飞检中实验室缺陷之文件及记录管理缺陷
药品GSP飞行检查缺陷项

药品GSP飞行检查缺陷项1.私设中药饮片仓库,存储条件不符GSP要求,库内计算机库存管理系统与公司计算机库存管理系统不一致。
2.计算机系统显示冷藏库中有大量低温保存药品,现场检查冷藏库为空库,经查,该宗药品存放于企业阴凉库中,企业对上述事实存在隐瞒行为。
3.质量负责人未能在职在岗履行职责。
4.企业自行设定温湿度探头温湿度上限,数值显示与实际温度不符,部分温湿度探测终端超温时不报警。
5.新版GSP认证时提出的多条缺陷问题在此次检查中仍然存在;非药品库中存放有大量药品,且库房破损,有漏水情况;未入采购系统且未经验收的货物进入整货区,且无有效标识;阴凉库中“五区”划分不明确,未严格按照划分区域存放货物,零货、整货混放、混垛、药品存放地面现象严重。
3.GSP管理混乱:新任命的质管部长未进行岗前培训和健康查体;待验区有大量药品未及时验收,温度未进行有效控制,有些药品无随货同行单;药品未严格按照既定区划放置,也未按照五距进行管理;阴凉库发货区预冷用的冷柜内存放有多种冷藏药品。
4.温湿度监测体系存在缺陷:温湿度自动监测系统历史数据的温湿度涉嫌与实际不符,且其中的数据可以手工修改,冷库的历史数据有超温现象;飞检当天全部阴凉库温度超标;未见保温箱的温度记录仪;温湿度自动监测系统未进行介质备份;冷库、冷藏车、保温箱和温湿度自动监测系统均未按制度规定进行定期验证,相关设备也未按制度规定进行定期校准。
2.赠品库内存有作为赠送药品的血塞通、卡托普利等多种处方药和甲类非处方药,且无来源证明。
1.擅自设置不符合GSP要求的库房储存药品(现场整改到位,并在检查结束的第三天向省局提请增设新仓库的申请)。
2.温湿度监控系统断电后未见声光报警、未定期验证、断电后未见不间断电源供电;温湿度监控系统测点终端安装位置不符合规定;3.未对冷库、保温箱进行定期验证以及极高温、极低温验证,温湿度监控系统未见冷藏车的温度数据,未对冷藏车、保温箱进行定期检查及维修,未建立记录;4.常温库内药品与非药品混放,药品库存放中药饮片,阴凉库温度超标后未采取调控措施等。
GMP飞检缺陷

GMP飞检缺陷六安华源制药有限公司一般缺陷10项:1、产品工艺规程中未列入原辅料、包装材料的生产厂家信息。
2、现场检查时玻瓶车间制水岗位人员检测pH值操作不规范;输液类产品批生产指令中缺少指令日期、物料批号和生产厂家等信息;A线灌装百级层流有半扇档风板门已坏未及时修理安装;配液补料缺少称量记录。
3、起泡点试验标准操作规程(SOP-03-218-06)规定0.22μm的滤芯每天生产前和结束后各做一次起泡点试验,但企业每天生产多个批次产品时,规程未对生产结束后起泡点试验不合格所生产产品的风险评估及处理作出规定。
4、塑瓶车间制塑料瓶未实行批号管理,企业未制定先生产先灌装,且在5日内用完的管理程序。
5、成品阴凉库面积偏小,使需要阴凉保存的成品存放在普通库。
6、质检中心培养室生化培养箱(Spx-250B-Z型)为企业自校,但未按时间定期校准,且企业未制定生化培养箱自校操作规程。
7、盐酸氨溴索(检品编号:JCz15007)检验报告书原始记录吸收系数测定只称取一份样品,未做平行样测定。
8、葡萄糖氯化钠注射液(检品编号:SY50070)检验报告书原始记录中重金属检查项未记录甲、乙、丙三管的制备操作。
9、替硝唑(检品编号:JCz150006)检验报告书原始记录炽灼残渣项未记录炽灼温度和时间。
10、乳酸左氧氟沙星氯化钠注射液(检品编号:SY150088)检验报告书原始记录有关物质项下供试品溶液量取体积数记录被涂改。
同路生物制药有限公司1、注射用水系统部分使用点通过热交换器降温致部分管路回水温度短时间内低于70℃,企业未对水系统的此种运行情况进行风险评估。
2、部分灯检不合格品未按不合格品管理程序和偏差管理程序处理,如狂犬病人免疫球蛋白(批号20130712)、乙型肝炎人免疫球蛋白(批号20140901)、人血白蛋白(批号20150517)、破伤风人免疫球蛋白(批号20141203)、静注人免疫球蛋白(批号20150407)等存放在3﹟待检冷库内,无台账。
药品生产企业GMP飞检常见160项缺陷项汇总

药品生产企业GMP飞检常见160项缺陷项汇总一、采购进货查验管理1、未查验某些原料供应商的生产许可证、产品合格证明;2、原料进货查验把关不严,产品检验报告项目记录不全;3、未建立进货查验记录或无原始检验记录;4、原材料查验记录,缺少审核人员签名;5、未按照企业进货查验制度进行进货查验;6、某原料的采购记录中缺少生产企业名称,无法实现有效追溯。
7、进货查验记录和凭证未按规定期限保存;8、部分原材料有供方的资质复印件,但无检验合格证明材料;9、未提供部分进口原料国内供应商的相关许可证件;10、某原辅料进货查验记录和凭证保存期限不满保质期后六个月;11、缺原料验收合格报告及相关记录;12、提供的部分原辅料及包装材料的供应商生产许可证已过期或未提供;13、某产品进货查验时缺少农药残留项目的记录。
二、产品追溯及产品召回1、没有不安全产品召回管理制度、计划、公告和召回产品处置记录;2、不安全产品无召回计划;3、不合格产品无安全防范措施记录;4、某些批次的不合格品有不合格品处置记录,但没有生产记录;5、产品召回记录批次未能与生产产品批号相对应;6、未提供某抽样产品不合格召回相关记录或召回记录不全;7、未提供不合格品的处置记录或记录不全;8、召回的不合格品储存记录不完整,缺少规格;9、未建立客户投诉处理机制,缺乏处理客户投诉的相关记录。
三、仓储及销售管理1、未能提供食品原辅料、食品添加剂、食品相关产品的贮存、保管记录和领用出库记录;2、原辅料存放,未与墙壁、地面保持适当距离;3、部分库房入口防鼠板较低或无防鼠设施。
4、某仓库换气扇的电源线断裂,不能正常运行;5、成品库顶棚局部区域及南墙玻璃窗不能有效防止阳光直射;6、产品出厂销售没有记录产品的规格、生产日期(或生产批号)、销售日期等基本信息;7、销售记录项目不完整,缺少购货者联系方式;8、原辅料领用出库记录的数量与进货验收记录不符(出库数量大于进货数量);9、某原材料无领用出库记录;10、包装材料领用出库记录不完整;11、原料库的贮存没有设置通道,无法做到先进先出;12、部分原料存放于厂区通道中;13、部分包材未离墙存放;14、原料仓库、半成品仓库、成品仓库未划定不合格品存放区域或合格品区及不合格品区域划分不明显;15、仓库各功能区未明确标识,库存原辅料标签未进行名称、规格、保质期、贮存条件等信息标识;16、原辅料和包材仓库未能完全分开;17、原辅料未按规定分区、分类存放,未与墙壁、地面保持适当距离;18、成品库缺少代码、收发双方核实签字的信息;19、未能提供相应年份产品销售台账;20、未能提供相应产品贮存、运输及交付控制记录;21、半成品库、成品库通风设施不够;22、原料库中存放的某材料已过保质期;23、原辅料仓库部分物料缺标识,半成品混放;24、半成品冷藏库内半成品未与地面保持适当的距离;25、仓库中产品标签与产品实质不相符。
飞检中常见不合格项

GSP飞检中常见不合格项1、仓库温湿度自动监测系统未设置远程报警功能;2、冷藏车、保温箱等运输设备验证项目不全,未对其经营品种的储存温度区间进行验证,如前列地尔注射液;3、企业使用的千方百剂医药管理系统操作日志可删除,无法保证数据真实、准确和可追溯;4、企业购进个别品种未对首营企业进行认真审核;5、企业温湿度超标后未及时采取措施;6、质量管理部门未对财务的账户、票据、资金流向的规范管理进行督促;7、企业发电机不能满足仓库控制温度的用电需求。
8、企业温湿度自动监测系统未按日备份以保证数据安全、完整;9、药品到货时,收货人员未严格按照要求核实运输温度并记录,未对照随货同行单和采购记录做到票账货相符;10、现场检查抽取品种的部分供货单位随货同行单样式与档案留存样式不一致,如山西同达药业有限公司;11、企业温湿度超标后未及时采取措施。
12、部分配送交接手续不完整,无销售客户收货签字或盖章。
13、个别冷链品种交接单记录不完整,如无日期、车牌号等内容。
14、少数品种未按批号堆放;15、培训档案不全;16部分首营企业审批滞后;17、销售员授权委托书未及时登记,有重号现象。
18、中药材库出现储存化学药品现象。
19、2号冷库6月22日温度超标约1小时,设备维护记录为当天进行冷库验证,但验证报告和各项记录中,未见温度超标期间是否储存冷链药品。
20、该企业计算机系统中显示培力(南宁)中药配方颗粒(骨碎补等几十个品种)2016年有销售出库,配送方式不符合省食品药品监督管理局的有关规定。
21、不合格区的药品:复方右旋糖苷40注射液(A141012-2,数量443瓶),未及时在计算机系统中按规定进行处理。
22、供货方质保协议对方只盖章未签字。
23、计算机系统质量负责人权限设置不正确,计算机系统运行数据未按日备份24、对供货企业资质审核不到位,部分企业质量保证协议签订不规范。
25、计算机系统无法及时调取数据;26、7-8月份温湿度记录无法调取;27、阴凉库超温时无法触发短信报警;28、向药品零售企业销售药品未做到发票随货同行;29、部分药品未严格按照制度要求验收。
GMP飞检实验室缺陷汇总

一、实验数据真实性缺陷1、查看6批五维他口服溶液含量测定高效液相图谱,检测前未进行色谱条件与系统适用性实验,检测时对照品和样品配制1份、进2针。
2、物料部分检测项目委托鄂州市食品药品检验检测中心检验,但现场不能提供相关检测报告。
3、QC实验室检验台账、取样台账,QA变更台账、偏差台账均为Word 格式的电子版本,储存上述台账的电脑无权限管理设置。
经现场核对,中药材检验台账中部分批次检验结果项目为空白,检验台账中部分中药材批次信息与原辅料台账中的相应信息不一致。
4、检查发现,企业用于申报生产的9个批次的人血白蛋白长期稳定性考察3个月、6个月、加速试验6个月大部分铝离子实际检测结果高于药典规定的不得高于200µg/L的标准,与注册申报数据不符。
5、上市后持续稳定性数据可靠性问题。
检查人血白蛋白上市后持续稳定性考察数据发现:铝离子检测数据与报告数据不一致,实际检测数据不符合标准,企业存在修改样品名、删除检测记录重新检测等问题,比如20130503批,实际检测结果为408µg/L,报告为143µg/L。
激肽释放酶原激活剂(PKA)含量实际值与记录值不一致,实际值不符合药典规定(≦35IU/ml)。
6、显微鉴别原始记录只有文字表述,无照片、影像或描图数据,现场未发现水合氯醛,企业亦提供不出配制记录,未配备检验样品对应的显微图谱,实验人员未开展显微鉴别培训,现场询问实验人员不能回答实验操作要求,企业无法证明实际开展了显微鉴别实验。
中检院穿心莲对照药材现批号为121082-201505,而企业批号为121082-200302。
7、未能提供检验原始记录。
未能提供160201批女宝胶囊部分原药材检验原始记录;未能提供150601批白芍(用于保胎灵生产)和140401批白芍(用于保胎灵、女宝胶囊生产)的检验原始记录;未能提供140401批和150701批阿胶(用于保胎灵、女宝胶囊生产)的检验原始记录;未能提供131101批穿山龙(用于接骨续筋片生产)和150901批焦槟榔委托检验报告和委托检验合同。
某药厂飞检缺陷项

某药厂飞检缺陷项
主要缺陷
1.质量管理和质量控制人员不能有效履行职责,对部分购进的
中药材未经全检即投入生产;XX色度检查无标准比色液对照仍下合
格结论;XX成品检验报告中含量测定未附无纸质和电子HPLC色谱图;XX重金属及有害元素、农药残留原始记录无原始数据,也未按要求
委托检验;
2.红花、人参、沉香、黄芩、砂仁、百部、豆蔻、鸡血藤、乌药、小茴香、黄精等个品种均不是GMP认证检查范围和在市局备案的品种,企业制订了工艺规程,购买了对照品,但扩大生产范围未经许可已进行了生产。
一般缺陷9项
1、麸炒白术批生产记录无炼蜜记录及蜜制麸皮的制备记录,蜜、
麸皮和白术使用量与工艺规定不一致;
2、《2016年设备年度预防性检修计划表》无起草人、审批人、
批准人签字;
3、《XX加工工艺规程》规定干燥温度为60℃,批生产记录中记
录干燥温度为80℃;
4、阴凉成品库中存放的XX无留样;
5、药材供应农户供应商档案中未包括农户所供应全部中药材品种;
6、《物料代码及进厂编号管理规程》编制日期、审核日期、批准
日期均未填写;
7、委托检验未按规定在市局备案;
8、成品质量标准和原料质量标准中部分品种未按《中国药典》2015年版四部《药材和饮片鉴定通则》增加二氧化硫残留的测定;9、未进行生产工艺再验证。
国家局飞检不合格项汇总

十万级洁净生产车间内的组装间天花板有漏水和维补痕迹,有锈迹。
12
《空调净化系统清洁维修保养管理制度》(文件编号:Q/SYS/SB/ZD-2008-124)规定初效每个月清洗一次,中效三个月清洗一次,高效过滤器每年监测完整性,但企业实际对初、中效每年更换,实际也未监测高效过滤器的完整性;企业生产体外诊断试剂产品具有不连续性,空气净化系统停机时间较长,企业尚未对不同停机时间后再次开启空气净化系统所需采取的措施进行相应的验证和规定。
39
现场检查洁净区内镜片分拣、内包间时正值下班时间,工人已离开,剩余半成品无防护措施,企业未按规定清场。
40
查实验室现场,阳性对照间为正压设置。
41
检验室一更换鞋处放置一标识有“待洗无菌服”字样的塑料桶,桶内装满未经防护处理的工作服。企业洁净服管理规定,万级无菌服应在万级洗衣机清洗,十万级应在十万级洗衣间清洗。查该公司实验室无菌服清洗记录,确认实验室无菌服在十万级生产车间内的洗衣间进行清洗,未执行有关规定。
65
空气压缩系统(YEP000020)压力表(PO21、P022)未校准。
66
微生物限度室内的微粒测试仪所使用的纯化水,从周一(4月10日)放置进微生物限度室,存放至今。
67
复合膜袋技术规格书未规定初始污染菌、细菌内毒素等要求,且未针对采购物品去除细菌内毒素处理制定相关作业指导书。
61
待检产品未按规定放置在待验区,无标识,无请检单、出库单等单据。成品库台帐不能反映入、出库情况,仅能反映库存数量。
62
空调系统标准操作程序规定压差不超过初始压差2倍,实际执行初中效压差10~150,中高效压差30~250。
63
注射用水点存在死水段,且未明确规定取水时如何避免死水的措施。
实验室日常检查中发现的管理缺陷

实验室日常检查中发现的管理缺陷一、实验数据真实性缺陷1、实验记录缺失。
被查企业生产的口服溶液在测定高效液相图谱前,未进行色谱条件与系统适用性实验,检测时对照品和样品配制1份、进2针。
2、检测报告缺失。
被查企业物料部分检测项目委托鄂州市食品药品检验检测中心检验,但现场不能提供相关检测报告。
3、台账存储电脑无权限设置。
被查企业QC实验室检验台账、取样台账,QA变更台账、偏差台账均为WOrd格式的电子版本,但储存上述台账的电脑无权限管理设置。
4、实际检测结果与注册申报结果不符。
被查企业用于申报生产的9个批次的人血白蛋白在长期稳定性考察中,大部分铝离子实际检测结果高于规定的“不得高于200Hg/L”的标准,与注册申报数据不符。
5、上市后持续稳定性数据可靠性与实际检测不符。
被查企业的人血白蛋白后持续稳定性铝离子实际检测数据不符合标准,企业存在修改样品名、删除检测记录重新检测等问题。
比如,实际检测结果为408μg∕L,报告为143μg∕L:激肽释放酶原激活剂(PKA)含量实际值与记录值不一致,实际值不符合药典规定(W35IU∕ml). 6、无中间品配制记录。
飞检人员未在被查企业发现生产所需的水合氯醛,企业亦无法提供其配制记录。
7、未能提供检验原始记录。
被检企业未能提供多批次原材料的检验原始记录或委托检验报告、委托检验合同。
8、计算机化系统内数据删改权限设置不合理。
被检企业化学实验室电脑、液相色谱仪软件各级别帐号权限混乱,分析员帐号进入色谱软件系统不能修改检验方法和积分参数,管理员帐号却可以。
9、无包材检验数据记录。
被检企业无法提供相关包材检验数据,调查发现QC人员未按SOP对物料检验对部分包材不取样,不检验,直接发放检验合格报告。
10、相关批次检验图谱雷同。
被检企业的西咪替丁原料药红外鉴别图谱201706048批、201706055批和201704003批雷同;201706017 批、201706015 批雷同;201704024 批、201704025 批和201706037 批雷同。
检查中质量控制与质量保证缺陷汇总PPT(共 33张)

另外,报告中显示"普鲁卡因青霉素溶剂残留正丁醇1、2
月份数据偏高、波动明显,可能与提高投料量或工艺控制
有关,应查找原因并采取措施",但企业未在报告中阐述
调查分析情况及纠正预防措施。
14
• 批号为20130922的血栓通注射液稳定性考察试验中第3月数据出 现偏差,但企业未给予关注和调查。
10
在数据管理方面存在缺陷。 1)SPX-250B-2型生化培养箱性能再确认中的
温度检测电子数据未进行备份; 2)高效液相色谱仪、原子吸收仪工作站虽
设定密码,但密码在仪器操作人员中共同 使用,无法防止电子数据被修改或删除; 3)SZA620型热风循环干燥机再验证报告 [PQP-0712(002-003)]所附原始记录中,悬浮粒子 测定数据非原始打印数据(为手工抄写),细菌 内毒素原始测定数据未归入报告中。
2)经检查组对企业电脑进行数据恢复,进一步检查发现该企业分别于 2014年和2015年生产促肝细胞生长素溶液7批和16批,但企业无法提供相 应的批生产、批检验等原始记录,生产过程无法追溯。
3)两台高效液相色谱仪(HPLC)未安装审计追踪功能,未设置登录用户 及权限,计算机操作系统时间有修改痕迹。(1)编造HPLC的电子数据。 检查发现实验室的两台HPLC中存有批号为140701、140901、141201、 141202、150201等5批促肝细胞生长素提取物溶液图谱。(2)篡改紫外 分光光度计(UV)的电子数据。将140701批促肝细胞生长素提取物溶液13
中药企业飞检缺陷汇总
湖北康源药业有限公司上述行为已违反《中华人民共和国药品管理法》及药品GMP相关规定,湖北省食品药品监管局已收回该企业相关《药品GMP证书》,国家食品药品监督管理总局要求湖北省局进一步监督企业召回相关产品,对企业涉嫌违法违规生产的行为立案调查。
序号
企业名称
检查时间
通报时间
发现问题
处理措施
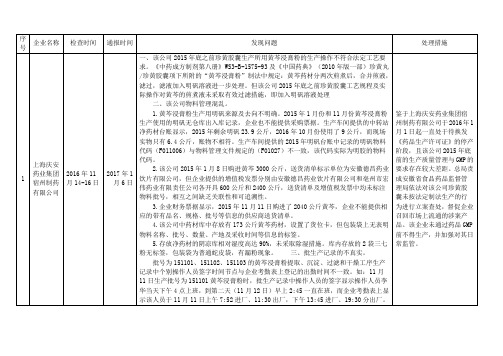
1
上海庆安药业集团宿州制药有限公司
2016年11月14-16日
2017年1月6日
一、该公司2015年底之前珍黄胶囊生产所用黄芩浸膏粉的生产操作不符合法定工艺要求。《中药成方制剂第八册》WS3-B-1575-93及《中国药典》(2010年版一部)珍黄丸/珍黄胶囊项下所附的“黄芩浸膏粉”制法中规定:黄芩药材分两次煎煮后,合并煎液,滤过,滤液加入明矾溶液进一步处理。但该公司2015年底之前珍黄胶囊工艺规程及实际操作对黄芩的煎煮液未采取有效过滤措施,即加入明矾溶液处理
2.该公司2015年1月8日购进黄芩3000公斤,送货清单标示单位为安徽德昌药业饮片有限公司,但企业提供的增值税发票分别由安徽德昌药业饮片有限公司和亳州市宏伟药业有限责任公司各开具600公斤和2400公斤,送货清单及增值税发票中均未标注物料批号,相互之间缺乏关联性和可追溯性。
3.企业财务票据显示,2015年11月11日购进了2040公斤黄芩,企业不能提供相应的带有品名、规格、批号等信息的供应商送货清单。
2、在存放上述分类账的档案柜中发现了两本《批生产指令西区》,指令时间分别为2016年1月至6月、2016年3月至6月。
32条国家局飞行检查确认与验证缺陷汇总及分析(GMP)

32条国家局飞行检查确认与验证缺陷汇总及分析1、产品工艺验证不符合要求现场检查发现企业生产的盐酸小檗碱7种批量,但仅对其中3种批量进行验证。
点评:确认与验证附录第十九条规定,…工艺验证应当包括首次验证、影响产品质量的重大变更后的验证、必要的再验证以及在产品生命周期中的持续工艺确认,以确保工艺始终处于验证状态。
所以,任何使用新批量进行商业化生产前均应进行工艺验证,盐酸小檗碱片增大生产批量或减小生产批量对制粒后、混合后颗粒的均一性产生影响,应进行工艺验证。
2003版药品生产验证指南中提到批量大小变化小于等于10%,属于较小变动,一般不需再验证!2、产品验证不符合要求(1)痛风舒片混合粉实际生产批量与工艺验证批量不一致。
点评:GMP第一百四十九条规定,应当根据验证的结果确认工艺规程和操作规程。
确认与验证附录第二十二条规定,工艺验证批的批量应当与预定的商业批的批量一致。
怎么个死法,任选一条!(2)氨咖黄敏胶囊总混质量均一性验证未确认取样位置和取样量。
点评:确认与验证附录第二十五条规定,工艺验证方案应当至少包括以下内容:…(十)取样方法及计划;应在工艺验证方案中明确氨咖黄敏胶囊总混质量均一性取样计划,包括取样位置、取样点数量、取样量、取样工具等,并根据取样点位置对样品进行编号,以便检验结果的追溯。
按照2015版中国药典0941含量均匀度检查法执行,取上中下各取3个样品+中心点,共10个样品。
(3)五维他口服液验证批未体现取样检测情况,缺失部分过程;未按要求检测配料的样品性状、相对密度和主药含量,报告中也无含量检测数据。
点评:GMP第一百四十八条规定,确认或验证应当按照预先确定和批准的方案实施,并有记录。
确认或验证工作完成后,应当写出报告,并经审核、批准。
确认或验证的结果和结论(包括评价和建议)应当有记录并存档。
(4)痛风舒片工艺规程的变更无相关研究数据或验证数据支持。
点评:GMP第一百四十二条规定,当影响产品质量的主要因素,如原辅料、与药品直接接触的包装材料、生产设备、生产环境(或厂房)、生产工艺、检验方法等发生变更时,应当进行确认或验证。
2016年飞行检查缺陷项汇总

2016年飞行检查发现缺陷项汇总一、严重缺陷1、**00401,在财务部的出纳组发现大量个人缴纳保证金的收款收据,且大多金额与实际拟购药品金额一致;采购的随货同行单中药品品种、数量及金额与收款收据中保证金金额一致;全部销售完毕后,保证金退回个人;企业会计凭证中的山东增值税普通发票94份(票号05093533--05093662)显示从张※※、车※※、王※※、黄※※四人处收购金银花、灵芝、生地,发票金额共937605元,企业不能提供上述四人的任何资料,也未提供上述产品的销售渠道证明,现用的计算机系统及保留的旧计算机系统均无任何记录;私设仓库储存胶体果铋胶囊(批号151101)220盒等大量药品。
2、**00402,企业阴凉库有三个温湿度探头,分别显示19.2℃、20.6℃、19.3℃;实际墙上空调显示器示数分别显示为31℃、30℃、30℃,一小时后,示数变为30℃、29℃、29℃。
温湿度探头数值不真实;企业的温湿度自动监测系统的温湿度传感器监测值具有修正、调整功能,涉嫌温湿度监测数据不真实。
3、**06601,在阴凉库零货区N-H-4-3、整货区H-1-1-1存放的对乙酰氨基酚片(生产厂家,※※※※制药有限公司;批号,151101;库存,2件*240盒+80盒),企业不能提供任何采购票据,现用的计算机系统及保留的旧计算机系统均无任何记录;在财务部计算机“进项”电子表格中发现磷酸氢钙、氯化钙、甘露醇、氢氧化镁、维生素E等品种,该企业不能提供任何采购票据,现用的计算机系统及保留的旧计算机系统均无任何记录。
注射级氯化钙在计算机系统中显示库存为100公斤,而实际库房内未发现实物;业购进药品未索取留存购货发票。
4、**09101,企业销售的部分药品开具发票中的购货单位名称与付款人名称不一致。
二、主要缺陷1、*00802,2015年12月计算管理系统软件由“药神系统”变更为“海典系统”,2016年3月质量部长变更,企业均未开展专项内审。
2017年4月医疗器械飞检缺陷项目总结

2017年4月国家局共飞检了6家医疗器械单位,其中5家为体外诊断试剂企业,现将这5家体外诊断试剂的缺陷项进行了汇总分析:5家单位的缺陷项目一共有72条,其中机构与人员7条、厂房与设施11条、设备12条、文件管理7条、设计开发5条、采购8条、生产管理8条、质量控制10条、销售和售后服务1条、不合格品控制1条、不良事件监测、分析和改进2条。
机构与人员的缺陷除了体检外,其他项目不论是培训还是人员操作均体现在与规定不符,可以挂靠到培训不到位,实际上还是在管理中没有做到写我所做,做我所写。
厂房与设施的缺陷除了硬件上的设计缺陷外,还体现在文件缺失和管理职责不明,需要各企业首先按照法规的要求对硬件设施进行维护与管理,并在日常管理时制定明确的文件制度,有效的对厂房和设施进行管控。
设备的缺陷一个是初中效的压差监控,好多企业都有遗漏,另外一个主要是计量和验证,尤其是计量院出具的计量报告,需要对计量结果进行有效性确认,因为报告一般不会明确是否合格,只是给你一个偏差范围,其次的验证主要体现在方法验证和效果验证,就是说需要对你文件规定的方法出具一个合理的数据分析报告,表明你的文件规定和方式方法可以达到你的预期效果,每个规定都要有出处(如空调停机后恢复的评估、消毒效果)。
文件管理的缺陷有两条都跟收集外来文件有关,需要时刻关注新的法规标准更新,其次就是文件管理的规定一定要细化,该如何进行文件升级、如何进行文件上内容的修改,如何回收作废文件和记录,并且对细化的内容培训到位,能够良好的实施。
设计开发的缺陷主要为设计评审的缺失,该有的文件一定要有,该有的记录一定要存在,针对冻融次数这个问题,我们需要明确,在工艺中不论是时限还是次数,对产品质量属性存在直接影响的,都应该有测试或者验证,来表明我们这个参数的制定是合理的。
采购的缺陷除了两条为缺失文件标准外,另外的几条均是文件与实际不匹配,没有按照规定的程序或者标准执行。
生产管理的缺陷为关键工序或者特殊工序的影响因素评估不到位,近效期物料的使用未评估,其他内容也是与文件规定的操作方式不一致。
飞检中实验室缺陷之标准品及对照品管理缺陷

飞检中实验室缺陷之标准品及对照品管理缺陷
标准品及对照品的管理缺陷
◆1、被查企业未按《质量控制室标准品/对照品/对照药材的管理规定》中“配制好的对照品溶液可存放6个月”的规定,对对照品溶液进行稳定性考察,抽查部分对照品溶液稳定性研究方案,发现其仅进行3个月稳定性考察。
◆2、被查企业在产品成品检验中使用的含量测定对照品未进行标定,并用于了产品放行检验。
◆3、被查企业在对照品过期的情况下,未对过期对照品的特性量值及适用性进行验证便用于产品放行检验。
◆4、被查企业的部分标准品、对照品领用台账中无相关领用记录。
◆5、被查企业无对照品溶液配制记录(仅记录取样量)以及领用记录,同时在检验原始记录中仅记录中检院对照品的批号,未体现对照品溶液的相关信息,难以溯源。
◆6、被查企业的试剂、对照品无购进和使用记录。
◆7、被查企业QC实验室使用过期的溶液标准样品。
◆8、被查企业的质量控制部采用的进口检验对照品,未与法定对照品进行比对、标化即投入使用。
◆9、被查企业未对部分对照品溶液存放期限进行考察。
环境监测飞行检查常见问题及法律责任

4、涉嫌造假 (1)质控数据长时间不变 。(甚至标准曲线) (2)样品交接记录与分析记录的一致性等 。 (3)同一检测人员在不同报告或记录里的笔迹不同 。 (4)同一采样/质量监督人员同时在不同地方的采样并签字。 (5)有数据报告,无分析原始记录。 (6)无仪器使用记录、试剂领用、使用记录。 5、记录的保存。资质认定要求不低于6年,但是。。。。。。
2019年1月,南昌市生态环境 局按照江西省生态环境厅要求 对公司开展执法检查。经现场 检查实验室和仪器设备,调阅 人员资料,抽查检测报告、原 始记录等材料后,发现银河公 司存在纸质原始记录与电子存 储记录不一致、伪造监测时间 、伪造签名等问题,涉嫌弄虚 作假。
查实相关问题后,南昌市生态环境局根据《江西省深化环境 监测改革提高环境监测数据质量实施方案》等文件要求,将 银河公司及法定代表人、授权签字人、技术负责人、质量负 责人、检测部负责人、相关采样及分析审核人员列入不良记 录名单,监测数据弄虚作假行为向社会公开,并上报省公共 信用信息平台,同时对银河公司参与政府购买环境监测服务 及监测数据弄虚作假人员在生态环境监测行业从业予以限制 。
租赁的仪器设备未重新检定// 校准;或检定 / 校准证书上的单位名称非使 用的检验检测机构 。同一台仪器设备不得同一时期在两家机构租赁、使用。 (借用外携仪器、拿样去其他机构分析)(人员劳动关系)
(二)仪器设备不能满足标准要求
7、量值溯源未按规定 (1)未做检定或校准、期间核查。仪器设备的量值溯源是保证其测量准确 性的主要手段之一,按照检验检测机构资质认定评审准则要求,仪器设备使 用前应经过检定或校准,必要时,在二次检定或校准之间应进行期间核查, 以保证数据的准确性。但部分机构未按规定将仪器送计量部门进行检定或校 准,造成仪器失准。 (2)校准参数不满足要求。
试验缺陷管理制度

试验缺陷管理制度一、制度目的为了规范试验活动中的缺陷管理,提高试验结果的可靠性和有效性,确保试验活动安全顺利进行,特制订本制度。
二、适用范围本制度适用于实验室试验、工业生产试验、产品检测试验等科研试验活动。
三、缺陷管理流程1. 缺陷发现试验活动中任何人员均有权发现并报告缺陷,包括实验人员、观测人员、仪器设备管理员等。
2. 缺陷报告发现缺陷的人员应当立即向试验负责人或主管部门报告,并填写缺陷报告单,详细描述缺陷的性质、位置、影响等信息。
3. 缺陷评估试验负责人或主管部门收到缺陷报告后,应当立即进行评估,确定缺陷的严重程度和影响范围。
4. 缺陷处理根据评估结果,试验负责人或主管部门应当制定缺陷处理方案,包括暂停试验、更换设备、修复设备等措施,确保缺陷得到及时有效处理。
5. 缺陷记录对于已经处理的缺陷,应当及时进行记录,包括缺陷的性质、处理措施、处理结果等信息,并进行归档保存。
四、缺陷分级根据缺陷的严重程度和影响范围,将缺陷分为不同级别,包括严重缺陷、一般缺陷和轻微缺陷。
1. 严重缺陷:对试验结果或人员安全产生重大影响的缺陷,应当立即暂停试验,并采取紧急措施处理。
2. 一般缺陷:对试验活动产生一定影响的缺陷,应当及时处理,确保试验活动能够继续进行。
3. 轻微缺陷:对试验活动影响较小的缺陷,可以在不影响试验结果和人员安全的情况下继续试验,并在试验结束后进行处理。
五、责任分工1. 试验负责人:负责制定和执行试验缺陷管理制度,对试验活动中的缺陷负总责,并确保缺陷得到及时有效处理。
2. 实验人员:负责发现和报告试验活动中的缺陷,并积极配合试验负责人或主管部门进行处理。
3. 仪器设备管理员:负责对试验设备进行定期检查和维护,及时发现并处理设备中的缺陷。
六、培训和教育为了提高试验人员的缺陷管理意识和能力,应当定期进行相关培训和教育,包括缺陷的识别、报告、处理等内容,确保各试验人员能够熟练掌握缺陷管理技能。
七、监督和检查试验负责人或主管部门应当定期对试验活动中的缺陷管理情况进行监督和检查,确保本制度的有效实施,并对违反本制度规定的行为进行相应处理。
GMP飞检缺陷项目

GMP飞检缺陷项目汇总1i>山东和*医疗技术有限公司品种:冠状动脉药物涂层支架系统检查发现一般不符合项6项。
一、厂房与设施方而1.企业外购的裸支架在洁净区超净台内采用易挥发的丙酮进行超声波清洗,淸洗区域距离万级下百级的药物喷涂区约2米,两个区域间无隔断,企业未评估裸支架淸洗对药物支架喷涂可能存在的污染。
2.药物支架产品涂覆工序在10,000级下的局部100级洁净室内进行,但企业未提供百级洁净区内使用的、药物涂覆过程中与产品使用表而直接接触的压缩空气尘埃粒子数等指标的检测数据;药物涂覆时使用高压氮气混合药液直接喷涂支架,企业提供的高压氮气验证报告未记录具体制造商和规格、级别等信息,且未规左日常检测周期和检测项目,未提供日常检测记录。
二、设计开发方而3.《药物混合工艺作业指导书》规定,聚合物取出后,剩余聚合物包装好,并用封口机封口进行保存,放置在低温冰箱内(实际存放在-20°C冰柜中),但未在文件中规定可重复冻融开封使用次数、使用期限:《支架喷涂、称量工艺作业指导书》用适量无水乙醇漂洗管芯、将适量药液倒入一只洁净烧杯中,未明确具体工艺参数。
三、生产管理方而4.《药物混合工艺作业指导书》中规定,聚合物称取前应连续三次称量裁剪好的称量纸,天平读数误差在土0.003mg以内,确左天平已经平稳,但企业未提供相关原始数据记录;《支架喷涂、称量工艺作业指导书》中规左裁管长度误差±5mm・但企业实际记录的数据仅精确到厘米。
5.《火菌工艺作业指导书》中规定的火菌条件:柜体内温度设定30°C,柜体内湿度230%RH, 抽査某火菌批次产品火菌湿度为62.5%RH,但企业提供的冠状动脉药物涂层支架系统环氧乙烷火菌确认报告中确认的最终火菌湿度为73.4%RH,火菌确认湿度与作业指导书不一致,且实际灭菌湿度低于火菌确认湿度。
四、不良事件监测、分析和改进方面6.抽査2019年省局飞行检査中发现的部分不合格项整改情况,企业已分析问题发生原因,采取整改措施,但未提供《纠正/预防措施报告单》,未评估整改措施有效性,不符合企业《纠正和预防措施控制程序》的规定。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
飞检中实验室缺陷之文件及记录管理缺陷
前言:随着数据完整性理念的深入人心,实验室数据越来越受到监管部门和企业重视,实验室数据完整性已然成为了新的监管趋势。
为方便大家自查和改进,小编整理了FDA 及国内飞行检查公布的实验室管理缺陷,供大家参考。
文件及记录管理缺陷
◆1、被查企业阴凉留样室的现场留样样品销毁单、常温留样室使用的温湿度记录表使用已被废弃的版本,且缺少文件编号。
◆2、被查企业实验室未建立样品接收、分样、领用、暂存记录,对部分原料在实验室中的粉碎、剩余样品、废弃处理等未建立记录。
◆3、被查企业实验室未按照企业制定的《实验室原始数据的管理规定》对薄层色谱照片进行电子数据管理,仅由实验员手机拍照后打印,原始电子照片未进行统一存储和备份。
◆4、被查企业计算机中存在部分难以溯源文件,且追溯仪器使用记录未见登记、未出报告。
◆5、被查企业实验室高效液相色谱仪的《仪器使用日志》每天使用一张记录页,且每张记录页仅记录1-2条仪器使用信息,剩余部分留空,记录不连续,存在潜在修改的可能性。
◆6、被查企业除批生产记录、检验记录外,其它的辅助记录未受控发放;已作废的文件无作废标识。
◆7、被查企业的两份相同版本号的标准中规定的同一产品的检测项目不同。
◆8、被查企业部分辅助记录未进行归档,且生产记录中均存在遮盖原始数据修改记录的情况。
◆9、被查企业精密仪器室现场高效液相色谱仪使用未经审核、批准的手写操作规程。
◆10、被查企业未建立计算机化系统管理程序,未对计算机化系统验证、关键数据管理、系统安全管理及清单管理等制定相关的管理规程。
◆11、被查企业原子吸收光谱仪计算机系统无审计追踪功能,未使用密码来控制系统登录。
◆12、被查企业的测定数据非原始打印数据,部分原始测定数据未归入相关报告中。
◆13、被查企业原始记录数据丢失无法溯源。
(1)原料检验原始记录丢失。
(2)相关检验用仪器高效液相色谱仪和天平无仪器使用记录。
(3)气相色谱仪中无相应数据,无仪器使用记录。
(4)气相色谱仪工作站内的数据丢失,且未备份。
◆14、被查企业质控实验室检验设备使用记录不规范不完整。
(1)菌种灭活记录未记录设备编号。
(2)超净工作台使用记录上无设备唯一性标识。
(3)使用电热恒温干燥箱对培养皿进行灭菌,未记录灭菌时间。
◆15、被查企业在成分含量测定的原始检验记录中,高效液相色谱图无进样时间、操作人等信息。