3.2.5 自由基聚合反应速率
自由基聚合速率控制

Rp
(MMA) 3
(S) 2
c(I) 图2.10 聚合速率Rp与引发剂浓度c(I)的关系 1-MMA,ABIN,50℃; 2-St,BPO,60℃; 3-MMA,BPO ,50 ℃
c(M)
图 2.11甲基丙烯酸甲酯自由基聚合 初速与单体浓度的关系曲线
2.7 自由基聚合反应的速率
3. 聚合温度对聚合速率的影响 kd 1 / 2 令 k = kp ( ) kt 1 1 ( E E kd 1 / 2 Ad 1 / 2 p 2 d 2 Et ) / RT k kp ( ) Ap ( ) e kt At
*
Hale Waihona Puke **图2.13自动加速现象示意图
fkd 1 / 2 Rp = kp ( ) c(M)c(I)1 / 2 kt
kp /( kt )
1/ 2
2.7 自由基聚合反应的速率
在低转化率情况下,聚合体系处于稳态,但在高转化率时,聚 合体系偏离稳态 , 聚合体系出现自动加速现象。直到聚合后期, 聚合速率才渐渐减慢。在很长一段时间内二者变化方向相反 , 二者叠加的结果,使聚合转化率-时间关系曲线呈S型。
四、自由基聚合过程中聚合速率的变化 ⒈ 正常聚合的速率”和“自动加速”现象 自由基聚合过程中的聚合速率由“正常聚合的速率”和 “自动加速”(autoacceleration)两部分叠加构成。 正常聚合的速率: 在低转化率情况下,聚合体系处于稳态时的速率。 正常聚合的速率遵循式(2.20)
fkd 1 / 2 Rp = kp ( ) c(M)c(I)1 / 2 kt
① ② (2.28)
取Ed1=130, Ed2=105,并设聚合温度为T=323K(50℃),气体常 数R=8.31J/mol.K,得 k2 k2 (130 -105)× 103 105 lg = = 2.02 k1 k1 2 ×2.303×8.31×323
自由基聚合反应简介

高分子化学
3.5 影响自由基聚合反应的因素
2、匀速聚合 当引发剂的半衰期适当时,正常聚合速率的衰减与凝胶效应的 自动加速部分可互补,从而可做到匀速反应。 3、前快后慢的聚合反应 当采用引发剂活性很高时,聚合 初期引发剂大量分解,使正常聚 合速率很大,聚合中后期,因引 发剂残留量少,正常聚合速率下 降幅度大,以致于凝胶效应的自 动加速部分还不足以弥补,故总 聚合速率降低,甚至为零。
CH2 CHR + O 苯醌
CH O CH2 CHR O CHR + HO HO CH2 CHR O O
O
CH2 CHR O
O
(17)
生成的自由基由于有苯环的强共振作用而稳定,引发活性低。
极性效应对苯醌(缺电子物质)的阻聚作用有显著影响。
18
高分子化学
3.6 阻聚和缓聚
• 芳族硝基化合物
(iii) 硝基化合物:硝基苯、多硝基苯
高分子化学
3.5 影响自由基聚合反应的因素
(3)链自由基卷曲、包埋的程度以及聚合速率的大小受聚合物在单 体或溶剂中溶解性能的好坏的影响。通常,自动加速现象在良溶剂 中较少出现,在非溶剂(沉淀剂)中出现得早、显著,在不良溶剂 中自动加速现象介于以上两种情况。
图2 溶剂对MMA聚合时自动加速效应的影响 (1~3—采用非溶剂;4~5—采用不良溶剂;8~10—采用良溶剂)
氯化铁不仅阻聚效率高,并能化学计量地消灭一个自由基,因 此,可用于测定引发速率。
22
高分子化学
自由基聚合反应机理

自由基聚合反应机理1. 引言自由基聚合反应是一种重要的有机化学反应,广泛应用于聚合物的合成和有机合成领域。
自由基聚合反应的机理对于合理设计反应条件和控制反应过程具有重要意义。
本文将简要介绍自由基聚合反应的机理及相关的反应条件和控制方法。
2. 自由基聚合反应的基本概念自由基聚合反应是指通过自由基的聚合反应来合成聚合物的过程。
在自由基聚合反应中,自由基分子通过聚合反应连续添加到聚合物链上,从而实现聚合过程。
聚合物链的生长是以共轭双键或其他反应位点为基础的。
3. 自由基聚合反应的机理自由基聚合反应包括引发步骤、传递步骤和终止步骤。
下面将逐个介绍这些步骤的机理。
3.1 引发步骤在自由基聚合反应中,反应的开始需要引发剂作为引发步骤的催化剂。
引发剂会被激活形成自由基,通常是通过热量、光或化学剂的作用来实现。
引发剂能够引发起反应的原因在于它能够提供链建立反应起点所需的自由基。
3.2 传递步骤在自由基聚合反应的传递步骤中,自由基分子会逐一添加到聚合物链的末端,并延长聚合物链的长度。
这个过程中,自由基通过与单体分子反应,将自由基转变为共轭双键或其他反应位点,从而继续聚合的过程。
3.3 终止步骤自由基聚合反应的终止步骤是不可逆的,通过各种反应途径来消除自由基,结束聚合反应。
终止步骤可以分为自发性终止和人为控制的终止。
4. 自由基聚合反应的控制方法为了获得所需的聚合物特性,需要对自由基聚合反应进行控制。
下面介绍几种常用的控制方法。
4.1 温度控制温度是自由基聚合反应的主要控制因素之一。
通常情况下,聚合反应速率随温度的升高而加快。
通过控制反应温度,可以调节聚合反应的速率和产物分子量分布。
4.2 引发剂选择不同的引发剂会对自由基聚合反应的速率和选择性产生影响。
选择合适的引发剂可以实现更高的反应活性和选择性。
4.3 单体选择单体的选择性也是自由基聚合反应的重要控制因素之一。
通过选择不同的单体,可以合成出具有不同结构和性质的聚合物。
第3章 自由基-3

3.5 自由基聚合反应速率
S型
中 期 诱 导 期 后 期 诱 导 期 ︵ 零 速 期 ︶
率自 由 时基 间聚 关合 系反 曲应 线转 图化
转 化 率
聚合过程
初 期 ︵ 匀 速 期 ︶ 中 期 ︵ 加 速 期 ︶ 后 期 ︵ 减 速 期 ︶
诱导期:初级自由基为阻聚杂质所终止,无聚合物形成,聚合速率为零。 初期:单体开始正常聚合,转化率在5%~10%以下(研究聚合时)或10%~20%(工 业上)以下阶段称初期;此时转化率与时间近似呈线性关系,聚合恒速进行。 中期:转化率达10%~20%以后,聚合速率逐渐增加,出现自动加速现象,直至转化 率达50%~70%,聚合速率才逐渐减慢。 后期: 自动加速现象出现后聚合速率逐渐减慢,直至结束,转化率可达90%~100%。
0.5级—双基终止时的引发剂浓度的反应级数; 1级— 单基终止时的引发剂浓度的反应级数。
(即:对于链转移那样的“单基终止”反应,动力学方程 中 引发剂浓度具有1次方。)
高分子化学
3.5 自由基聚合反应动力学
(b)对单体浓度一次方的偏离:
对引发效率低的聚合反应,初级自由基与单体的引发反应 较慢,与引发剂的分解速率相当,链引发速率则与引发剂和单 体浓度都有关,应表示为:
(4)
I— 引发剂; [ ]— 浓度; M— 单体; R. — 初级自由基; d — 分(decomposition); i — 引发(initiation) k— 速率常数。 kd :10-4~10-6s-1; f :0.6~0.8; Ri :10-8~10-10mol/(L.s)
高分子化学
链增长反应 propagating reaction
3.6 聚合物的平均聚合度
高分子化学——第三章-自由基聚合

平面形
三角锥形
(3) 自由基的化学反应
a. 自由基的偶合和歧化反应 b. 加成反应 c. 氧化-还原反应
CH3CH2CH2 CH3(CH2)4CH3
偶合反应
CH3CH2CH2
CH3CH2CH2
CH2 CH X
OH Fe2+ OH
CH3CH2CH3+ CH3CH CH2 歧化反应
CH3CH2CH2 CH2 CH 加成反应
C=O双键具有极性,羰基的π键异裂 后具有类似离子的特性,可由阴离子或 阳离子引发剂来引发聚合,不能进行自 由基聚合。
●杂环(heterocyclics):如环醚、环 酰胺、环酯等
C-Z单键不对称,异裂后具有类似于离 子的特性,可由阴离子或阳离子引发剂 来引发聚合,不能进行自由基聚合。
(二)乙烯基单体对聚合机理的选择
X
+ Fe3+ 氧化-还原反应
二、 自由基的活性
自由基的活性与其结构有关,主要影响因素 是共轭效应,极性效应和空间位阻。
高活性自由基
中活性自由基
低活性自由基
3.2 自由基聚合的基元反应(重点)
(Elementary reaction) 1)链引发反应(chain initiation) 2)链增长反应(chain propagation)
逐步聚合反应中每一个官能团都是具有 相同反应活性的“反应中心”。
一、单体聚合的可能性
★ 热力学可能性(thermodynamic feasibility) △G<0
单烯类、共轭双烯类、炔类、羰基化合 物等一般都属于热力学可能的单体。
★ 动力学可能性(kinetics feasibility) 引发剂、温度等动力学条件
第2.5节 自由基聚合反应速率
根据稳态原理,引发速率=终止速率, 根据稳态原理,引发速率=终止速率,Ri= Rt, 稳态原理 链终止速率
d[M•] 2 Rt = − = 2kt [M•] dt
R [ M •] = i 2k t 1/ 2
(2(2-22) (2(2-23)
∴自由基浓度
链引发速率
R i = 2 fkd [I]
为了简化推导过程,自由基聚合反应暂时 为了简化推导过程, 只考虑双基终止的情况。 只考虑双基终止的情况。以自由基消耗速率表 示的链终止反应速率 链终止反应速率为 示的链终止反应速率为: 双基偶合终止
M n • + M m • → M n+m
2
R tc = 2 k tc [M • ]
双基歧化终止
M n • +M m • → M n + M m
Rd = −
d [ I] = k d [ I] dt
d [R •] = 2k d [ I] dt
d [M•] d [ R •] Ri = = = 2 k d [ I] dt dt
基于生成单体自由基的速率远远大于引发剂分 解速率,链引发速率完全由引发剂分解速率决定。 解速率,链引发速率完全由引发剂分解速率决定。
4
后期——减速期 减速期 后期
随着单体和引发剂的不断消耗,体系黏度 随着单体和引发剂的不断消耗, 更大,单体浓度减少。 更大,单体浓度减少。聚合反应速率呈现逐渐 减小的趋势。 减小的趋势。通常情况下如果需要达到很高的 转化率,此阶段将消耗很长时间。 转化率,此阶段将消耗很长时间。
二、 聚合动力学研究方法 聚合动力学主要研究聚合速率、分子量、 聚合动力学主要研究聚合速率、分子量、引发剂 浓度、单体浓度、聚合温度等因素间的定量关系。 浓度、单体浓度、聚合温度等因素间的定量关系。 聚合速率可以用单位时间内单体消耗量或聚合 物生成量来表示。测定方法有直接法和间接法2 物生成量来表示。测定方法有直接法和间接法2类。 最常用直接法是沉淀法测定聚合物量。在聚合瓶中 最常用直接法是沉淀法测定聚合物量。 沉淀法测定聚合物量 进行聚合,定期取样,加沉淀剂使聚合物沉淀, 进行聚合,定期取样,加沉淀剂使聚合物沉淀,后经分 精制、干燥、称重等手续,求得聚合物量。 离、精制、干燥、称重等手续,求得聚合物量。 间接法系测定聚合过程中比容、粘度、折光率、 间接法系测定聚合过程中比容、粘度、折光率、 介电常数、吸收光谱等物性的变化,间接求聚合物量。 介电常数、吸收光谱等物性的变化,间接求聚合物量。 膨胀计法, 最常用的是比容的测定——膨胀计法,即利用聚 膨胀计法 最常用的是比容的测定 合过程中的体积收缩与转化率的线形关系。 合过程中的体积收缩与转化率的线形关系。
自由基聚合—聚合速率

= kp[M] [M•]
= kp[M]
Ri 2kt
1/2
[M•] = (Ri /2kt)1/2
Ri = 2fkd[I]
将取决于温度 和各反应活性
0.5级反应是双基终止的 结果(当链自由基活性末端
受到包埋,难以双基终止, 往往单基终止和双基终止并 存,对[I]的反应级数介于0.5 ~1.0之间)
聚合总速 率方程
3.4 温度T对R的影响
在上述速率方程中虽然没有出现温度(T)因子,但是 它通过速率常数k来影响R
k = Ae-E/RT
聚合总速率常数k和 各基元反应速率常数 与温度T的关系遵循 Arrhenius方程式
k = kp (kd /kt)1/2
其中的关键因子 是反应活化能E
可推算出E与Ed、 Ep、Et之间的关系
kp约 102 ~ 104 l/mol·s [M·]约10-7 ~ 10-9 mol/L [M]取 1 ~ 10 mol/L
Rp约10-4 ~ 10-6 mol/L·s
式中:p— 链增长(propagation)
链终止反应
链终止标志着自由基的消失,所以,终止速率可以以因 终止反应而引起的自由基消失速率表示
❖ 诱导期 — 初级自由基为阻聚杂质终止,无聚合物形成,聚
合速率为零。
❖ 聚合初期 — 单体开始正常聚合时期,通常将转化率在5
%~10%以下的阶段称做初期;在这一阶段,转化率与时 间近似呈线性关系,聚合基本以恒速进行。
❖ 聚合中期 — 在转化率达10%~20%以后,聚合速率逐渐增
加,出现了自动加速现象,直至转化率达50%~70%,聚合 速率才逐渐减慢。这一阶段称为聚合中期。
链引发 速率方程
Ri
自由基聚合反应动力学

主要研究:聚合反应速率、分子量与引发剂浓度、 单体浓度、聚合温度等因素间的定量关系 聚合过程的速率变化常用转化率-时间曲线表示。
.
第一阶段: 诱导期, X=0。
转
化
率1 2
3
4
x%
聚合时间/min
产生的原因:引发剂产生的初级自由基被阻 聚杂质所消耗。诱导期的长短取决于阻 聚杂质的多少。
1
)2
2kt
R pkP [M ]M []kp[M ]R (i2kt)12
Rpkp[M](2f2kkdt[I])12
R 总 R PR iR Pkp.(fktk d)12[I]12[M ]
3.2.3.4 温度对聚合速率的影响
▪ 聚合反应速率常数与温度的关系遵守 Arrhenius方程:
▪
k = Ae-E/RT
C H 3
2
.. C H 3
CO+O H
C H 3
叔丁基过氧化氢 (t-BHP)
C H 3
C H 3 COOH
C H 3 .
.. C H 3
C H 3 CO+O H
C H 3
== == == ==
(2) 无机过氧类 过硫酸钾、过硫酸铵等
. . K OO S O OOO O SO K t5 1 2 = 0 1 ℃ 3 0 h 2 K OO S O O 或 2 K + 2 OO S O O
.
2 聚合反应总速率
Rd[dM t ]Ri2Rp
聚合物分子的长链原理:
单体主要消耗于链增长,引发反应所消耗的 单体可忽略不计。
R Rp
.
3.2.
1
第3章自由基聚合反应

tr,I Mx I Mx R
k
Rtr,I ktr,I [M ][I]
Rtr,S ktr,S[M ][S]
Rp Rt Rtr Rp C ( D) Rt Rtr 2
tr,S Mx S Mx S
k
聚合度的表达式应是:
高分子化学
3.5 聚合中期聚合反应速率
聚合初期的恒速阶段一般可持续到转化率达10~15%。
fkd 1/2 Rp k p [M ] [I ] kt
1/2
自动加速效应(autoacceleration)——聚合反应速率 自动加快的现象称为自动加速现象。
一般单体的聚合体系都会存在自动加速现象,差别是 体系不同,出现的早晚和程度不同。 主要原因 3.5.3.1 凝胶效应 ——由于反应过程体系粘度的增加引起的自动加速现象称为 凝胶效应(gel effect)。
Rp k p [M ][M ]
Rt 2kt [M ]2
ktr , M ktr , I [ I ] ktr , S [ S ] ktr , P [ P ] 2 kt R p 1 C ( D ) 2 2 2 k p [M ] kp k p [M ] k p [M ] k p [M ] Xn
凝胶效应的主要原因在于链终止反应是受扩散控制的反应。
高分子化学
第3章 自由基聚合反应
3.5-3.9
对于正常的双基终止,链自由基双基终止过程可以分为 三步:链自由基质心的平移、链段重排(控制步骤)和双基 碰撞发生反应。体系黏度是影响的主要因素。
fk R p k p [ M ] d [ I ]1/2 kt
k p [M ] (2kt )1/2 Ri1/2
自由基聚合反应

过氧化酯 RCO-OO-R’
过氧化二酰
RCO-O-O-COR’
过氧化二碳酸酯 ROOC-O-O-COOR’
常用的过氧化二苯甲酰(BPO)分解反应机理如下:
其它常见的有机过氧化物引发剂如叔丁基过氧化氢、过氧化 苯甲酸叔丁酯等的分解反应分别如下所示:
叔丁基过氧化氢:
过氧化苯甲酸叔丁酯:
(2)偶氮类引发剂 为带吸电子取代基的偶氮化合物,分对称和不对称两大类:
无机过氧化物包括过氧化氢、过硫酸钾、过硫酸铵等,其分解
反应机理如下:
HO OH
2HO.
O
O
KO S O O S OK
O
O
. O
_
2KO S O (SO4)
O
其中,H2O2 分解活化能高达 218kJ/mol,需要在高温下才能 分解,因此很少单独使用。
有机过氧化物:
烷基过氧化氢 RC-O-O-H
二烷基过氧化物 R-O-O-R’
1分子量调节剂只需加入少量的链转移剂如十二烷基硫醇便可明显降低分子量而且还可通过改变其用量来调节分子量因此这类链转移剂又叫分子量调节剂与聚合度的定量关系以歧化终止为链转移剂浓度2调节聚合调节聚合在活性溶剂中进行聚合合成极低分子量的聚如乙烯在四氯化碳中聚合在适当条件下可获得聚合度为10左右的所谓人造石蜡
由于其给电子效应部分地抵消了吸电子效应,使其吸电子 效应减弱,该类单体一般难以进行阴离子聚合,而只能进行自 由基聚合,如:
H2C CH Cl
氯乙烯
H2C CH
O C CH3 O
乙酸乙烯酯
(e) 具有共轭体系的烯类单体
p电子云流动性大,易诱导极化,可随进攻试剂性质的不同 而取不同的电子云流向,因此视引发条件不同而可进行阴离子型、 阳离子型、自由基型等各种链式聚合反应。如苯乙烯、丁二烯等:
自由基聚合反应1

取代基与聚合反应类型简列:
10
11
12
3.2 连锁聚合反应热力学(了解)
一、聚合热
1
1
13
△G旳符号取决于△H、△S旳正负及大小。
14
当温度在25~100℃内: -T△S=105×298~125×373 =31~47 KJ/mol
△G =△H-T△S <0
从热力学角度分,聚合多能自动进行。
定义为聚合反应旳临界上限温度或极限温度25
3.3 自由基聚合反应机理
一、自由基旳产生及其活性 自由基:
凡带有未配对独电子旳原子、分子或原子团叫自由基。
1.自由基旳产生方式:均裂和电子转移
弱共价键旳均裂
过氧化苯甲酰(BPO)
26
单电子转移旳氧化还原反应
27
2. 自由基旳活性
影响自由基旳活性旳两个主要原因:
1,1-二取代
1,2-二取代
5
唯一旳例外是当取代基为F时,它旳一、二、 三、四取代乙烯都能够参加聚合反应
聚四氟乙烯
6
二、取代基旳电负性和共轭性决定烯烃旳聚 合反应类型
(1)带吸电子取代基旳烯烃——能够进行自由基型 和阴离子型两种聚合反应
7
(2) 带推(供)电子取代基旳烯烃——能够进行阳 离子型聚合
44
4 氧化还原体系
过氧化氢-亚铁离子: 异丙苯过氧化氢-亚铁离子: BPO-叔胺体系:
45
氧化还原体系特点:
• 引起温度低 • 引起效率相对较低
46
二、引起剂分解动力学
结论:引起剂旳分解属于单分子一级分解,其 浓度降低速率旳自然对数与反应时间成正比。 47
引起剂活性高下旳判断
半衰期:反应物浓度降低二分之一所需旳 时间
第3章自由基聚合反应

Y CH2=C X
3. 1,2-二取代以及三、四取代烯烃原则上都不能聚合
H C == C X Y H
Z C == C X
H Y
Z C == C X
W Y
1,2-二取代
三取代
四取代
空间位阻影响聚合
唯一的例外是当取代基为体积很小的F时,它的 一、二、三、四取代乙烯都可以参加聚合反应。
如 CF2 = CF2 和CF2 = CFCl 就可以聚合
不活泼单体的引发效率低,活泼单体的引发效率高
影响引发剂效率的因素: ◆ 诱导分解反应
R. + C6H5CO4CC6H5 = C6H5COOR + C6H5COO.
诱导分解与生成单体自由基的引发是一对竞争反应:
R. +BPO = C6H5COOR+C6H5COO. R.+M = RM. ◆ 笼蔽效应 溶液聚合反应中,低浓度引发剂分子及初级自由基 处于高黏度聚合物溶液包围之中,部分初级自由基 无法与单体接触而向引发剂或溶剂转移,从而使引 CH CH CH 发效率降低。 H C C N N C CH
BF3 +H2O
CH3
_
CH + .... [BF 3OH] -
丁二烯、异戊二烯同样也可进行3种聚合反应
* 乙烯分子高度对称,聚合活性很低。取代基使烯
烃分子的对称性改变,从而导致其聚合活性增加。
* 取代基与聚合反应类型简列
_ NO2 ; _ CN; _ COOR; _ CH=CH2; _ C6H5; _ CH3; _ OR
特别提醒:带1个推电子甲基的丙烯却不能进行阳离子 聚合,而只能进行配位聚合。
3. 带共轭取代基的烯烃,可以进行自由基、阴离子和 阳离子等3种类型的聚合反应
自由基聚合反应速率

2. 链增长
RM
+M kp1
RM2
+M k p2
RM3
+M k p3
RMn
通式为: Mn + M
kp Mn+1 kp 为增长反应速
率常数
3.2.5 自由基聚合反应速率
三、基元反应及速率常数
3. 链终止
Mn + Mm ktc
Mn+m
ktc 为偶合终止反应速 率常数
Mn + Mm ktd Mn + Mm ktd 为歧化终止反应速 率常数
• 终止反应同时含有偶合及歧化时
kt = ktc + ktd kt 为终止反应速率常数
3.2.5 自由基聚合反应速率
四、聚合反应初期动力学(速率表达式)
• 三个基本假定:等活性、聚合度很大、稳态
• 聚合反应速率可用单体消耗的速率表示。在自由
基聚中,引发和增长是消耗单体的步骤,因此:
R
≡
−
d [M ]
第三章 连锁聚合反应
第三章 连锁聚合反应 3.2.5 自由基聚合反应速率
一、聚合速率及其测定方法
C%
t
图 转化率~聚合时间关系
一、聚合速率及其测定方法
• 转化率随聚合时间变化的测定:
• 分为直接法和间接法两类: • 常用的直接法为沉淀法
– 一定温度下聚合,定时取样,求得不同 t 时 的聚合物量。
– 也可通过分析单体的浓度而求得某时刻的转 化率。
• 间接方法,即测定聚合体系的比容、粘度、折 光率、吸收光谱等物理化学性质的变化,推算 出反应体系中单体浓度的减少,或聚合物量的 增加。
一、聚合速率及其测定方法
间接方法
(完整版)高分子化学复习简答题(三)---自由基聚合(精)

高分子化学复习简答题(三)---自由基聚合学校名称:江阴职业技术学院院系名称:化学纺织工程系时间:2017年3月10日1、自由基聚合反应转化率-时间曲线特征。
答:诱导期:初级自由基为阻聚杂质所终止,无聚合物形成,聚合速率零。
若严格取除杂质,可消除诱导期。
初期:单体开始正常聚合,转化率在5%~10%以下(研究聚合时)或10%~20%(工业上)以下阶段称初期;此时转化率与时间近似呈线性关系,聚合恒速进行。
中期:转化率达10%~20%以后,聚合速率逐渐增加,出现自动加速现象,直至转化率达50%~70%,聚合速率才逐渐减慢。
后期: 自动加速现象出现后聚合速率逐渐减慢,直至结束,转化率可达90%~100%。
2、自由基聚合与缩聚反应的特征比较。
答:自由基聚合:(1)由基元反应组成,各步反应的活化能不同。
引发最慢。
(2)存在活性种。
聚合在单体和活性种之间进行。
(3)转化率随时间增长,分子量与时间无关。
(4)少量阻聚剂可使聚合终止。
线形缩聚:(1)聚合发生在官能团之间,无基元反应,各步反应活化能相同。
(2)单体及任何聚体间均可反应,无活性种。
(3)聚合初期转化率即达很高,官能团反应程度和分子量随时间逐步增大。
(4)反应过程存在平衡。
无阻聚反应。
3、为什么自由基聚合时聚合物的相对分子质量与反应时间基本无关,缩聚反应中聚合物的相对分子质量随时间的延长而增大?答:自由基聚合遵循连锁聚合机理:链增加反应的活化能很低,Ep=20~34KJ/mol,聚合反应一旦开始,在很短的时间内(0.01s~几秒)就有成千上万的单体参加了聚合反应,也就是生成一个相对分子质量几万~几十万的大分子只需要0.01s~几秒的时间(瞬间可以完成),体系中不是聚合物就是单体,不会停留在中间聚合度阶段,所以聚合物的相对分子质量与反应时间基本无关。
缩聚反应遵循逐步聚合机理:单体先聚合成低聚体,低聚体再聚合成高聚物。
链增加反应的活化较高,Ep=60KJ/mol生成一个大分子的时间很长,几乎是整个聚合反应所需的时间,缩聚物的相对分子质量随聚合时间的延长而增大。
自由基聚合速率

第32页,共38页。
影响自加速现象的因素
聚合物在单体或溶剂中溶解性能的优劣对链自由基的卷曲、 包埋的影响很大,
苯乙烯是聚苯乙烯的良溶剂,链自由基链段扩散重排较容 易,因此出现自加速现象要晚。
若聚合物不溶于其单体或溶剂中,长链自由基边生成边沉
淀出来,构成非均相体系,称为沉淀聚合。聚合一开始即 出现自加速现象。例如丙烯腈、氯乙烯的聚合均属此类。其 自由基寿命很长,例如四氟乙烯在 50℃水中聚合时,自由基
第3页,共38页。
转化率随聚合时间变化的测定
直接法: 沉淀法
一定温度下聚合,定时取样,求得不同t时的聚 合物量
也可通过分析单体的浓度而求得某时刻的转 化率
间接法: 测定聚合体系的比容、粘度、折光 率、吸收光谱等物理化学性质的变化,推 算出反应体系中单体浓度的减少,或聚合 物量的增加
第4页,共38页。
第13页,共38页。
自由基聚合速率方程
因此总的聚合速率的普适方程为
当用引发剂引发时,将式
代入上式
得
引发剂浓度的平 方根成正比
聚合速率
单体浓度的一次方成正比
第14页,共38页。
第15页,共38页。
对引发剂反应级数介于0.5~1.0之间
0.5级和1.0级是双基终止和单基终止的两 种极端情况
往往是单基终止和双基终止并存,其对引 发剂浓度的反应级数介于0.5~1.0之间
系数 2 表示终止反应同时消失两个自由基
第11页,共38页。
(2)自由基聚合速率方程
为简化动力学方程的处理,在总速率方程 的推导时,作了四个假定:
①假定链转移不影响聚合速率,链终止反应 为双基终止. 聚合速率是由链引发、链增长 和链终止三种基元反应所决定,
自由基聚合反应的速率.ppt

dc(R • )
>>
dt
dt
引发反应速率方程为 Ri = 2 fkdc(I)
2.7 自由基聚合反应的速率
2. 链增长反应的速率方程
RM·+M
kp1
RM1M· Rp1
RM1M·+M kp2 RM2M· Rp2 …………
RMn-1M·+M kpn RMnM· Rpn
Rp1 = kp1c(M)c(M1• ) Rp2 = kp2c(M)c(M•2 )
·
**
*
图2.13自动加速现象示意图
Rp
=
kp (
fk d kt
)1/ 2 c(M)c(I)1/ 2
kp /( kt )1/ 2
2.7 自由基聚合反应的速率
在低转化率情况下,聚合体系处于稳态,但在高转化率时,聚 合体系偏离稳态,聚合体系出现自动加速现象。直到聚合后期, 聚合速率才渐渐减慢。在很长一段时间内二者变化方向相反, 二者叠加的结果,使聚合转化率-时间关系曲线呈S型。
❖ 1. 引发剂浓度c(I)对聚合速率的影响(图2.10)
❖ 2. 单体浓度c(M)对聚合速率的影响(图2.11)
❖ 3. 聚合温度对聚合速率的影响
❖ 4. 引发剂种类对聚合速率的影响
2.7 自由基聚合反应的速率
Rp
Rp
(MMA) 1 (MMA) 3
(S) 2
c(I)
图2.10 聚合速率Rp与引发剂浓度c(I)的关系 1-MMA,ABIN,50℃; 2-St,BPO,60℃; 3-MMA,BPO ,50 ℃
…………
Rpn = kpnc(M)c(M•n )
2.7 自由基聚合反应的速率
➢ 假定①——官能团等活性理论的假定:
自由基聚合过程中反应速度和聚合物分子量

自由基聚合过程中反应速度和聚合物分子量自由基聚合是一种常见的加聚反应,它是指由自由基作为活性种,与烯类单体发生连锁反应,形成高分子的过程。
自由基聚合的反应速度和聚合物分子量是两个重要的参数,它们反映了聚合反应的动力学和热力学特征,也影响了聚合物的性能和应用。
本文将介绍自由基聚合过程中反应速度和聚合物分子量的影响因素和控制方法。
反应速度是指聚合反应的进行速率,它与聚合反应的转化率和反应时间有关。
反应速度受到以下因素的影响:聚合温度。
聚合温度是影响反应速度的主要因素,一般来说,升高聚合温度,可以增加分子的热运动,提高引发剂的分解速率,增加自由基的浓度,促进链增长反应,从而提高反应速度。
但是,过高的聚合温度,也会加速链终止和链转移反应,降低聚合反应的选择性,导致聚合物分子量的降低和分布的变宽,甚至引起反应失控和爆聚。
因此,聚合温度应根据不同的单体和引发剂的特性,选择适当的范围,以保证反应速度和聚合物质量的平衡。
引发剂的种类和浓度。
引发剂是自由基聚合的启动剂,它可以在一定的温度下分解生成自由基,引发单体的聚合。
引发剂的种类和浓度决定了自由基的数量和活性,从而影响反应速度。
一般来说,引发剂的分解温度越低,分解速率越快,生成的自由基越多,反应速度越高。
引发剂的浓度越高,自由基的浓度越高,反应速度越高。
但是,引发剂的种类和浓度也会影响聚合物分子量,因为引发剂的残基会存在于聚合物的链端,增加聚合物的分子量分布,降低聚合物的均一性。
因此,引发剂的种类和浓度应根据不同的单体和聚合条件,选择合适的类型和用量,以保证反应速度和聚合物质量的平衡。
单体的种类和浓度。
单体是自由基聚合的原料,它与自由基发生链增长反应,形成聚合物。
单体的种类和浓度影响反应速度,主要是通过影响链增长反应的速率常数和反应级数。
一般来说,单体的反应活性越高,链增长反应的速率常数越大,反应速度越高。
单体的浓度越高,链增长反应的反应级数越大,反应速度越高。
第三章_自由基聚合-速率方程

链引发反应
I kd 2R ki
R
+
M
RM
初级自由基生成速率: Rd =
d [R.] / d t = 2 kd [ I ]
单体自由基生成速率: d [R.] / d t = 2 kd [ I ] 单体自由基生成速率: Ri=d [R.] / d t = 2 f kd [ I ]
条件: 1. 引发剂分解速率 远小于单体自由 基的生成速率; 2. 初级自由基或 引发剂的分解并 不完全参与引发 反应。
引发反应
H2 C
St
CH
H2 C
H C
H2 C
St
CH
H2 C
H C
H2 C
H C
H2 C
CH
增长反应 (St)
n
H 3C
2 H 3C
C NC
H2 C
H C
H2 C
H C
H2 C
偶合终止
CH
H2 C CH
歧化终止 n
H 3C H2 C H C H2 C H C H2 C H2 C CH
H 3C
C NC
单体浓度越高越容易出现自动加速现象;溶剂或单体对 聚合物的溶解性越小越容易导致自动加速效应。 减缓自动加速作用:提高温度,使用良溶剂,降低单体 浓度等。
1.诱导期
2.匀速期 (初期)
3.加速期 (中期)
4.减速期 (后期)
自由基聚合转化率-时间曲线
对分子量影响:
自动加速除使聚合反应速率显著上升外,由于终止反应 速率减慢,相应地链自由基的寿命增加,可结合更多的 单体,从而自动加速作用使聚合产物分子量显著增加。 凝胶效应的规律: 在一些聚合产物在反应过程中从体系中沉淀出来的非均 相聚合体系中,如乳液聚合、气相聚合、交联聚合、固 相聚合等,由于活性中心可能被包裹,导致链终止反应 难以进行,也可发生自动加速作用。
自由基聚合反应速率

(单位:mol 引发剂 / s.)
初级自由基生成速率: d [R.] / d t = 2 kd [ I ] (单位:mol 引发剂 / s.) 单体自由基生成速率: Vi == d [M.] / d t = d [R.] / d t = 2 kd [ I ] (单位:mol 引发剂 / s.) 将引发效率考虑进去,则: Vi == 2f kd [ I ]
3.5 自由基聚合反应速率
3.5.1 聚合反应动力学过程
1.诱导期; 2.匀速期; 3.加速期; 4.减速期
图3-5 自由基聚合转化率-时间曲线
3.5.2 聚合反应初期动力学
1、3基元反应动力学方程 链引发:
I kd
. 2R
. R+M
ki
. RM
引发剂分解速率: - d [ I ] / d t = 2 kd [ I ]
引发剂、单体、自由基的浓度通常大约在
10-2; 100; 10-7~10-9mol /L
Vp == kp[M][M.] == 102×1×10-7 == 10-5
Vt == kt [M.]2 == 106×10-7×10-7 == 10-8 原来虽然链终止速率常数大于链增长速率常数,但是 由于自由基浓度远低于单体浓度,所以链终止速率仍 然远小于链增长速率。
在0→t, [M]0→[M]范围内对上式定积分,即得
ln [ M ]0 [M]
= f kd k ( = p kt
1/2 kp ′ [I] t
) [ I] t
1/2
1/2
式中kp′== kp(f kd / kt )1/2
定义为聚合反应的总速率常数,它包含引发、增长、 终止3个速率常数对聚合反应综合速率常数的贡献
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
[M ] = ∑ [RM ]
• • i
⎛ d [M ] ⎞ • R p ≡ −⎜ = k p [M ]∑ RM i = k p [M ] M • ⎟ 则: ⎝ dt ⎠ p
[
]
[ ]
四、聚合反应初期动力学(速率表达式)
Mn + Mm ktc Mn+m
Mn + Mm ktd Mn + Mm
• 稳态假设理论:自由基浓度在最初是增大的,但
R
+
M
ki
RM
1 2
• 聚合反应总速率
以
Ri = 2 fk d [I]
⎛ Ri ⎞ R = R p = k p [M ]⎜ ⎟ ⎜ 2k ⎟ ⎝ t⎠ 代入,得:
1 2
⎛ fk d ⎞ ⎟ Rp = k p ⎜ ⎜ k ⎟ ⎝ t ⎠
[I] [M ]
1/ 2
此式描述了单体-引发剂体系及双基终止的 自由基连锁聚合的普遍规律。
一、聚合速率及其测定方法
2. 膨胀计法:利用聚合过程中反应体系的体积
收缩与转化率的线性关系:
ΔV 1 聚合反应转化率: C % = V0 K
ΔV为体积收缩值 V0 为原始体积
转化率为100%时的体积变化率 K: V m − Vp K= × 100% Vm
测定不同反应时刻的反应体系体积的收缩值,就 得到转化率随时间的变化结果,得到聚合速率。
第三章 连锁聚合反应
第三章
连锁聚合反应
3.2.5 自由基聚合反应速率
一、聚合速率及其测定方法
C%
t
图
转化率~聚合时间关系
一、聚合速率及其测定方法
• 转化率随聚合时间变化的测定:
• 分为直接法和间接法两类: • 常用的直接法为沉淀法 – 一定温度下聚合,定时取样,求得不同 t 时 的聚合物量。 – 也可通过分析单体的浓度而求得某时刻的转 化率。 • 间接方法,即测定聚合体系的比容、粘度、折 光率、吸收光谱等物理化学性质的变化,推算 出反应体系中单体浓度的减少,或聚合物量的 增加。
瞬间就达到稳态,此时自由基浓度为定值,在聚 合过程中不再变化。既引发速率 Ri 与终止速率 Rt 相等,构成动态平衡。
Ri = Rt = 2 k t M
[ ]
2
• 2
系数 2 表示自由 基成对地消灭
[ ]
⎛ Ri ⎞ • ⎟ M =⎜ ⎜ 2k ⎟ ⎝ t⎠
1
四、聚合反应初期动力学(速率表达式)
I kd 2R
+
R
M
ki
ki为引发步骤的速率常数
RM2
+
2. 链增长
RM
M k p1
M k p2
RM3
+
M k p3
RMn
通式为: Mn + M
kp
Mn+1
kp 为增长反应速
率常数
3.2.5 自由基聚合反应速率
三、基元反应及速率常数
3. 链终止
Mn
Mn
+ Mm
+ Mm
ktc
ktd
Mn+m
Mn + Mm
ktc 为偶合终止反应速 率常数 ktd 为歧化终止反应速 率常数
一、聚合速率及其测定方法
间接方法
1. 化学及光谱分析
例: 用溴滴定未反应的双键。 用FTIR、UV、NMR测定单体或聚合物与双 键有关的吸收信号的减弱或增强。当此二者信 号不相重叠时,光谱法可得精确的结果。 例,苯乙烯聚合,用NMR法,单体的 C CH2 (5.23ppm)和 C CH (6.71ppm)减弱,聚 合物的 CH2 及 CH(1.44及1.8ppm)增强。
d [M ] R≡− = Rp dt
d [M ] R≡− = Ri + R p dt
单体分子数很少,可以忽略不计,Ri << Rp ,因 此聚合反应速率就等于增长反应速率:
四、聚合反应初期动力学(速率表达式)
Mn + M kp Mn+1
• 等活性理论:链自由基的活性与链长基本无关,即
各步链增长的速率常数相等 kp1=kp2=kp3=…=kpn=kp , kp 和 kt 与自由基大小无关。 令:
•
终止反应同时含有偶合及歧化时
kt = ktc + ktd
自由基聚合反应速率
四、聚合反应初期动力学(速率表达式)
• •
三个基本假定:等活性、聚合度很大、稳态 聚合反应速率可用单体消耗的速率表示。在自由 基聚中,引发和增长是消耗单体的步骤,因此:
• 聚合度很大:与增长反应相比,引发反应消耗的
3.2.5 自由基聚合反应速率
二、聚合反应过程
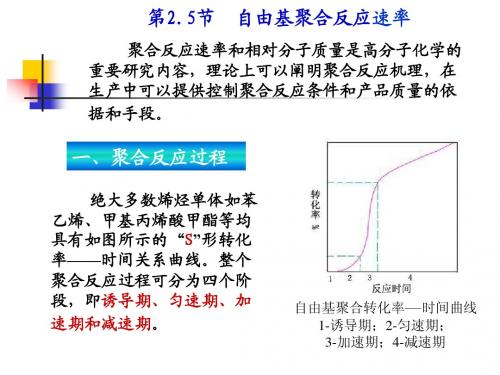
1. 诱导期----零速期 2. 初期----匀速期 3. 中期----加速期 4. 后期----减速期
图2-3 自由基聚合转 化率--时间曲线
3.2.5 自由基聚合反应速率
三、基元反应及速率常数
1. 链引发
I kd
+
2R
kd 为引发剂裂解速率常数
RM