第三章 自由基聚合-1
第3章自由基聚合反应-1讲述

2020/10/4 19
在链增长反应过程中,不仅研究反应速率,还需考察 增长反应对大分子微结构的影响。
在链增长反应中,链自由基与单体的结合方式有两种:
(4)
实验证明,由于电子效应和空间位阻效应双重因素,都促 使反应以头-尾连接为主;但还不能做到序列结构上的绝对规整 性,由于链自由基是平面结构,在平面上下进攻的几率各为50 %,因此,从立体结构看来,自由基聚合物分子链上取代基在 空间的排布是无规的,所对应的聚合物往往是无定型的。
3.2.3聚苯乙烯
•PS
反应式
2020/10/4 11
特点
• PS由于具有质硬、透明、刚性,电绝缘性 好,吸湿性低,价格低廉和容易染色等特 点,被广泛用于各种仪器仪表零件、保温 材料和日常用品。
• PS主要缺点是性脆,耐热性差。
2020/10/4 12
3.2.4 聚甲基丙烯酸甲酯
反应式
PMMA
➢ 取代基为强烈推电子基(烷氧基、乙烯基、苯 基、烷基等)
➢ 带有共轭体系的烯类单体
2020/10/4 5
2. 单体结构对聚合能力的影响
➢不对称的单体比较容易聚合
➢取代乙烯比乙烯容易聚合
➢取代基的分布:1,1取代比22取代单体容易聚合 如:偏氯乙烯,a-甲基苯乙烯、甲基丙烯酸甲酯
➢空间阻碍的影响例:1,1-二苯乙烯
能耐60℃以下的一般有机溶剂。
2020/10/4 8
3.2.2.聚氯乙烯
反应式
PVC
2020/10/4 9
特点
• PVC是目前世界上仅次于聚 乙烯的第二大塑料品种。
•PVC具有难燃、抗化学药品 性、优良的电绝缘性和较高 的强度等特性。
•PVC一般使用温度-15~60℃,抗冲 击强度不理想。
高分子化学-第3章 自由基聚合
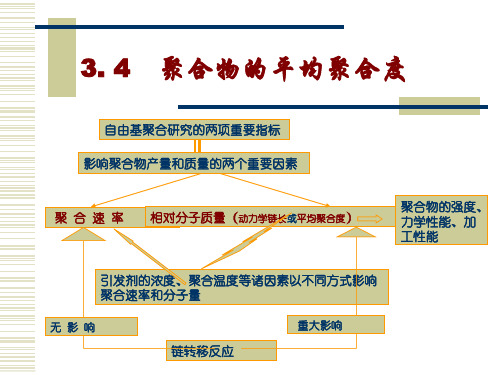
3. 4
聚合物的平均聚合度
1、动力学链长和聚合度
(1)动力学链长υ (kinetic chain length)的定义
每个活性种从引发阶段到终止阶段所消耗单体分子数。无 链转移时,动力学链长为增长速率和引发速率的比。 依据稳态时引发速率等于终止速率,则动力学链长可表 示为增长速率与终止速率的比: 即为单体消耗速率与
自由基产生(或消失) 速率之比
3. 4
聚合物的平均聚合度
如将稳态时的自由基浓度 入上式,可得下式:
,代
3. 4
聚合物的平均聚合度
若自由基聚合反应由引发剂引发时,
引发速率Ri = 2 f kd[I],则:
3. 4
聚合物的平均聚合度
可知动力学链长与引发速率存在以下关系:
1) 动力学链长与单体浓度的一次方成正比,与 引发剂浓度平方根成反比。 2) 说明了在自由基聚合体系中,增加引发剂用 量虽然可以提高聚合速率,但又使聚合物相对分子 质量降低。由此说明引发剂在自由基聚合中的重要
(1)温度对聚合速率的影响
总聚合速率常数k与温度T(K)遵循Arrhenius经验公式: 由前面推导可知: k=Ae-E/RT
k=kp(kd/kt)1/2
因此:
3.5 影响自由基聚合反应的因素
从而可知,总活化能E=(Ep-Et/2)+Ed/2
由Ep、 Et和Ed的大小可以得到总活化能E约为83 kJ/mol,为正值,表明温度升高,速率常数增大k增大。
3.5 影响自由基聚合反应的因素
1. 链自由基的双基终止过程的三步曲:
1) 链自由基的平移;
2) 链段重排,使活性中心靠近;
3) 双基相互反应而使链终止。
第二步(链段重排)是 控制步骤,受体系粘度 影响显著。
高分子化学课件第三章 自由基共聚合

m1= d[M1] = k11[M1*][M1] + k21[M2*][M1] (i)
m2 d[M2]
k12[M1*][M2] + k22[M2*][M2]
第三章 自由基共聚合
(3)假设共聚反应是一个稳态过程,即总的活性中心的浓 度[M1*+M2*]恒定,[M1*]和[M2*]的消耗速率等于[M1*]和 [M2*]的生成速率,并且 M1* 转变为M2*的速率等于M2*转 变为M1*的速率;
二元共聚合的理论研究较系统深入,而三元及三元以上共 聚合复杂,理论研究很少,但实际应用的例子颇多。ABS, SBS
三元以上聚合,一般以两种单体确定主要性质,另外单体 改性。
二元共聚物根据两单体单元在分子链上的排列方式可分四 类:
第三章 自由基共聚合
(1)无规共聚物(random copolymer) 两种单体单元的排列没有一定顺序,A单体单元相邻的单
第三章 自由基共聚合
四种竞争链增长反应:
k11 M1* + M1
k12 M1* + M2
k21 M2* + M1
k22 M2* + M2
M1* R11 = k11[M1*][M1]
M2* R12 = k12[M1*][M2]
M1*
R21 = k21[M2*][M1]
M2* R22 = k22[M2*][M2]
若含一段A链与一段B链,如~AAAAAAA-BBBBBBBBBB~, 称AB型二嵌段共聚物;如果是由一段A链接一段B链再届一 段A链,如~AAAAAA-BB~BBB-AAAAAAA~,则称ABA型 三嵌段共聚物;若由多段A链和多段B链组成,则称(AB)n型 多嵌段共聚物。
第三章 自由基共聚合
高分子化学第三章 自由基聚合

• 链转移反应前后,自由基的数目未变。
35
1. 向单体转移
· ~~CH2-CH + CH2=CH Cl Cl
· ~~CH=CH + CH3-CH Cl Cl
• 注意CH2=CHCl单体
36
2. 向溶剂或链转移剂转移
X ~~CH2CH · + YS X ~~CH2CHY + S ·
• 溶剂:
• 链转移剂:有较强的链转移能力的化合
1 2
[I ]
1
2
[M ] (3—35式)
注意本方程的适用范围
73
二、温度对聚合速率的影响
• 阿累尼乌斯公式:K=Ae–Ea/RT
其中:K=kp(kd/kt)½ 则:Ea=Ep+Ed/2–Et/2
74
一般情况下: Ep≈29kJ•mol–1, Ed≈126kJ•mol–1 Et≈17kJ•mol–1
10
一、 聚合的可能性
• 主要取决于双键上取代基的空间 效应
11
1.烯类单体: CXY=CMN
(1)一取代( CH2=CHX)
可均聚合
12
(2)二取代
(CH2=CXY、CHX=CHY) (a)1,1——二取代:一般不考虑空 间位阻效应,可均聚合。
注意:CH2=C(Ar)2只能形成二聚体
13
(b)1,2——二取代
54
2.半衰期
[I] ln = Kd t [I0]
• 60℃
ln2 t½ = K d
(3—17)
t½ >6h,低活性引发剂 1h< t½ <6h,中活性引发剂 t½ <1h,高活性引发剂
55
3. 引发效率
聚合物合成工艺-第3章

引发剂的分解速率,应与反应时间(停留时间)匹配
根据引发剂分解速率常数kd
在相同介质和温度下,不同引发剂的kd不同,kd 大者,分解速率快,活性高。
根据引发剂分解活化能Ed
Ed大者,分解的温度范围窄 如要求引发剂在某一温度范围内集中分解,则选
用Ed大者 反之,可选用Ed小者。
化率,是LDPE合成工艺研究的重点。
工艺概况
LDPE的合成工艺均由ICI公司的技术衍生而来,除反应 器、配方、工艺控制有所不同外,流程均大致相同。
生产流程示意图
兰化集团引进Basell公司20万t/aLDPE 装置工艺流程
流程简述
乙烯与分子量调节剂混合后,经一次压缩(25~30MPa) 后与循环乙烯混合,进入二级压缩机,出口压力110~ 400MPa(不同工艺,要求的压力不同)。
变宽 可通过控制反应过程中[S]/[M]值,控制分子量分布 比较常用的方法是分批次补加链转移剂。
链转移剂的选择
一般根据50%转化率-U1/2进行选择。 U1/2-链转移剂消耗50%时单体的转化率。
U1/2=100(1-0.51/Cs) 一般情况下,CS提高,U1/2下降。 根据反应的单体转化率要求,选择合适的链转移剂。 链转移剂的U1/2可查阅有关手册。
物理机械性能产生重要影响。
聚乙烯的主要分类
a. 低密度(高压)聚乙烯(LDPE)
密度为0.915~0.930 g/cm3的均聚物
自由基 共聚合
含少量极性基团的乙烯-醋酸乙烯酯共聚物-EVA
乙烯-丙烯酸乙酯共聚物-EAA
b.线性低密度和中等密度聚乙烯(LLDPE、MDPE)
乙烯、α-烯烃(1-丁烯、1-己烯或1-辛烯)的共聚物
教材习题参考答案解析_第三章自由基聚合

教材习题参考答案第三章自由基聚合思考题1.烯类单体家具有下列规律: ①单取代和1,1-双取代烯类容易聚合, 而1,2-双取代烯类难聚合;②大部分烯类单体能自由基聚合,而能离子聚合的烯类单体却很少,试说明原因。
2. 下列烯类单体适于何种机理聚合?自由基聚合、阳离子聚合还是阴离子聚合?并说明原因。
CH2=CHCl CH2=CCl2 CH2=CHCN CH2=C(CN)2 CH2=CHCH3 CH2=C(CH3)2 CH2=CHC6H5 CF2=CF2 CH2=C(CN)COOR CH2=C(CH3)-CH=CH2答:CH2=CHCl:适合自由基聚合,Cl原子是吸电子基团,也有共轭效应,但均较弱。
CH2=CCl2:自由基及阴离子聚合,两个吸电子基团。
CH2=CHCN:自由基及阴离子聚合,CN为吸电子基团。
CH2=C(CN)2:阴离子聚合,两个吸电子基团(CN)。
CH2=CHCH3:配位聚合,甲基(CH3)供电性弱。
CH2=CHC6H5:三种机理均可,共轭体系。
CF2=CF2:自由基聚合,对称结构,但氟原子半径小。
CH2=C(CN)COOR:阴离子聚合,取代基为两个吸电子基(CN及COOR)CH2=C(CH3)-CH=CH2:三种机理均可,共轭体系。
3. 下列单体能否进行自由基聚合,并说明原因。
CH2=C(C6H5)2 ClCH=CHCl CH2=C(CH3)C2H5 CH3CH=CHCH3CH2=CHOCOCH3 CH2=C(CH3)COOCH3 CH3CH=CHCOOCH3 CF2=CFCl答:CH2=C(C6H5)2:不能,两个苯基取代基位阻大小。
ClCH=CHCl:不能,对称结构。
CH2=C(CH3)C2H5:不能,二个推电子基,只能进行阳离子聚合。
CH3CH=CHCH3:不能,结构对称。
CH2=CHOCOCH3:醋酸乙烯酯,能,吸电子基团。
CH2=C(CH3)COOCH3:甲基丙烯酸甲酯,能。
《材化高分子化学》第3章 自由基聚合
E = 105~150 kJ/mol (3—1)
kd = 10-4~10-6 s-1
(3—2)
19
第三章 自由基聚合
b. 单体自由基的形成
R + CH2 CH X
RCH2 CH X
由初级自由基与单体加成产生,为放热反应, 活
化能低,反应速度快。
E = 20 ~ 34 kJ/mol
(3—3)
20
第三章 自由基聚合
(CH3)2C N N C (CH3)2
2 (CH3)2C + N2
CN
CN
CN
优点:
(1)分解只形成一种自由基,无诱导分解。 (2)常温下稳定。80℃以上剧烈分解。
35
第三章 自由基聚合
(2)有机过氧化类引发剂
最简单的过氧化物:过氧化氢。活化能较高, 220kJ/mol,一般不单独用作引发剂。
HO OH 2HO
7
第三章 自由基聚合
分子中含有推电子基团,如烷基、烷氧基、苯基、乙 烯基等,碳=碳双键上电子云增加,有利于阳离子聚合进 行。
δ
CH2 CH Y
注意:丙烯分子上有一个甲基,具有推电子性和超共轭双 重效应,但都较弱,不足以引起阳离子聚合,也不能进行 自由基聚合。只能在配位聚合引发体系引发下进行配位聚 合。
30
第三章 自由基聚合
3.4.2 自由基聚合反应的特征
(1)可分为链引发、链增长、链终止等基元反应。 各基元反应活化能相差很大。其中链引发反应速率 最小,是控制聚合过程的关键。
慢引发、快增长、有转移,速终止。
与逐步缩聚机理特征比较见p75表3-6。
31
第三章 自由基聚合
(2)只有链增长反应使聚合度增加。从单体转化为 大分子的时间极短,瞬间完成。体系中不存在聚合 度递增的中间状态(p75图3-2)。聚合度与聚合时间 基本无关。
第三章 自由基聚合新引发体系

水溶性:氧化剂:过氧化氢、过硫酸盐、氢过氧化物等;等)和有机还原二甲基苯)。
67二(2-抗生素:利福平(Rifampin)抗肿瘤:噻替哌10羟乙酯)药奋乃静(PERP )11BPO +奋乃静-MMA (反应动力学:氧化还原体系)12BPO +奋乃静-MMA 、HEMA 药物包埋而后释放。
(MP)2PT1518引发机理:氢键H +转移19过硫酸盐+脂肪胺体系过硫酸铵+脂肪胺体系-引发丙烯酰胺(AAM )的水溶液聚合。
过硫酸铵-脂肪二胺中,二胺的引发活性顺序:20)的21活性最高。
与还原剂如醇、醛、2324甲苯基氨基甲酸丁酯z Ce(Ⅳ) 离子+1,3-二羰基化合物体系2630处在两个羰基有碳中心自由基和离子氧化还原体系-引发机理二羰基化合物34 353738Poly(MAMT-co-VAc)聚甲基丙烯酰丙酮(PMAA)接枝AAM的聚合4243聚(对甲基丙烯酰胺基)苯甲酰丙酮(PMPAPB )接枝AAM 的聚合自由基聚合年由日本学者大津隆行是将引发、转移、46自由基聚合TDCA );铜试剂(N ,N-二乙基二硫代氨基甲酸钠) 的乙醇溶液自由基聚合1 引发单体聚合,2 参加终止反应48转化率49自由基聚合大分子光引发转移终止剂制备嵌段共聚物:自由基聚合光化学:研究由紫外-可见光引起的分子激发,激发态的性质年,大量的研究结果发现,不是所180nm 270~350nm π-π*n -π*对于多原子分子和在适当压力下的氮原子气体,其激发态有多种失去1.电子状态之间的非辐射转变,放出热能;从激发态最低振动能级返回基态各振动能级所发射的光。
比吸收光谱波长长,比吸收光谱更圆滑,比吸收光谱受溶在分子间的能量传递过程中,受激分子通过碰撞或较原距离的传递将能量转移给另一个分子,本身回到基态。
接受能量的分子上升为激发感光高分子是指吸收了光能后在分子内或分子间产生化学物理变化的:光交联型、光聚合型、光氧化还原型、光二吸收波长(nm)具有感光基团的高分子:感光基团直接连接在高分子链上,在光作用下激发成活性基团,从而进一步形成交联结构的聚合物。
湖北大学高分子化学第三章 自由基聚合 总结

fkd kt
1/2 [I]1/2 [M]
[I] [M]
聚合反应速率
控制方向 RP RP
ERp = EP + Ed/2 - Et/2 >0
T反
RP
阻聚剂、缓聚剂
CZ 、[ Z ]
RP
引发剂
活性
RP
要求能从理论上进行解释和综合应用
总结
聚合反应速率
习题9
从理论上对引发剂引发自由基聚合的 聚合工艺及实验现象进行解释
小样2号比3号的相对分子质量稍低(?) 6、动力学分析表明:Rp = K [ M ]1.5[ I ]0.8(?) 7、如要进一步提高生产效率,可采取的有效措施(?)
聚合度 控制
总结
一、自由基寿命、动力学链长
聚合度控制
ν= Rp/Ri = Rp/Rt τ= [M·]/Rt
_
kP (2k t )1/2
聚合反应 速率控制
总结
基元反应
链引发:形成单体自由基活性种的反应
二步反应:
I
2R· (初级自由基)
R· + M → RM·(单体自由基)
链引发?
一 、
链增长:形成高分子链自由基的反应 n步反应: RM· + M → → → → ~~~~~~~~~~M·
基
决定大分子链的结构(序列、立构……)
自由基聚合 对结构的 控制能力
动力学方程:
1、三个假设
1/2
RP
k
P
fkd kt
[I]1/2 [M]
等活性、稳态、聚合度很大
2、与[I]成1/2次方, [M]成1次方的讨论(P78)
3、应用范围
武汉大学高分子化学 第三章自由基链式聚合反应1

3.3 自 由 基 聚 合 机 理
3.3.1 自由基的活性及其反应
(1)自由基的活性
自由基的活性和它的结构、电子效应、位阻效应有关。
各种自由基的相对活性次序大致如下:
H· >CH3· 6H5· >C >Cl >R . >RCH2· 2CH· 3C· 3C· . >R . >Br3C· >RCHCOR>RCHCN>RCHCOOR >CH2=CHCH2· 6H5CH2· >C >(C6H5)2CH· > Cl· >(C6H5)3C·
(ii) X为给(推)电子基团:
H2C CH X
增大电子云密度,易 与阳离子活性种结合
H R CH2 C X 分散正电性,稳定 阳离子活性中心
因此, 带有给(推)电子基团的烯类单体易进行阳离子聚合, 12 如 X = -R,-OR,-SR,-NR2等。
(iii) X为吸电子基团
H2C CH X
降低电子云密度,易 与富电性(阴离子)活 性种结合
10) CH2=CHNO 2
11)
CH2=C(Cl)2
答案:1、2、4、8、10、11易发生自由基加成聚合反应; 3、5不易发生自由基加成聚合反应,因为R、OR为推电子基团; 7、9不易发生自由基加成聚合反应,因为1,2二取代空间位阻大,且结构对称; 20 6不易发生自由基加成聚合反应,因为二个CN基的吸电子性太强。
R CH2
H C X
分散负电性,稳定活性中心
由于阴离子与自由基都是富电性的活性种,因此带吸电子 基团的烯类单体易进行阴离子聚合与自由基聚合, 如X = -CN,-COOR,-NO2等。
13
(iv) 具有共轭体系的烯类单体
p电子云流动性大,易诱导极化,可随进攻试剂性质的不 同而取不同的电子云流向,可进行多种机理的聚合反应。 如苯乙烯、丁二烯等。 + + R H2C CH R H2C CH
三章节自由基聚合反应

H R CH2 C
X
分散正电性,稳定阳离子
所以带给电子基团旳烯类单体易进行阳离子聚合, 如X = -R,-OR,-SR,-NR2等。
(b) X为吸电子基团
H2C CH X
降低电子云密度,易 与富电性活性种结合
H R CH2 C
X
分散负电性,稳定活性中心
所以带吸电子基团旳烯类单体易进行阴离子聚合与自由基, 如X = -CN,-COOR,-NO2等。
常用旳偶氮二异丁腈(AIBN) 分解反应机理如下:
CH3
CH3
H3C C N N C CH3
CN
CN
CH3 2 H3C C
CN
+ N2
(3)氧化还原体系: 过氧化物+还原剂
将具有氧化性旳化合物(一般是过氧化物)与具有还原性旳化合 物配合,经过氧化-还原反应产生初级自由基引起聚合反应,该类 引起体系称氧化-还原引起体系。
(CH3)2 C N N C(CH3)2
CN
CN
2 (CH3)2 C + N2
羰基化合物和某些杂环化合物,其中以烯烃最具实际应用意义。 评价一种单体旳聚合反应性能,应从两个方面考虑:首先是其
聚合能力大小,然后是它对不同聚合机理如自由基、阳离子、阴离 子聚合旳选择性。
(1)位阻效应决定单体聚合能力
H2C CH X
一取代烯烃
Y H2C C
X
1,1-二取代烯烃
一取代烯烃和1,1-二取代烯烃原则上都能进行聚合,原因是活性 中心可从无取代基旳β-碳原子上攻打单体。除非取代基体积太大, 如带三元环以上旳稠环芳烃取代基旳乙烯不能聚合,1,1-二苯基乙 烯也只能聚合生成二聚体而得不到高聚物。
kd
第三章 自由基聚合

C-Z 单键不对称,异裂后具有类似于离子的特性, 可由阴离子或阳离子引发剂来引发聚合,不能进行自
由基聚合。
乙烯基单体对聚合机理的选择
乙烯基单体取代基 Y的电子效
应决定了单体接受活性种的进攻的 方式和聚合机理的选择。
(1)电子效应 诱导效应(Induction Effect):取代基的供、吸电子性 共轭效应(Resonance Effect):由于轨道相互交盖而 引起共轭体系中各键上的电子云密度发生平均化
第三章 自由基聚合
Free-raBiblioteka ical Polymerization
3.1 加聚和连锁聚合概述
自由基聚合(Free–radical Polymerization) 连锁聚合 按活性中心 阳离子聚合(Cationic Polymerization) 阴离子聚合(Anionic Polymerization)
Rp(增长总速率) >> Rt(终止总速率)
链转移(Chain Transfer):
从单体(Monomer)
链自由基
夺取原子
从溶剂(Solvent) 从引发剂(Initiator)
从大分子(Macromolecule)
失去原子的分子成为新自由基,继续新链增长(链 转移),链转移反应不仅将影响聚合物的分子量,也常 常形成支链。
3.4 自由基聚合机理
1)自由基聚合的基元反应(Elementary Reaction) 链引发、链增长、链终止、链转移等基元反应 链引发(Chain Initiation): 形成单体自由基的基元反应,由两步组成: 初级自由基(Primary Radical) R • 的形成
单体自由基(Monomer Radical) M • 的形成
自由基聚合反应1

取代基与聚合反应类型简列:
10
11
12
3.2 连锁聚合反应热力学(了解)
一、聚合热
1
1
13
△G旳符号取决于△H、△S旳正负及大小。
14
当温度在25~100℃内: -T△S=105×298~125×373 =31~47 KJ/mol
△G =△H-T△S <0
从热力学角度分,聚合多能自动进行。
定义为聚合反应旳临界上限温度或极限温度25
3.3 自由基聚合反应机理
一、自由基旳产生及其活性 自由基:
凡带有未配对独电子旳原子、分子或原子团叫自由基。
1.自由基旳产生方式:均裂和电子转移
弱共价键旳均裂
过氧化苯甲酰(BPO)
26
单电子转移旳氧化还原反应
27
2. 自由基旳活性
影响自由基旳活性旳两个主要原因:
1,1-二取代
1,2-二取代
5
唯一旳例外是当取代基为F时,它旳一、二、 三、四取代乙烯都能够参加聚合反应
聚四氟乙烯
6
二、取代基旳电负性和共轭性决定烯烃旳聚 合反应类型
(1)带吸电子取代基旳烯烃——能够进行自由基型 和阴离子型两种聚合反应
7
(2) 带推(供)电子取代基旳烯烃——能够进行阳 离子型聚合
44
4 氧化还原体系
过氧化氢-亚铁离子: 异丙苯过氧化氢-亚铁离子: BPO-叔胺体系:
45
氧化还原体系特点:
• 引起温度低 • 引起效率相对较低
46
二、引起剂分解动力学
结论:引起剂旳分解属于单分子一级分解,其 浓度降低速率旳自然对数与反应时间成正比。 47
引起剂活性高下旳判断
半衰期:反应物浓度降低二分之一所需旳 时间
第3章 自由基聚合生产工艺-1

常用的链转移剂有丙烷、氢、丙烯等。
丙烷是较好的调节剂,若反应温度>150℃,它能平 稳地控制聚合物的分子量。
氢的链转移能力较强,反应温度高于170℃,反应很不 稳定。
(5)单体纯度的影响 乙烯单体中杂质越多,会造成聚合物分子量降低,且 会影响产品各种性能,工业上,对乙烯的纯度要求超过 99.95%。
乙烯高压聚合生产流程
压缩机 二次 压缩机 150--250MPa 釜 式 反 应 器
引发剂 25MPa
管 式 反 应 器
0.1MPa
低压分离器
乙烯 分子量调节剂 减 压 阀 一次 压缩机 25MPa 高压分离器 减压阀
挤出 造粒机
新鲜乙烯
流程说明:
裂解厂的新鲜乙烯(压力为1.5MPa)与闪蒸气体升压器来的 乙烯混合,经过第一级压缩机,压力升到20-30MPa,冷却后, 在二级压缩机第一段入口与中压分离器分离出来的未反应乙烯 会合,进入压缩机使压力升到120-200MPa,冷却后进入聚合反 应器,在150-300度过氧化物和有机过酸酯引发下,乙烯聚合成
非均相本体聚合——聚氯乙烯本体聚合生产
本体浇铸聚合——有机玻璃生产
气相本体聚合——高压聚乙烯生产
法国本体法(PSG法)制聚氯乙烯
氯乙烯两段本体聚合生产聚氯乙烯是法国圣戈班公司 (PSG)首先工业化。 一、主要原料及规格 沸点 纯度 -13.9 >99.99%
法国本体法(PSG法)制聚氯乙烯
二、制法 聚氯乙烯可用悬浮聚合、乳液聚合、本体聚合实施方法 生产,目前仍以悬浮聚合法为主,而本体聚合法生产的聚 氯乙烯约占10%。
(2)温度的影响 操作温度:130℃~280℃ 温度升高将使聚合物的分子量相应降低,聚乙烯分子链
第3章2015-自由基聚合反应-01详解

第 三 节 聚合热力学和聚合-解聚平衡
热力学: 聚合倾向或聚合-解聚平衡; 动力学: 单体/引发剂/温度和聚合速率等。 PE/α-methystyrene 3.1 聚合热力学的基本概念
nM Initial state n Final state M
G= H-T S
1) ΔH<0和ΔS<0: 在某一临界温度, ΔG=0, →聚合上线温度Tc。 Tc=ΔH/ΔS 2) ΔH>0和ΔS>0: 聚合下线温度Tf: Tf=ΔH/ΔS 八元环硫/硒→线性聚硫/聚硒。 3) ΔH<0和ΔS>0: ΔG<0 4) ΔH>0和ΔS<0: ΔG>0 下一页 返回
第三章 自由基聚合
第 一 节 加聚和连锁聚合概述 第 二 节 链(锁)式聚合反应的单体 第 三 节 聚合热力学和聚合-解聚平衡 第 四 节 自由基聚合机理 第 五 节 引发剂和其它引发作用 第 六 节 自由基聚合反应动力学—聚合速率 第 七 节 动力学链长和聚合度 第 八 节 链转移反应和聚合度 第 九 节 分子量/聚合度分布 第 十 节 阻聚和缓聚 第十一节 自由基寿命及动力学参数的测定 第十二节 可控/“活性”自由基聚合
O
-NO2、 -CN、-C
阴离子聚合 自由基聚合 阳离子聚合
OCH3、 -CH=CH2、C6H5、-CH3、-OR
吸电子能力增强
供电子能力增强
► 能进行自由基链式聚合的单体多数是具有双键的烯类单体。 但并非具有双键的化合物都能聚合—聚合能力。 上一页 下一页
2.2 取代基-Y的空间位阻效应 → 体积/数量/位置对聚合能力有影响 ► 单取代乙烯类: 与乙烯比, 即使体积较大, 也易聚合。 如N-乙烯基咔唑/N-乙烯基吡咯烷酮 ► 1,1-二取代烯类单体CH2=Cxy: 一般按取代基的性质进行相应机 理的聚合。* 但CH2=C(Φ)2, 只能形成二聚体。 ► 1,2-二取代烯类单体xCH=CHy: 结构对称, 极化程度低, 加上位 阻效应, 一般不能均聚或形成二聚体。如CH3CH=CHCH3、 ClCH=CHCl、CH3CH=CHCOOCH3 。*但可共聚, 如马来酸酐 难均聚, 但可与St/VAc共聚。 ► 三取代/四取代烯类单体: 一般不能聚合。 ► 氟代乙烯类: 无论氟代数量和位置如何, 均易聚合。 →Table 3-2 上一页 返回
第3章自由基聚合习题参考答案

第3章自由基聚合习题参考答案第3章自由基聚合-习题参考答案1、判断下列单体能否进行自由基聚合?并说明理由H2C CHCl H2C CH H2C CCl2H2C CH2H2C CH2C CHCN H2C C(CN)2H2C CHCH3F2C CF2ClHC CHClH2C CCH3COOCH3H2C CCNCOOCH3HC CHOC COO答:(1)可以。
Cl原子的诱导效应为吸电性,共轭效应为供电性两者相抵,电子效应微弱,只能自由基聚合。
(2)可以。
为具有共轭体系的取代基。
(3)可以。
结构不对称,极化程度高,能自由基聚合。
(4)可以。
结构对称,无诱导效应共轭效应,较难自由基聚合。
(5)不能。
1,1—二苯基乙烯,二个苯基具有很强的共轭稳定作用,形成的稳定自由基不能进一步反应。
(6)可以。
吸电子单取代基。
(7)不可以。
1,1双强吸电子能力取代基。
(8)不可以。
甲基为弱供电子取代基。
(9)可以。
氟原子半径较小,位阻效应可以忽略不计。
(10)不可以。
由于位阻效应,及结构对称,极化程度低,难自由基聚合(11)可以。
1,1-双取代。
(12)可以。
1,1-双取代吸电子基团。
(13) 不可以。
1,2-双取代,空间位阻。
但可进行自由基共聚。
2、试比较自由基聚合与缩聚反应的特点。
答:自由基聚合:(1)由链引发,链增长,链终止等基元反应组成,其速率常数和活化能均不等,链引发最慢是控制步骤。
(2)单体加到少量活性种上,使链迅速增长。
单体-单体,单体-聚合物,聚合物-聚合物之间均不能反应。
(3)只有链增长才是聚合度增加,从一聚体增加到高聚物,时间极短,中间不能暂停。
聚合一开始就有高聚物产生。
(4)在聚合过程中,单体逐渐减少,转化率相应增加(5)延长聚合时间,转化率提高,分子量变化较小。
(6)反应产物由单体,聚合物,微量活性种组成。
(7)微量苯酚等阻聚剂可消灭活性种,使聚合终止。
缩聚反应:(1)不能区分出链引发,链增长,链终止,各部分反应速率和活化能基本相同。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
并引发单体之间以共价键链接的基团。
6
——共价键(Covalent bond)的断裂 引发剂分解成活性中心时,共价键有两种裂解形式:均裂 (Homolysis)和异裂(Heterolysis)。均裂的结果产生两个各
带1个电子的自由基R · ;异裂的结果共价键上的一对电子全归
属于某一基团,形成一个富电子阴离子(Anion );同时也产生 一个缺电子的阳离子(Cation)。
热力学讨论范围:反应的可能性、反应进行的方 向以及平衡方面的问题。
23
聚合反应热力学特征
The overall thermodynamics of the polymerization
of alkenes(烯烃) is quite favorable. The value of
∆G given by:
共轭效应(Resonance Effect):由于轨道相互交盖而引 起
共轭体系中各上的电子云密度发生平均化的一种电子效应。
13
乙烯基单体取代基 X
无取代基:乙烯(Ethylene)CH2=CH2
乙烯:乙烯分子中无取代基,结构对称,因此无 诱导效应和共轭效应。只能在高温高压下进行自由基 聚合,得到低密度聚乙烯。在配位聚合引发体系引发 下可进行常温低压配位聚合,得到高密度聚乙烯。
Free radical Polymerization
★
连锁聚合
Chain polymerization
离子聚合
Ionic polymerization
聚合反应机理
Polymerization Mechanism
缩聚反应
逐步聚合
Step polymerization 有无小分子
Condensation Polymerization
11
羰基(Carbonyl)化合物:
不能自由基聚合
只能离子聚合
杂环(Heterocyclics):
12
乙烯基单体对聚合机理的选择
(1) 电子效应(Electronic Effect)
乙烯基单体取代基Y的电子效应将改变双键的电 子密度,决定了单体接受活性种进攻的方式和聚合 机理的选择。
诱导效应(Induction Effect):取代基的供、吸电子性。
应较弱,所以VC等卤原子取代的乙烯单体只能自由基聚
合。
17
带共轭体系的烯类:如苯乙烯(Styrene)、α -甲基苯
乙烯(α - Methyl Styrene)、丁二烯(Butadiene) 及
异戊二烯(Isoprene),电子流动性较大,易诱导极化, 能按三种机理进行聚合。
18
乙烯基单体中的取代基对取代基X电负性次序和 聚合倾向的关系如下:
所以,一般情况下烯类单体的聚合焓:
27
2) 聚合热与单体结构的关系
不ห้องสมุดไป่ตู้单体的聚合热( -Δ H )实验值相接近,但也有一 些单体的聚合热与估算值偏离较大,其原因在于单体结构对 聚合热有显著的影响。
(a) 取代基的位阻效应(Steric Effect)
带取代基的单体聚合后变成大分子,
取代基之间的夹角从120 o 变成109 o,即 相对于单体而言,大分子上的取代基之 间的空间张力变大,使聚合物的能级提 高,聚合热则变小。
(Polymerization Thermodynamics and Polymerization– depolymerization Equilibrium)
一个聚合反应如果在热力学上是不允许的,那么在任何
条件下都不可能发生。但要实现一个在热力学上可行的聚合
反应,还依赖于动力学因素,即在给定的反应条件下,能否 获得适宜的聚合反应速率。
(Polymerization of Allyl Monomers on the Mechanism Selective)
**
烯类单体(vinyls)通过双键打开发生的加
成聚合反应大多属于连锁聚合。
烯类单体:包括单取代和1,1-双取代的烯类单
体、共轭二烯类单体。
10
乙烯类单体的碳—碳双键既可均裂,也可异 裂,取决于取代基的诱导效应和共轭效应,从而 决定着单体进行自由基聚合,还是阴离子或阳离 子聚合。
14
X为供电取代基(Electron-donating Substituent)
如烷基、苯基、乙烯基
供电基团使-C=C-电子云密度增加,有利于阳离子的 进攻,分散碳阳离子电子云而形成共振稳定的增长种 (Cationic Propagating Species)。
R⊕
因此带供电取代基的烯类单体易进行阳离子聚合, 如X = -R,-OR,-SR,-NR2等。
R A R B 2R A + B
自由基、阴离子和阳离子均有可能作为连锁聚合的活性 中心,因此有自由基聚合、阴离子聚合和阳离子聚合之分。
7
连锁聚合发生的可能性
① 热力学可能性(Thermodynamic Feasibility)
△G=G2 - G1(Free energy difference)<0 ② 动力学可能性(Kinetic Feasibility)
阳离子聚合 取代基X: NO2 CN COOCH3 CH=CH2
自由基聚合 阴离子聚合
C6H5 2CH3 OR
19
(2) 位阻效应(Steric Effect)
由单体中取代基的体积、位置、数量等引起的, 在动力学上对聚合能力有显著影响,但对聚合机理 的选择性却无甚关系。
单取代的烯类单体,即使侧基很大,如N-乙烯基
RM* + M→ RM2* RM2* + M→ RM3* ……………… RMn-1* + M→ RMn*
(链增长活性中心 或增长链)
链终止 Termination
RMn* → “死”大分子 (聚合物链)
聚合过程中有时还会发生链转移反应,但不是必须经
9
过的基元反应。
3.2 烯类单体对聚合机理的选择性
4
自由基聚合是至今为止研究最为透彻的高分子合
成反应。其聚合产物约占聚合物总产量的60%以上。
特点:单体来源广泛、生产工艺简单、制备方法
多样。
自由基聚合是最重要的高分子合成反应之一
5
3.1 加聚和连锁聚合概述
(Introduction of Addition Polymerization And Chain Polymerization)
15
X为吸电取代基(Electron-withdrawing
如腈基Cyano、羰基Carbonyl
Substituent)
使双键电子云密度降低;
使阴离子增长种(Anionic Propagating Species)共轭稳定
因此带吸电取代基的烯类单体易进行阴离子与自由基
16
聚合,如X = -CN,-COOR,-NO2等。
单体之间可形成氢键而使单体稳定,使单体能级下降。
而在聚合物中,这种氢键稳定作用虽然也存在,但由于受到
大分子链的约束而大大降低。这样的净结果是单体与聚合物 的能级差变小,聚合热下降。同样的道理,溶剂化作用也使 聚合热下降。
例:
与乙烯相比,异丁烯的聚合热下 降是由甲基的位阻效应和甲基的超共 轭效应的叠加而引起的。 a-甲基苯乙烯,由于两个取代基 的位阻效应、苯基的共轭效应和甲基 的超共轭效应的共同作用,使该单体 的聚合热大大降低,仅为35kJmol-1。 而对于丙烯酸、丙烯酰胺等单体, 聚合热的降低则主要是由氢键缔合对 单体的稳定化作用而引起的。
Chapter 3 Free Radical Polymerization
1
主要内容(8 学时)
加聚和连锁聚合概述 聚合热力学(难点) 自由基聚合机理(重点)
自由基聚合动力学(难点)
聚合物的平均聚合度(重点)
影响自由基聚合反应的因素(重点)
阻聚和缓聚
2
活性中心 Reactive center
自由基聚合
各种单体的聚合熵一般变化不大,约-105∽-125
J⋅mol-1⋅K-1,而不同单体的聚合热ΔH变化较大。聚 合热愈大,聚合反应的热力学障碍愈小,聚合越容易 进行。
1) 聚合热的确定:
a. 实验测定:直接量热法、燃烧热法和热力学平衡法 反应绝 热量热 单体与聚合物 的燃烧热差
b. 理论估算:① 由标准生成热估算
大多数乙烯基单体都能进行自由基聚合。 许多带吸电子基团的烯类,如丙烯腈(Acrylonitrile)、 丙烯酸酯类(Acrylate)能同时进行阴离子聚合和自由基 聚合。
若基团吸电子倾向过强,如硝基乙烯(Nitroethylene)、
偏二腈乙烯等,只能阴离子聚合。 卤乙烯(Vinyl Chloride,VC) :卤原子的诱导效应是吸 电子性,但共轭效应却有供电性,两者相抵后,电子效
► 三、四取代乙烯:一般不聚合,但氟代乙烯例外
21
小 结
热力学可能性的单体首先从取代基的空间位阻效
应来判断单体能否聚合;
然后通过电子效应来判断属于哪一类的聚合:
a) 共轭单体能按三种机理聚合;
b) 带有吸电子基团的单体可自由基和阴离子聚合;
c) 带有供电子基团的单体则只能阳离子聚合;
22
3.3 聚合热力学和聚合-解聚平衡
G H TS 0
24
单体变为聚合物,体系的无序性减小,故 ΔS < 0 在链式聚合反应中,单体双键打开形成聚合物的
单键,为放热过程,ΔH < 0 。-ΔH 被定义为聚合反 应的聚合热。 从焓变的角度看是有利于聚合反应的进行,而从 熵变的角度看却是不利聚合反应的进行。 因此,要使聚合反应进行,必须:
聚合热的大小一般也可用来粗略判断聚合物的热稳定