prepared media培养基是否合格,对微生物的生长、分离、鉴定及检验结果的正确与否起着至关重要的作用,因此,对培养基的质量控制十分必要。
引用标准 EP2.6.12和2.6.13

目的:建立微生物限度检查法标准操作程序。
2 范围:用于原辅料、包装材料、中间产品以及成品的微生物限度检验。
3 责任:质量部。
4 内容4.1 引用标准: EP2.6.12和2.6.134.2 方法提要:通过对试样在不同条件下的微生物培养,检查试样受到微生物污染程度情况。
微生物限度检查法系检查非灭菌原料药及其原、辅料受到微生物污染程度的方法,检查项目包括细菌数、霉菌数、酵母菌数及控制菌的检查。
微生物限度检查应在环境洁净度10000级下的局部洁净度100级的单向流空气区域内进行。
检验全过程必须严格遵守无菌操作,防止再污染。
单向流空气区域、工作台面及环境应定期按《医药工业洁净室(区)悬浮粒子、浮游菌和沉降菌的测试方法》的现行国家标准进行洁净度验证。
供试品检查时,如果使用了表面活性剂、中和剂或灭活剂,应证明其有效性及对微生物的生长和存活无影响。
除另有规定外,本检查法中细菌培养温度为30℃~35℃,霉菌、酵母菌培养温度为20℃~25℃。
检验结果的报告以1g、1mL、10 mL或10cm2为单位报告。
4.3样品的制备取样方法:产品取样必须依照规定的采样方法取样。
在每批样品中取三个样品并分别检查。
4.3.1水溶性产品:在pH=7.0氯化钠-蛋白胨缓冲液中稀释10g 或10ml产品。
一般准备1:10稀释倍数,作为供试液。
不过,根椐产品的特性或必要的敏感性也可做成其它的比例的。
如果已知此产品有抗微生物活性,可加入灭活剂到稀释液中。
如果需要调节pH值,调至pH值约为7,如果需要进一步稀释,用相同稀释液进行系列10倍稀释。
4.4阴性对照为了确定实验条件对检测结果无影响,要用稀释剂代替供试液进行阴性对照试验。
应无菌生长。
对于不合格的阴性对照结果应进行调查。
4.5培养基促生长试验(阳性对照)适用于每一批购买的现成培养基,每一批使用脱水培养基制备的培养基或者按照指定成分配制的培养基。
按照表1所示,接种少量的菌种(不多于100 CFU)于大豆酪蛋白胰酶消化物肉汤和大豆酪蛋白胰酶消化物琼脂培养基中,每一份菌种接种于各自的培养基上。
初始污染菌计数方法适用性试验方案 ——供试品释出物的抑菌试验

XXXX产品初始污染菌计数方法适用性试验方案——供试品释出物的抑菌试验NO. BCM/BT-1.目的确定从产品释放到悬液中的物质对脆弱的微生物生存能力的影响。
1.适用范围适用本公司内部所有不同材料产品、包装材料的初始污染菌计数方法适用性试验——供试品释出物的抑菌试验。
1.参照依据******1.试验样品的确定4.1试验样品列表4.2样品选择说明1.仪器设备电子天平、电热恒温干燥箱、压力蒸汽灭菌器、恒温水浴锅、摇床、漩涡振荡器、超净工作台、生物安全柜、生化培养箱、霉菌培养箱、集菌仪1.试剂胰酪大豆胨琼脂培养基、沙氏葡萄糖琼脂培养基、胰酪大豆胨液体培养基、沙氏葡萄糖液体培基、蛋白胨、吐温80、0.9%无菌氯化钠溶液1.辅料0.45μm滤膜、无菌泵管1.标准菌株金黄色葡萄球菌[CMCC(B) 26003]铜绿假单胞菌[CMCC(B) 10104]枯草芽孢杆菌[CMCC(B) 63501]白色念珠菌[CMCC(F) 98001]黑曲霉[CMCC(F) 98003]1.供试品释出物的抑菌试验9.1洗脱液0.1%蛋白胨水溶液(含0.01%吐温80)9.2菌液制备9.2.1试验用菌株的传代次数不得超过5代,计数方法适用性试验用菌株见表1:表1无菌氯化钠溶液制成适宜浓度的菌悬液;黑曲霉的新鲜培养物加入3~5ml含0.05%(ml/ml)聚山梨脂80的0.9%无菌氯化钠溶液,将孢子洗脱。
然后用适宜的方法吸出孢子悬液至无菌试管内,用含0.05%(ml/ml)聚山梨脂80的0.9%无菌氯化钠溶液制成适宜浓度的黑曲霉孢子悬液。
菌液制备后若在室温下放置,应在2小时内使用;若保存在2~8℃,可在24小时内使用。
黑曲霉孢子悬液可保存在2~8℃,当天制备当天使用。
9.2.3菌液计数按Q/SOP-###《菌种保存、传代、使用、销毁标准操作规程》的规定执行。
9.3样品处理选择适宜的方法进行洗脱方法1 :a.洗脱液用量:150ml洗脱液+150ml洗脱液冲洗b.洗脱设备名称/参数:摇床振荡,频次100,振荡15分钟c.洗脱液器皿:500ml玻璃瓶d.洗脱方法描述:将产品接种到150ml洗脱液中,放置于摇床振荡,频次100,振荡15分钟,备用e.计数方法:薄膜过滤法方法2:a.洗脱液用量:150ml洗脱液+150ml洗脱液冲洗b.洗脱设备名称/参数:漩涡混合器,振荡3分钟c.洗脱液器皿:500ml玻璃瓶d.洗脱方法描述:将产品接种到150ml洗脱液中,,用漩涡振荡器,振荡3分钟,备用。
药品生产监督质量控制实验室管理检查重点内容和检查方法指导原则

药品生产监督质量控制实验室管理检查重点内容和检查方法指导原则1.质量控制实验室布局检查方法:结合有关验证方案、记录,现场检查实验室布局和设施。
检查内容:1.1质量控制实验室是否与生产区分开;设在生产区内中间控制实验室是否对生产也无不利影响,中间控制检验结果的准确性是否受生产操作的影响。
1.2质量控制实验室是否有足够的空间用于样品处置、检验和实验用物料、留样、稳定考察样品存放及记录的保存。
1.3生物检定、微生物和放射性同位素的实验室是否彼此分开;有菌实验室(微生物限度检查、菌种传代、阳性菌接种)是否与无菌检查室分开。
1.4阳性菌室是否配备生物安全柜,是否设置直排风保持与其他区域的负压。
1.5 是否设置专门的仪器室,使灵敏度高的仪器免受静电、震动、潮湿、高温或者其他外界因素影响。
1.6产生挥发性气体或者高温的设备是否设置局部排风。
1.7用于滴定液标定储存、稳定性考察药品贮存等对温湿度有特殊要求的房间或者设备,温湿度及其控制精度范围是否符合《中国药典》的规定。
2.质量控制实验室文件管理2.1文件种类检查方法:抽查物料、成品质量标准文件,审查内容是否完整、正确,检查相关检验记录看质量标准是否被正确执行。
检查内容:质量控制实验室是否建立下列详细文件:2.1.1质量标准;2.1.2取样操作规程和记录,检验操作规程和检验记录2.1.3试剂、试液、培养基配制操作规程和记录,滴定液配制标定操作规程和和记录2.1.4检验仪器操作、清洁、维护、校准的操作规程和记录;2.1.5检验报告或证书,必要的环境监测操作规程、记录和报告;必要的检验方法验证报告和记录。
2.2文件、记录管理的基本要求检查方法:选取部分产品、物料,结合现场检查审核相关文件和检验记录。
检查内容:2.2.1药品的每批检验记录是否包括中间产品、待包装产品和成品的质量检验记录,记录是否可以追溯该批药品所有相关的质量检验情况;2.2.2 是否用便于趋势分析的方法保存某些数据(如检验数据、环境监测数据、制药用水的微生物监测数据);2.2.3 记录所有计算均应当严格核对,记录内容是否经过复核,确保结果与记录一致。
药品生产监督检查重点内容和检查方法指导原则

药品生产监督检查重点内容和检查方法指导原则本指导原则,分为通则和分则两个部分,通则部分是指药品生产质量和管理管理的通用要求,分则部分列举了不同类别药品生产和质量管理的特殊要求。
监督检查时,应根据不同的药品类别,将两部分的相关内容结合实施检查。
第一部分通则(一)证照合法性1.许可证合法性检查方法:检查《药品生产许可证》、《医疗机构制剂许可证》正副本和其变更记录及相关批件。
检查内容:1.1《药品生产许可证》(《医疗机构制剂许可证》)企业(制剂室)负责人、生产(配制)地址、生产(配制)范围等许可事项是否变更,变更是否按照规定经省局批准。
1.2《药品生产许可证》(《医疗机构制剂许可证》)企业(医疗机构)名称、企业类型(医疗机构类别)、法定代表人、注册地址等登记事项是否变更,变更是否按照规定报省局备案。
1.3《药品生产许可证》、《医疗机构制剂许可证》是否在有效期内。
2.药品批准证明文件合法性检查方法:检查药品(医疗机构制剂)注册批件、再注册批件、注册补充申请批件,必要时查询国家局数据库。
检查内容:2.1 药品批准证明文件中载明的药品(制剂)名称、剂型、规格、药品有效期、生产(配制)地址等信息是否发生变更,变更是否经过国家局或者省局批准。
2.2 药品批准证明文件是否在有效期内,到期是否通过再注册。
3.药品GMP证书合法性检查方法:检查GMP证书和GMP认证申报材料。
检查内容:3.1制剂剂型、中药饮片炮制范围、原料药品种及其生产车间是否取得相应GMP证书。
3.2中药饮片生产品种是否在GMP认证申报范围内,超出范围的品种是否通过市局组织的现场检查。
3.3 药品GMP证书是否在有效期内。
(二)机构和人员1.组织机构检查方法:检查组织机构框图,抽查关键岗位职责和有关管理文件。
检查内容:1.1是否设立了与生产品种相适应的质量管理、生产管理、设备管理、仓储管理等部门。
1.2质量管理部门是否独立设置(不得隶属于其他部门,也不得归属质量管理负责人或者企业负责人以外的其他人员领导)。
微生物限度检测相关问答

微生物限度1. 包装材料的大肠埃希菌检查?答:根据包装材料的不同,大肠埃希菌的检查方法大致有两种,一种是将浸提液合并后,取一部分,接种胆盐乳糖培养基进行检查;另一种是将浸提液合并后,薄膜过滤,然后按规定的方法检验。
2. 抗真菌药品的微生物限度检查法答:这类产品的霉菌和酵母菌计数方法需要进行调整,可以根据产品剂型的不同,选择薄膜过滤法或培养基稀释法等方法。
3. 为什么在日常样品检测中还需要做阳性对照(国外不需要)?答:为了确保每一次检测对可能存在的微生物都是有效的。
4. 对于同一个品种,药典为什么不规定统一的检测方法?答:本版药典进行过这样的尝试,也有某些品种已经收载了统一的检测方法,如注射用头孢类抗生素的无菌检查。
之所以没有大量收载,主要是由于检测方法还没有经过必要的复核。
将微生物检验的统一方法收载在品种的各论项下,始终是努力的方向。
5. 梭菌检查中需置厌氧条件下培养48小时,是否指在厌氧培养箱中?答:需要在厌氧培养装置中进行培养,未必一定是厌氧培养箱。
6. 控制菌检查方法验证时,采用多种方法均未检出控制菌,如何处理?答:在确保所使用的各种方法都没有错误的情况下,如果仍无法使试验菌生长,则应该采用薄膜过滤法进行检查。
理由是该方法能够最大程度地去除产品的抑菌作用。
7. 常规的监督抽样检品(包括原料)在进行微生物限度检查时,是否一定要进行活螨检查并在原始记录与报告书中体现?答:需要进行检查。
可以在原始记录中体现,报告书上可以不出现,除非当发现有活螨检出。
8. 药包材微生物限度检查规定了合格质量水平,通常产量下取样8个,要求8个瓶子均符合规定,如何进行?答:每个瓶子分别进行实验。
9. 大肠埃希菌具体操作规程?答:参见中国药品检验标准操作规程。
10. 动物组织及动物类原药材的提取物入药,是否需要检沙门菌?答:需要进行沙门菌检查。
11. 关于中国药典菌落计数结果的判断还存在疑义,望能举例说明。
答:不清楚所指的存在疑义是指哪方面。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
不同药品包装材料微生物限度检测方法适用性验证作者:朱莲花凌蕾来源:《上海医药》2017年第09期摘要目的:选择适宜方法对多种直接接触药品的包装材料微生物限度进行检查,并对方法的适用性进行验证。
方法:对固体药用聚丙烯瓶等数种药品包装材料按照相应药品包装材料国家标准和中国药典2015年版四部附录通则的要求进行微生物限度方法适用性验证。
结果:金黄色葡萄球菌、铜绿假单胞菌、枯草芽胞杆菌、白色念珠菌和黑曲霉五种标准菌株在菌落计数法验证试验中的回收率为60%~150%,符合药典规定。
在控制菌的方法验证试验中,均可检出相应的试验菌。
结论:选定的方法适用于不同的药品包装材料的微生物限度检测。
关键词药品包装材料微生物限度检查适用性验证中图分类号:R951 文献标识码:A 文章编号:1006-1533(2017)09-0073-03Applicability verification of microbial limit test method for different drug packaging materialsZHU Lianhua*, LING Lei(Shanghai Food and Drug Packaging Material Control Center,Shanghai 201203, China)ABSTRACT Objective: To verify the applicability of the appropriate method for microbial limit test on different drug packaging materials. Methods: The microbial limit test and the validation test for the determination of the microbial contamination on pharmaceutical packaging materials such as the solid medicinal polypropylene bottle were performed based on the regulations of the National Standard Packaging Method and Chinese Pharmacopoeia 2015 edition. Results: The recovery rates of five standard strains Staphylococcus aureus, Pseudomonas aeruginosa, Bacillus subtilis,Candida albicans and Aspergillus niger were in the range of 60% ~150% by the colony count method, which meet the requirements of Chinese Pharmacopoeia. The corresponding test strains cold be detected by the validation test of control bacteria when they were incubated for a certain time at appropriate temperature. Conclusion: The selected method can be applied to the microbial limit test for the determination of the microbial contamination on different drug packaging materials.KEy WORDS drug packaging material; microbial limit test; applicability verification直接接触药品的包装材料及容器既具备了盛药品的功能,也因为其与药品的接触而可能影响药品的质量。
微生物污染是药品包装容器可能给药品质量带来的风险之一,因此,为了确保药品的安全有效,必须严格控制包装容器的微生物污染程度[1-2]。
药品包装材料微生物限度检验主要是通过对附着在容器或材料表面的微生物进行采集和检验[3-4],而药品包装容器的材料组成成分、生产工艺、贮存条件等各种因素都会对附着的微生物数量和种类带来影响[5-6]。
同时,检验方法中对样品表面附着微生物的采集方法——即供试液制备过程中涉及的提取介质及具体制备方法都可能对微生物造成一定的损伤从而影响微生物生长,可能给检验的准确性带来影响。
微生物检验结果的准确性还受到其他多种因素的影响,如样本分布均匀性、样品溶液制备过程中微生物收集的有效性、检验方法的灵敏度等,这些内容一般已经在标准方法中有详细的规定从而确保检验方法的科学性。
但各个实验室仍需对标准方法在本实验室的适用性进行验证,从而确保本实验室的人员操作、实验环境、培养基和试剂等各个环节不会对该方法的结果造成影响,从而保证检测结果的准确性[7-10]。
1 仪器与材料1.1 仪器生化培养箱Thermo 815(赛默飞世尔);高压蒸汽灭菌锅MLS-3780(日本三洋公司);ESCO生物安全柜Class II BSC(艺思高科技有限公司)。
1.2 实验用标准菌株生物梅里埃定量质控菌株BioBall MultiShot 550系列,包括金黄色葡萄球菌(Staphylococcus aureus,S. aureus)NCTC 10788(批号:B3651)、铜绿假单胞菌(Pseudomonas aeruginosa,P. aeruginosa)NCTC 12924(批号:B3653)、枯草芽孢杆菌(Bacillus subtilis,B. sub)NCTC 10400(批号:B3647)、白色念珠菌(Candida albicans,C. albicans)NCPF 3179(批号:B3663)、黑曲霉(Aspergillus niger,A. niger)NCPF 2275(批号:B3642)、大肠埃希菌(Escherichia coli,E. coli)NCTC 12923(批号:B3661)。
1.3 培养基及试剂胰酪大豆胨琼脂培养基(TSA)干粉、胰酪大豆胨液体培养基(TSB)干粉、沙氏葡萄糖琼脂培养基(SDA)干粉、麦康凯液体培养基干粉、麦康凯琼脂培养基干粉、溴化十六烷基三甲铵琼脂培养基干粉、甘露醇氯化钠琼脂培养基干粉,均来自于上海中科昆虫生物技术开发有限公司。
上述培养基适合性检查符合规定[11]。
1.4 实验样品本实验所用样品均为日常测试样品。
1.5 菌液的制备将不同种类的BioBall MultiShot 550标准菌株球分别倒入1.1 ml 14 d复溶液中,混匀,为实验用菌液(即500 cfu/ml标准菌株实验用菌液)。
1.6 供试液及滤膜的制备1.6.1 垫片、膜、药用瓶、铝管等药用聚酯/铝/聚丙烯封口垫片、外用贴膏剂用聚酯非织造布及复合膜、口服固体药用瓶、铝管及软膏管分别按国家药包材标准中YBB00132005-2015、YBB00132002-2015、YBB00082002-2015、YBB00162002-2015微生物检查项下的要求进行供试液制备后,分别取相当于技术标准中的一个单位(一片垫片、100 cm2的复合膜、一个瓶、一支铝管)的供试液进行薄膜过滤[12]。
1.6.2 气雾剂阀门取样品10个,置于锥形瓶中,加入氯化钠注射液100 ml,振摇1 min,即得1∶10供试液。
取供试品溶液10 ml进行薄膜过滤。
1.6.3 口服固体药用瓶盖取本品10个,将无菌棉签用氯化钠注射液沾湿,在瓶盖内侧擦拭,然后立即剪断后将棉签投入盛有100 ml氯化钠注射液的锥形瓶中,换1支棉签,同法操作(即每个瓶盖用2支棉签擦拭)。
将瓶快速摇晃1 min,即得1∶10供试品溶液。
分别取供试品溶液10 ml进行薄膜过滤。
1.6.4 滤膜的冲洗每张滤膜冲洗3次,每次的冲洗液体积为50 ml。
取滤膜照中国药典2015年版四部通则1105进行需氧菌总数、霉菌及酵母总数检查,照中国药典2015年版四部通则1106进行控制菌检查。
1.6.5 菌落计数和控制菌检验需氧菌、酵母及霉菌菌落计数及控制菌E. coli、S. aureus和P. aeruginosa检验均按文献操作[11]。
1.7 方法适用性验证需氧菌、霉菌及酵母总数计数及控制菌方法适用性验证试验按照文献操作[11]。
加入的标准规定菌种量为50 cfu,即上述1.5中的菌液0.1 ml。
回收率(%)=(试验组菌数–供试品对照组菌数)/菌液对照组菌数×100%。
当5种标准菌的回收率均为50%~200%时,判定为合格。
2 结果对8种药品包装材料的微生物限度试验中的需氧菌、霉菌及酵母的方法适用性验证结果显示,标准菌株的回收率均为63%~143%(表1~2);在控制菌的验证试验中,大肠埃希菌的试验组和菌液对照组在封口垫片、药用复合膜、气雾剂阀门、口服固体药用瓶及药用瓶盖包装材料上的试验结果均是阳性,而供试品对照组则为阴性;金黄色葡萄球菌和铜绿假单胞菌的试验组和菌液对照组在非织造布、气雾剂阀门、药用复合膜、铝管和软膏管上的试验结果均是阳性,而供试品对照组则为阴性。
这些结果表明,这三种控制菌的方法适用性验证结果均符合药典相关要求,而8种药品包装材料的微生物限度试验方法也均适用。
3 讨论本实验对直接接触药品的包装容器及材料中的封口垫片、非织造布、复合膜、气雾剂阀门、口服固体药用瓶、瓶盖、软膏管、铝管8个产品的检验方法进行了适用性验证。
在菌落计数法中,金黄色葡萄球菌、铜绿假单胞菌、枯草芽孢杆菌、白色念珠菌和黑曲霉5种标准菌株回收率均为63%~143%,符合2015年版《中国药典》四部通则1105中50%~200%的要求。
大肠埃希菌、金黄色葡萄球菌及铜绿假单胞菌的控制菌方法适用性验证也均符合2015年版《中国药典》四部通则1106的要求。
对不同的药品包装材料的产品,应选择合适正确的检验方法,并对所选的方法进行适用性验证,只有当方法适用性验证结果符合要求时,才能证明所选用的方法标准是合理的,检测该类产品的结果是准确有效的。
但在标准中对于同类品种不同生产企业生产时是否进行方法适用性的验证未作明确规定,有待今后进一步完善[13-14]。
参考文献[1] 李永安,蔡弘,金宏,等. 药品包装实用手册[M]. 北京:化学工业出版社, 2003.[2] 周健丘,梅丹. 药品包装材料对药品质量和安全性的影响[J]. 药物不良反应杂志,2011, 13(1): 27-31.[3] 张艳霞,陈光伏. 药品包装用复合膜与铝箔实施微生物限度检验的方法[J]. 人参研究,2010(2): 42-43.[4] 战宏利,马仕洪,戴翬,等. 口服液体药用塑料瓶微生物污染状况分析及试验方法改进[J]. 药物分析杂志, 2014, 34(6): 1100-1105.[5] 胡昌勤,林平华,许华玉. 实用药品微生物检验检测技术指南[M]. 北京:人民卫生出版社, 2015.[6] 马绪英,苏德模. 药品微生物学检验手册[M]. 北京:科学出版社, 2001.[7] 曹晓云,郭艳娟. 药品微生物限度检查方法学验证试验及其相关的技术问题[J]. 天津药学, 2006, 18(6): 49-51.[8] 严杰,罗海波,陆德源. 现代微生物学实验技术及其应用[M]. 北京:人民卫生出版社, 1997.[9] 朱亚虹,黄凯,曾环想. 药品微生物检测的质量保证[J].中国医药指南, 2010, 8(33): 176.[10] 刘金凤. 分析药品微生物限度检验误差影响因素[J]. 食品与药品检验, 2014, 24(2): 188-189.[11] 国家药典委员会. 中华人民共和国药典2015年版四部[M]. 北京:中国医药科技出版社, 2015:附录通则1105-1106.[12] 中国食品药品检定研究院. 国家药包材标准(2015年版)[M]. 北京:中国医药科技出版社, 2015.[13] 柴海毅. 浅析微生物计数方法的回收率[J]. 医药工程设计,2013, 34(1): 30-31.[14] 宁黎丽,成海平,魏农农. 微生物学检查方法验证与评价要求[J]. 药物评价研究,2009, 32(1): 6-9.。