如何利用软件获取和基因的互补配对结合位点.doc
GeneTools 操作指南(中文)

GeneTools 也提供了一系列的工具,可以然您更好的手动控制处理过程和编辑分析结 果。但是您并不需要经常使用这些工具,因为 GeneTools 提供的自动分析工具是如此强大和 灵敏,它们会很好的处理绝大多数的图片,极少数特殊图像可能需要您进行手动编辑、分析。
分子量分配。
GeneTools 软件自己内置了一个分子量标准库,您可以对分子量标准库进行编辑。如果
其中没有符合您要的分子量标准,您也可以根据自己的需求,创建适合自己的分子量标
准。
确定分子量标准条带,分配分子量:
1. 选择分子量标准泳道(Marker 条带)
2. 选择对应分子量标准的第一条带
5. 使用双向箭头选择于分子量标注的第一峰
GeneTools
入门指南
基因有限公司售后服务部编译
编者言 本手册系原版英文说明书编译而来,希望它能为您操作该软件时 带来帮助。由于译者的水平限制,手册中可能存在错误和遗漏,如与 英文说明书发生冲突,请英文原版说明书为准。本手册只作为您操作 的参考依据,不能作为其他评定标准。
基因公司售后部应用工程师:张传峰 2004-3-23
样本属性对话框会立即弹出
4 选择 Analysis Type 为 Gel(Genetools 默认的分析类型是 Gel,您也可以通过 Extra - configuration-Analysis type 改变默认的设置)。 5 选择电泳的方向,默认方向是 Down。 您不必要修改其它的设置,尤其是: l GeneTools 自动检测图像类型 l 您应该设置“Number of tracks”为 0,这样 GeneTools 会自动进行泳道定 义。 6 点击“OK”键 短暂的停顿以后,GeneTools 会自动进行泳道定位和条带定位。 结果会显示在凝胶窗口。 关于该命令和相关主题的介绍请参阅在线帮助的相关章节 Reference → Gel analysis → How to → Create and work with secure sample files : l 打开一个 secure sample file l 创建一个新的 secure sample file l 使用查询功能 l 获取图像创建 secure sample file
基因序列拼接器软件 使用说明书(软件操作文档)

基因序列拼接器软件使用说明书(软件操作文档)1. 引言基因序列拼接器软件(MergeSeq)基于研究者测序获得或从核酸数5据库(如GenBank等)中批量下载的fasta格式存储的基因片段,按照使用者指定顺序,将同一物种不同基因片段拼接成由多基因组成的长序列,用于不同物种间的分子系统发育分析。
1.1编写目的本说明书为在Linux/UNIX环境下使用MergeSeq软件的用户编写。
10它将指引使用者按步骤搭建该软件的运行环境,明确输入文件的录入格式,熟悉运行参数的配置规则,理解软件运行时出现的状态信息并掌握获取输出fasta格式文件的方法。
1.2项目背景非模式生物(如蜘蛛等)构建分子系统发育树一般选取低速进化15(Slow-Evolving)且有种属特异性的基因片段进行分析。
为提高结果的置信度,一般采取多基因组合分析的方法(Dimitrov et al., 2016; Wheeler et al., 2016)。
但在实际操作中,将同一物种不同基因片段拼接在一起是一件十分费事且容易出错的重复性操作。
尤其当某一物种某个基因数据缺失时,20必须在拼接后的长序列中填充与同一基因其他对齐序列等长的占位符以保证结果为对齐。
本项目基于测序或下载获得的fasta格式基因序列片段,按照研究者指定的基因排列顺序,自动将同一物种不同基因片段拼接成多基因长序列,并保证结果对齐,可直接用于不同物种间的分子系统发育分析。
251.3 定义(专门术语的定义和缩写词的原意)fasta格式(fasta format):fasta格式是一种基于文本用于表示核酸序列或多肽序列的格式。
其中核酸或氨基酸均以单个字母来表示,且允许在序列前添加序列名及注释。
该格式已成为生物信息学领域常用的30标准文件格式。
2. 软件性能2.1.数据精确度本软件不涉及数字的计算和处理,输入、输出数据的格式均为35UTF-8编码的文本类型文件。
2.2.时间特性本软件对输入数据、输出数据的处理时间由基因片段长度和运行软件主机性能决定。
分子生物学实验中的分析软件使用方法介绍

分子生物学实验中的分析软件使用方法介绍随着科技的发展和进步,分子生物学实验的数据量不断增加,对于这些大量的数据进行分析成为了科研工作者不可或缺的一部分。
为了更好地处理和解读这些数据,科研人员们使用各种分析软件来辅助他们的研究工作。
本文将介绍一些常用的分析软件及其使用方法。
一、基因序列分析软件基因序列分析软件是分子生物学实验中最常用的软件之一,它们用于分析DNA或RNA序列以及蛋白质序列。
其中,NCBI Blast是一种非常常用的基因序列比对软件,它可以通过将待比对的序列与已知的序列数据库进行比对,从而确定序列的相关性和相似性。
使用NCBI Blast,我们可以快速找到与我们研究对象相关的序列信息。
二、基因表达分析软件基因表达分析软件用于分析基因在不同组织或条件下的表达水平,以及基因调控网络等。
在这方面,R语言是一种非常强大的工具。
通过使用R语言中的各种包和函数,我们可以对基因表达数据进行聚类分析、差异表达分析、通路富集分析等。
同时,R语言还提供了丰富的数据可视化功能,可以帮助我们更好地展示和解读实验结果。
三、蛋白质结构分析软件蛋白质结构分析软件主要用于预测蛋白质的三维结构以及模拟蛋白质的动力学行为。
其中,Swiss-PdbViewer是一种常用的蛋白质结构可视化软件,它可以帮助我们观察和分析蛋白质的结构特征。
而GROMACS则是一种常用的分子动力学模拟软件,它可以模拟蛋白质在不同环境下的运动轨迹,帮助我们理解蛋白质的功能和机制。
四、基因组学分析软件基因组学分析软件主要用于处理和分析整个基因组的数据,包括基因组序列、基因组注释以及基因组变异等。
在这方面,Ensembl是一种非常常用的基因组分析软件。
它提供了大量的基因组数据和工具,可以帮助我们进行基因组注释、基因组比对以及基因组变异的分析。
五、细胞图像分析软件细胞图像分析软件用于分析和处理细胞图像数据,帮助我们了解细胞的形态和功能。
其中,ImageJ是一种非常流行的细胞图像分析软件,它提供了丰富的图像处理和分析工具,可以帮助我们进行细胞计数、细胞形态分析以及细胞追踪等。
miRNA的研究方法

科研(kē yán)中的miRNA研究(yánjiū)方法总结microRNA(miRNA) 是一类(yī lèi)长度在22nt左右(zuǒyòu)的内源非编码小RNA,广泛存在于动物、植物(zhíwù)、病毒等多种有机体中。
1993年,Lee等人在秀丽新小杆线虫(C.elegans)中发现了控制着线虫时序性发育的lin-4。
2000年,Reinhart等发现了另一个具有转录后调节功能的小分子RNA:let-7。
随后的几年时间里,许多研究人员相继发现了这类RNA,并将这些具有时空表达特异性的非编码小分子RNA命名为microRNA(miRNA)。
从长的初级miRNA(pri-miRNA)到形成成熟的单链miRNA到与靶标RNA结合,至少需要四步:首先是由核糖核酸酶ⅢDrosha和DGCR8组成的复合物将初始miRNA 转录本(pri-miRNA)加工成miRNA前体(pre-miRNA);接着由转运蛋白Exportin-5和Ran GTP酶将miRNA从细胞核转运到细胞质中;第三步是由另一种核糖核酸酶ⅢDrosha将miRNA前体剪切成成熟的miRNA双链,miRNA双链打开,其中一条进入到RNA诱导的沉默RISC复合体(RISC)中,该复合体还包含TarRNA结合蛋白(TRBP);最终RISC复合体根据miRNA与靶标RNA的互补配对,对靶基因进行剪切,或者翻译抑制。
图 1 miRNA生物学调控过程(摘自文献8)miRNA通过作用于相应靶mRNA,参与细胞增殖、凋亡、分化、代谢、发育、肿瘤转移等多种生物学过程。
预测超过1/3的人类基因都是保守的miRNA靶基因。
近些年,随着科学研究的进展,大量的miRNA已经被鉴定出来,截至目前发现的人的成熟的miRNA有2578条,小鼠的有1908条。
虽然miRNA的数量很多,但大多miRNA的功能并不为人所知。
基因结合位点查找

基因结合位点查找
摘要:
1.基因结合位点查找的定义和重要性
2.基因结合位点的查找方法
3.基因结合位点查找的应用
4.基因结合位点查找的挑战与未来发展
正文:
基因结合位点查找是生物信息学中的一个重要研究领域,它关乎基因表达调控、蛋白质相互作用以及表观遗传学等多个生物学层面的问题。
通过查找基因结合位点,我们可以了解基因如何在调控元件、启动子、增强子等区域上发挥作用,从而揭示生物过程的基因调控机制。
基因结合位点的查找方法主要有以下几种:
(1)基于基因组数据的查找方法:通过分析基因组序列特征,如启动子、增强子等区域,来预测基因结合位点。
这种方法主要依赖于基因组数据和生物信息学算法,如BPROM 和NNPP 等。
(2)基于实验数据的查找方法:通过实验技术,如ChIP-seq、ChIP-qPCR 等,直接检测蛋白质与DNA 的相互作用,从而获取基因结合位点信息。
这种方法具有较高的准确性,但实验操作较为复杂。
基因结合位点查找在生物学研究中有着广泛的应用,例如:
(1)研究基因调控机制:通过查找基因结合位点,可以揭示基因如何在调控元件、启动子、增强子等区域上发挥作用,从而揭示生物过程的基因调控机
制。
(2)疾病相关研究:某些疾病的发生与发展与基因调控异常密切相关,因此通过查找相关基因的结合位点,有助于深入了解疾病发生机制,为诊断、治疗提供新的思路。
(3)药物研发:基因结合位点查找可以为药物筛选和设计提供重要依据,通过靶向作用于特定基因的结合位点,可以提高药物的特异性和疗效。
尽管基因结合位点查找取得了显著进展,但仍面临着许多挑战,如数据分析方法的局限性、实验技术的不确定性等。
基因组序列比对分析及相关软件的使用

基因组序列比对分析及相关软件的使用基因组序列比对分析是一种常见的生物信息学分析方法,广泛用于研究DNA、RNA或蛋白质序列的相似性和差异性,以及基因组结构和功能等方面的研究。
下面将介绍基因组序列比对分析的基本原理和常用的比对软件的使用方法。
常用的比对软件:1. BLAST(Basic Local Alignment Search Tool)BLAST是一种常用的比对软件,可以快速比对两个序列之间的相似性。
BLAST将查询序列与参考序列进行比对,并给出一个比对得分(称为E值)来表示两个序列的相似性。
BLAST包含多种版本,如BLASTn用于DNA-DNA序列比对,BLASTp用于蛋白质序列比对等。
使用方法:b.准备查询序列和参考序列。
c.打开BLAST软件,选择相应的版本(如BLASTn)。
d.在查询序列窗口中输入查询序列,点击“运行”按钮开始比对。
e.在结果中查看比对得分(E值)和匹配的位置信息。
2. Bowtie / Bowtie2Bowtie和Bowtie2是一对基因组序列比对软件,用于比较长的DNA序列。
Bowtie使用索引来加快比对速度,可以在较短的时间内进行大规模比对。
Bowtie2相比Bowtie具有更高的准确性和更好的感受性。
使用方法:b.准备查询序列和参考序列。
c.构建索引文件,将参考序列转换为索引文件格式。
d. 打开终端或命令提示符窗口,输入相应的命令来运行Bowtie或Bowtie2e.在结果中查看比对得分、匹配的位置信息和SAM/BAM格式文件。
3. BWA(Burrows-Wheeler Aligner)BWA是一种用于DNA和RNA序列比对的软件,可以高效地进行大规模比对和可变位点检测。
BWA将参考序列转换为索引,然后将查询序列与索引进行比对,以找到最佳比对结果。
使用方法:b.准备查询序列和参考序列。
c.构建索引文件,将参考序列转换为索引文件格式。
d.打开终端或命令提示符窗口,输入相应的命令来运行BWA。
利用生物大数据技术分析转录因子结合位点的步骤说明

利用生物大数据技术分析转录因子结合位点的步骤说明在利用生物大数据技术分析转录因子结合位点的步骤中,首先需要了解转录因子及其结合位点的基本概念。
转录因子是一类调控基因表达的蛋白质,通过结合到DNA上的特定区域,即转录因子结合位点,来调控基因的转录活性。
通过分析转录因子的结合位点,可以揭示基因调控网络的组成和功能,进而深入了解生物过程中的细节及其相互关系。
接下来,将介绍利用生物大数据技术分析转录因子结合位点的步骤说明,包括获取转录因子结合位点数据、数据预处理、位点注释、转录因子结合位点的富集分析等。
第一步,获取转录因子结合位点数据。
生物数据库,如Encode、JASPAR、TRANSFAC等提供了大量的转录因子结合位点数据,可以通过这些数据库获取所需要的数据。
此外,还可以通过ChIP-seq、DNase-seq等高通量测序技术获得转录因子结合位点数据。
第二步,数据预处理。
由于生物大数据往往具有较高的维度和复杂性,处理这些数据的第一步是进行预处理。
预处理过程包括数据清洗、数据格式转换、数据质量评估等,目的是排除噪声和不可靠的数据,保留高质量的转录因子结合位点数据。
第三步,位点注释。
位点注释是将转录因子结合位点与基因组中的基因、剪接变异、启动子区域等进行关联,以了解这些位点的功能和可能的调控机制。
常用的位点注释工具包括Homer、BEDTools等,它们可以根据对应的基因组信息进行注释分析。
第四步,转录因子结合位点的富集分析。
富集分析是用来判断一组结合位点是否在某些功能上过于聚集或分散。
常见的富集分析方法包括基于超几何分布的富集分析和基于基因集合比较的富集分析。
这些方法可以帮助研究人员发现与特定生物过程或疾病相关的转录因子结合位点。
此外,还可以进行转录因子结合位点共现网络的构建。
共现网络分析可以帮助研究人员揭示转录因子之间的相互作用关系,进一步理解转录调控网络的复杂特性。
在进行以上步骤时,需要借助生物信息学工具和编程语言进行数据处理和分析。
如何使用生物大数据技术进行转录因子结合位点分析

如何使用生物大数据技术进行转录因子结合位点分析生物大数据技术是近年来在生物医学领域取得突破性进展的一项重要技术。
其中,转录因子结合位点分析是生物大数据技术中的一个关键研究方向。
转录因子是参与基因表达调控的重要蛋白质,而结合位点则是转录因子与DNA序列结合的特定区域。
通过分析转录因子结合位点的分布和功能,研究人员能够深入了解基因调控网络的构建和生物过程的调控机制。
下面将介绍如何使用生物大数据技术进行转录因子结合位点分析。
首先,在进行转录因子结合位点分析之前,研究人员需要获取转录因子的结合位点数据。
目前,公共数据库中已经收集了大量的转录因子结合位点数据,如ENCODE、Roadmap Epigenomics、Cistrome等数据库。
这些数据库提供的数据包括了不同细胞类型和生物过程中的转录因子结合位点信息。
研究人员可以通过访问这些数据库的网站或使用特定的API获取所需数据。
获取到结合位点数据后,下一步是进行数据预处理。
数据预处理的目的是将原始数据进行清洗和标准化,以确保后续分析的准确性和可靠性。
预处理的步骤包括数据去重、过滤低质量数据、修剪序列、统一坐标系统等。
这些步骤可以借助生物信息学工具和编程语言实现,如BEDTools、SeqKit、R语言等。
接下来,研究人员可以根据具体研究目的,进行转录因子结合位点的富集分析。
富集分析的目的是确定在某个生物过程或细胞类型中,哪些转录因子结合位点具有显著的富集表现。
为了实现这一目标,研究人员可以使用一些富集分析工具,如HOMER、GREAT、ChIPseeker等。
这些工具可以根据输入的结合位点数据和基因组注释信息,进行富集分析,并提供富集结果的可视化展示。
除了富集分析,研究人员还可以对转录因子结合位点进行进一步的功能分析。
功能分析的目的是探究转录因子结合位点在基因调控中的具体作用和机制。
研究人员可以使用一些基因组学工具,如SeqMINER、BEDTools、GREAT等,进行位点序列分析、DNA序列保守性分析、TF motif分析等。
python碱基互补配对

在生物信息学和分子生物学中,碱基互补配对是一个非常重要的概念。
它指的是DNA 和RNA中的碱基之间的一种配对关系,这种配对关系是:腺嘌呤(A)配对胸腺嘧啶(T),鸟嘌呤(G)配对胞嘧啶(C)。
这种配对关系是由DNA双螺旋结构模型的研究者们发现的。
在Python中,我们可以使用一个字典来表示这种配对关系。
字典的键是DNA中的碱基,值是与其配对的碱基。
例如:pythonbase_pair = {"A": "T", "T": "A", "G": "C", "C": "G"}这个字典表示的是:如果DNA中有腺嘌呤(A),那么在配对的RNA中就是胸腺嘧啶(T);如果DNA中有鸟嘌呤(G),那么在配对的RNA中就是胞嘧啶(C)。
依此类推。
如果我们想要找到一个特定的碱基的互补碱基,我们可以通过查找这个碱基在字典中的值来找到答案。
例如:pythoncomplement = base_pair["A"]print(complement) # 输出:T以上代码会找到碱基"A"的互补碱基,输出结果为"T"。
请注意,这个字典只包括了DNA和RNA中的主要碱基。
实际上,DNA中还存在其他一些次要碱基,如次黄嘌呤(I)和胸腺嘧啶(B),但它们并不参与主要的碱基互补配对。
同样,RNA中的尿嘧啶(U)也不在这个字典中,因为它与DNA中的胸腺嘧啶(T)配对。
如果需要考虑这些次要碱基的配对关系,那么这个字典就需要进行扩展。
利用生物大数据技术分析基因共表达网络的步骤说明

利用生物大数据技术分析基因共表达网络的步骤说明生物大数据的快速发展和广泛应用,为研究基因表达网络提供了宝贵的数据资源。
基因共表达网络分析是一种基于基因表达模式相似性的方法,用于揭示基因调控网络中的关键基因和信号通路。
以下是利用生物大数据技术分析基因共表达网络的步骤说明。
1. 数据收集与预处理首先,我们需要收集所需的基因表达数据。
这些数据可以来自公共数据库,如NCBI的Gene Expression Omnibus (GEO)、ArrayExpress等。
选择适当的实验数据集,确保样本数量足够,并且具备完整的样本信息和实验设计。
下载数据后,进行数据预处理,包括数据清洗、异常值处理、数据标准化等。
这一步骤的目的是消除数据中的噪声和偏倚,提高数据的可靠性和可比性。
2. 基因共表达计算基因共表达计算是构建基因共表达网络的核心步骤。
常用的计算方法包括相关系数、互信息等。
相关系数常用来衡量两个基因表达模式之间的相似性。
一般来说,Pearson相关系数和Spearman相关系数是常见的选择。
互信息是一种衡量两个基因表达模式之间的非线性关系的方法。
基于计算的结果,可以构建一个基因共表达矩阵。
3. 网络构建与可视化基于基因共表达矩阵,我们可以利用网络构建算法,如WGCNA、PEAR、MM算法等,构建基因共表达网络。
这些算法会将具有相关表达模式的基因连接在一起,形成一个网络图。
具体构建网络的方法可以根据研究的目的和数据特点进行选择。
构建完成后,我们可以利用可视化工具,如Cytoscape、Gephi等,将基因共表达网络进行可视化展示。
4. 基因功能注释与信号通路分析基因共表达网络的构建完成后,我们可以对网络中的基因进行功能注释和信号通路分析。
功能注释可以帮助我们理解网络中不同基因的生物学功能和相关性。
常用的功能注释工具包括DAVID、GSEA、STRING等。
信号通路分析可以帮助我们确定哪些信号通路在基因调控网络中起着重要的作用。
基因结合位点查找

基因结合位点查找摘要:一、基因结合位点查找的概述二、基因结合位点查找的方法与技术1.生物信息学方法2.实验方法三、基因结合位点查找的应用领域四、基因结合位点查找在医学研究中的重要作用五、未来发展趋势与展望正文:基因结合位点查找是研究基因与疾病、表型等关联的重要手段。
通过查找基因结合位点,可以揭示基因在生物体生长发育、生理功能及疾病发生发展中的作用机制,为遗传病诊断、药物研发和基因治疗提供重要依据。
一、基因结合位点查找的概述基因结合位点是指基因与其他生物大分子(如蛋白质、核酸等)相互作用的位置。
基因结合位点查找旨在发现和研究这些相互作用,以揭示基因在生物体内的功能及调控机制。
基因结合位点查找可为遗传病诊断、药物研发和基因治疗提供重要依据。
二、基因结合位点查找的方法与技术1.生物信息学方法:生物信息学方法主要通过计算机算法和数据分析,预测基因结合位点。
常用的生物信息学方法包括:基因组学、蛋白质组学、比较基因组学等。
2.实验方法:实验方法是通过实验技术验证生物信息学方法预测的基因结合位点。
常用的实验方法包括:ChIP-seq、ChIP-qPCR、高通量测序等。
三、基因结合位点查找的应用领域基因结合位点查找在多个领域具有广泛应用,如:医学、农业、生物工程等。
在医学领域,基因结合位点查找为遗传病诊断、药物研发和基因治疗提供重要依据;在农业领域,基因结合位点查找可助力优良品种的选育和栽培;在生物工程领域,基因结合位点查找有助于优化生物反应器和生物制品的生产过程。
四、基因结合位点查找在医学研究中的重要作用基因结合位点查找在医学研究中具有重要作用。
通过查找基因结合位点,可以揭示基因与疾病的关系,为遗传病诊断提供依据;同时,基因结合位点查找还可以指导药物研发,从而实现精准医疗。
五、未来发展趋势与展望随着生物技术的不断发展,基因结合位点查找技术将更加完善和高效。
未来,基因结合位点查找将在以下方面取得突破:1.高效准确的基因结合位点预测算法和数据分析方法的开发与应用。
tbtools基因结合位点

tbtools基因结合位点
一、概述
tbtools是一个用于分析和可视化基因组数据的强大工具包。
其中一个重要的功能是识别基因结合位点,即DNA序列上能够与转录因子结合的特定区域。
通过确定这些位点,研究人员可以更好地理解基因的表达和调控机制。
二、基因结合位点的定义
基因结合位点是指DNA序列上能够与转录因子特异性结合的区域。
这些区域通常具有特定的序列模式,能够与转录因子的DNA结合域相匹配。
转录因子是一种蛋白质,能够识别并结合特定的DNA序列,进而调控基因的表达。
三、tbtools在基因结合位点分析中的应用
tbtools通过一系列强大的功能,帮助研究人员识别和分析基因结合位点。
这些功能包括:
1.数据导入与预处理:tbtools可以方便地导入基因组数据,并对其进行清洗
和格式化,以确保分析的准确性。
2.序列模式匹配:tbtools利用各种算法和工具,如正则表达式、隐马尔可夫
模型等,对DNA序列进行模式匹配,以识别潜在的基因结合位点。
3.统计分析:通过对比不同条件下的基因表达数据,tbtools可以帮助研究人
员分析转录因子在特定生理或病理条件下对基因表达的调控作用。
4.可视化:tbtools提供了丰富的可视化功能,可以帮助研究人员直观地展示
基因结合位点的分布、转录因子的表达以及它们之间的关系。
四、总结
tbtools是一个功能强大的基因组数据分析工具包,尤其在基因结合位点分析方面表现出色。
通过使用tbtools,研究人员可以更深入地了解基因的表达和调控机制,为疾病诊断和治疗提供有力支持。
利用BLAST工具寻找新基因

案例分析
搜索结果返回页面 :
图中三项依次是图表摘要、描述和 比对信息,这三项在返回的页面中 默认是展开的,这里为了在同一截 屏显示,把这三项手动折叠了
案例分析
搜索结果返回页面 :
接下页
案例分析
搜索结果返回页面 :
参考文献上说这些匹配的
蛋白质可能: ①在一些数据库中得到的结 果完全匹配或者几乎完全匹 配。这就不是新基因了。 ②一些数据库的结果也非常 匹配,而该数据库中编码这 些蛋白质的DNA还没有被 注释过。这种情况可能是新 基因。 ③一些搜索结果并不是非常 匹配。这就需要依赖经验来 判断哪些数据库中的匹配是 真正的匹配,哪些不是。
参考文献
作 者:不详 作者单位:华中科技大学 生命科学与 技术学院 成稿日期:2011年1月6日 Reference 原文链接: /view/5bfd4c 87bceb19e8b8f6ba28.html
数据库和工具
我采用的数据库为美国国立生物技术信息中 心(The National Center for Biotechnology Information 网址: /)GenBank 数据库。 使用的工具为BLAST (Basic Local Alignment Search Tool 网址: http://blast.ncbi.nlm.nih.go v/)。
个人认为逐个序列的验证
是最经典而有效的方法: 具体来说,就是点击最右边 一列Accession,进入该基 因的详情页面,主要看 FEATURES,如果 FEATURES 下面只有 source(有的还有gap 或 misc_feature等 ),不含 有Protein、Region、Site、 CDS等注释属性,则可初 步判定它是未被注释的新基 因。
DNA 序列拼接软件

DNAStar→Seqman (subprogram of DNAStar)----------------------------------Sequencher 5.0 Demo序列拼接软件,还具有ORF分析、蛋白翻译、酶切位点作图、杂合子识别等分析功能。
-------------------------------------百度注册信息User’s name: 序列拼接Password: xulie123456---------------------------------用ContigExpress做序列拼接ContigExpress软件可用于做序列拼接,主要使用方法如下:解压缩下载的压缩文件contig.zip文件,保证文件CExpress.exeContigExpress软件可用于做序列拼接,主要使用方法如下:解压缩下载的压缩文件contig.zip文件,保证文件CExpress.exe,Gexudat.def 在同一个目录下,打开Cexpress.exe应用程序,进入ContigExpress操作界面。
点击菜单上的“Project”选择“Add Fragments”,一般我们发给您的是SCF 文件,如果您有其它格式的文件,也可以选择。
选择您存放SCF文件(即我们Email给您的测序结果的彩图文件)的目录,选择文件并打开,从而添加进ContigExpress软件。
在此以A、B两个序列为例,如果有多个序列的也可以同时添加进入。
选中要拼接的序列,再选菜单中的“Assemble”下的“Assemble Selected Fragments”命令,或用工具栏上的按钮。
若两个结果能够拼接起来的,会得到一个Assemble1下的contig1的结果。
双击contig1,打开拼接后的结果。
此时可能会因为两条序列的测序结果误差,会有不同的地方,在拼接图片框中的绿色竖杠就表示了这些不同的地方。
接着可点击绿色竖杠找到有误差的地方,进行修改。
基因预测软件,ORF Finder,详细使用方法
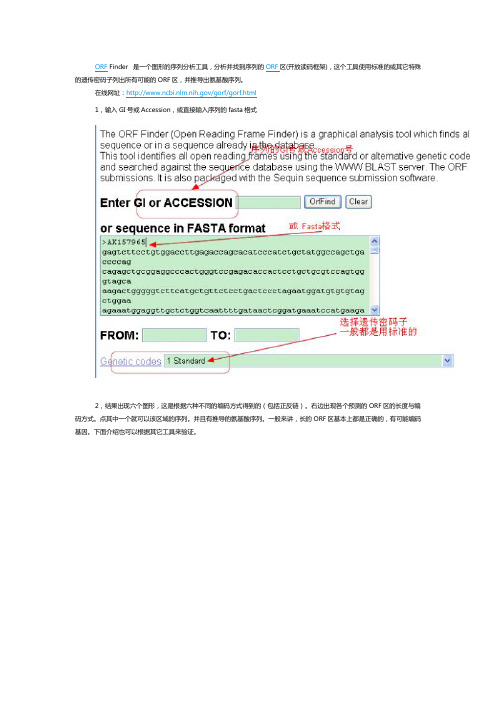
ORF Finder 是一个图形的序列分析工具,分析并找到序列的ORF区(开放读码框架),这个工具使用标准的或其它特殊的遗传密码子列出所有可能的ORF区,并推导出氨基酸序列。
在线网址:/gorf/gorf.html
1,输入GI号或Accession,或直接输入序列的fasta格式
2,结果出现六个图形,这是根据六种不同的编码方式得到的(包括正反链)。
右边出现各个预测的ORF区的长度与编码方式。
点其中一个就可以该区域的序列。
并且有推导的氨基酸序列。
一般来讲,长的ORF区基本上都是正确的,有可能编码基因。
下面介绍也可以根据其它工具来验证。
3,拿到氨基酸序列后,你可以直接做blastp,如果有匹配到,就是正确的ORF区了。
另外也可以用Pfam的方法,在Pfam数据库搜索。
如:/,输入氨基酸序列,仅仅是序列就可以了。
匹配到的话就会有结果,说明是属于那个家族的,能匹配就说明是正确的。
4,另一个工具是NCBI的CCD工具,网址:/Structure/cdd/wrpsb.cgi,同样输入氨基酸序列。
5,结果表明该ORF编码的蛋白是属于BTB家族的。
说明,你可以同时用几个工具来验证,如blastp,Pfam,CDD。
但有时的结果不一定全部一致。
这有可能是因为不能的数据库,存放的数据有些差异,算法也有些差异,是属于正常的。
基本上大部分都是可靠的。
基因结合位点查找

基因结合位点查找基因结合位点查找是一种重要的基因组学研究工具,用于确定转录因子与基因组DNA上的特定序列结合的位置。
这种技术可以帮助科研人员了解细胞中基因的调控机制,并进一步研究生物体的发育、疾病和其他生物学过程。
本文将详细介绍基因结合位点查找的原理、方法和应用。
基因结合位点(gene regulatory elements)是指参与基因调控的区域,通常位于基因的上游区域或内含子区域。
这些位点上的转录因子与特定的DNA序列结合,通过增强或抑制基因的转录来调控基因表达。
因此,确定基因结合位点是理解基因调控网络和预测转录因子靶点的重要一环。
基因结合位点查找的主要方法之一是染色质免疫共沉淀(ChIP)。
这种技术使用特定抗体与染色质上的转录因子结合,并通过免疫沉淀将转录因子结合的DNA分离出来。
然后,通过测序技术对这些DNA片段进行测序,并与参考基因组进行比对,以确定转录因子结合的具体位置。
基因结合位点查找的实验步骤通常包括以下几个关键步骤:1. 细胞处理:选择适当的细胞系,根据实验需要进行处理,例如药物处理或疾病模型建立。
2. 交联:使用交联剂将细胞中的DNA与蛋白质交联在一起,以保持转录因子与DNA的结合状态。
3. 破碎:将交联的细胞进行破碎,使得DNA片段化。
4. 免疫共沉淀:利用转录因子特异性的抗体与目标蛋白质结合,通过共沉淀技术将转录因子与结合的DNA片段一起沉淀下来。
5. DNA纯化:从免疫沉淀物中分离出DNA,并进行净化。
6. 序列测定:对纯化的DNA片段进行高通量测序,获得大量的测序数据。
7. 数据分析:对测序数据进行质量控制、比对和统计分析,确定转录因子结合位点的位置。
基因结合位点查找在许多生物学研究领域有着广泛的应用。
一方面,这项技术可以用来研究特定转录因子的作用机制,通过确定其结合位点,进一步了解其调控的靶基因和生物学功能。
另一方面,基因结合位点查找可以用于研究转录因子在不同细胞类型和不同环境条件下的调控网络,从而深入了解生物体的发育、疾病和进化等过程。
基因结合位点比对

基因结合位点比对是一种生物信息学方法,用于确定基因序列中与特定转录因子结合的区域。
以下是基因结合位点比对的基本步骤:
1. 获取基因序列:从NCBI等数据库中获取基因序列,选择相应的物种和基因。
2. 转录因子结合序列:获取特定转录因子的结合序列,这些序列通常是从实验中获得的。
3. 序列比对:使用生物信息学工具,如BLAST或BLAT等,将基因序列与转录因子结合序列进行比对。
4. 确定结合位点:通过比对结果,确定基因序列中与转录因子结合的区域。
这些区域通常称为基因结合位点。
5. 分析结合位点:对确定的结合位点进行分析,以了解其功能和作用机制。
需要注意的是,基因结合位点比对并不是绝对准确的,因为转录因子结合序列的识别是基于已知的结合模式进行的,而实际的结合模式可能存在差异。
此外,基因序列中可能存在多个与转录因子结合的区域,需要进行全面的比对和分析。
bedtoolsmiRanda—...
bedtoolsmiRanda—...问题的提出解决这个问题的思路其实很简单。
大家都知道核酸分子间相互结合的理论基础就是碱基互补配对,并且已经有了很好的基于序列信息预测结合位点的生物信息学软件,如miRanda。
该软件最初是为miRNA靶向mRNA设计的,需要miRNA序列和mRNA UTR序列作为输入。
在这个基础上,我们只要把mRNA相关序列换成环状RNA 的序列就解决问题了。
即最终的问题转换成了如何获取环状RNA全长序列。
首先要知道环状RNA在基因组上的位置这里推荐使用CIRCexplorer2流程完成环状RNA的鉴定工作(只针对RNA-seq分析)。
因为CIRCexplorer2给出了相当丰富的环状RNA位置信息,如下图:mark由于大多数环状RNA来源于可变剪切的外显子区域,我们在获得其首尾相接的位点后还需要考虑去除中间可能包含的内含子序列。
利用以上信息,我们只要摘取外显子序列就可以避免引入内含子区域序列。
下一步就是在CIRCexplorer2的输出基础上制作bed格式文件,主要目的是满足bedtools getfasta的输入要求。
假如有一批18个样本的CIRCexplorer2输出结果,每一个样本放置在自己的文件夹下面,如下图:markmark红框标记是C01号样本的CIRCexplorer2鉴定结果输出文件,具体文件格式和内容请参考CIRCexplorer2网站。
我们要做的就是从18个这样的文件中提取全部环状RNA的位置区间信息,并以bed文件格式输出。
实现方式用python,代码如下:import osimport globimport sysCIRCexplorer2_path = sys.argv[1]file_name = sys.argv[2]f = open(file_name, 'w+')circ_list = []for i in next(os.walk(CIRCexplorer2_path))[1]:for file in glob.glob(os.path.join(CIRCexplorer2_path, i, '*kno wn.txt')):with open(file, 'r') as filereader:for line in filereader:l = line.strip('\n').split()if len(l) <>10: continueif int(l[12]) <>2: continuecirc_id = '%s_%s_%s'%(l[0], l[1], l[2])if circ_id in circ_list:continueelse:circ_list.append(circ_id)f.write('%s\t%s\t%s\t%s\t%s\t%s\t%s\t%s\t%s\t%s\t%s\t%s \n' % (l[0], l[1], l[2], circ_id, l[4], l[5], l[6], l[7], l[8], l[9], l[10], l[11])) bedtools getfasta获取环状RNA全长序列执行完上一步中最后的python脚本,得到如下形式的示例文件:mark随后执行bedtools getfasta代码,从参考基因组中直接获取环状RNA全长序列。
GeneSnap操作指南

SynGene GeneGenius凝胶成像系统操作指南1、打开暗箱后侧的电源开关,暗箱前边左下角的电源指示灯显示绿色。
2、打开电脑,启动系统,按“GeneSnap”图标启动GeneSnap图像捕捉软件。
启动后界面如下:3、将暗箱门向上推开,根据使用的样品不同选择紫外透视仪或白光透射仪,放置样品至相应的透射仪上(为避免污染,建议将保鲜膜铺平在透射仪,再将样品放置其上,使用完毕将样品和保鲜膜一同弃置即可,否则每次用完后需清洁干净透射仪表面)。
4、根据机器配置不同,选择相应的光源(上左图,“No Light”无光源;“Transilluminator”紫外光源;“Epi long wave uv”365nm紫外投射光源(选配件),“Epi short wave uv”254nm 紫外投射光源(选配件),“Upper white”投射背景光源,仅用于调整样品位置;“Lower white”白光透射仪,需将透射仪放下);相应的滤光片(仅适用于配置滤光片轮的机器,上中图,随机器配置不同可能有不同滤光片,不带滤光片的机型该项为灰色,不需选择);根据样品不同,在上左图中选择相应曝光时间,蛋白样品时间较短,凝胶则需提高(一般在1秒左右)。
5、选定完毕,按上左图的绿色按钮开始实时预览,控制面板变成如上中图所示,同时屏幕将会显示暗箱中的图像,三个按钮可分别控制光圈、图像缩放、焦距调整。
在实际工作中,先适当调整光圈及曝光时间使样品可见(建议适当减少光圈,增大曝光时间,一般在1-2秒左右,在做凝胶时),使用缩放按钮使样品尽量占满屏幕,最后调整焦距使样品清晰可见,做综合调整即可获得适合图像,若效果合适,按下红色按钮即可将图像摄取下来。
6、摄取下来后在“File”菜单下的“Save”项保存文件(只能以“sgd”格式保存),假如要以其他格式(如BMP、TIF、JPG等格式保存),可以选择“File”下的“Export”项保存。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
如何利用primer5软件获取miRNA和基因的互补配对结合位点??
Primer5软件安装说明:
首先下载Primer5.0软件,该软件可以转化序列方向,引物预测,引物设计,引物质量检测,核酸序列转换为氨基酸序列等。
首次安装需要注册激活,压缩包中包含有注册机。
具体安装步骤如下:
1、解压压缩包:
2、压缩后在primer premier 5文件夹中打开primer premier 5.exe软件
3、打开软件后点击activate product,点击后会出现mechin ID
4、打开注册机,在mechine ID处输入上一步中出现的数字,点击generate,在Activation key
中会出现一排数字,该数字即软件激活码,填入上一步中激活码处即可激活。
案例说明:
利用primer软件对miRNA与lncRNA结合位点预测实例,以lncRNA461和miR-141-3p为例
1.获取lncRNA和miRNA序列:
LncRNA序列获取:在NCBI中,选择Nucleotide,输入long intergenic non-protein coding RNA 461,选择序列,进入页面后点击FASTA,即可以获得lncRNA461序列,复制备用。
在miRBase中,检索miR-141-3p,点击get sequence,获得miR-141-3p的序列,miRNA序列中包括U碱基,但是软件不能识别U碱基,因此,需要手动将U替换为T。
直接利用word 中的替换功能,即可将所有的U替换为T,将替换后的序列保存备用。
2.打开primer premier5.0软件,点击左上角File,然后点击New,点击DNA sequence,出现下图对话框:
3.在上图对话框中,粘贴lncRNA461序列,粘贴时会出现四个选项(只能用ctrl+v进行粘贴),选择第一个As Is,点击OK
3.点击中间对话框中左上角Primer按钮
4.在新弹出的对话框中点击Edit Primers
5.删除新弹出对话框中红色序列部分
6.在删除后的方框中贴入已经转换过的miRNA序列,在弹出的对话框中选择第四个选项,点击OK
7.先点击Analyze,然后点击Prime
8.最终结果如下,该结果表示在lncRNA461的2300位左右,miRNA-141-3p能够与它发生结合。
注意事项:
1.输入序列时注意序列方向。
2.由于miRNA中T变为了U,该软件不能识别U碱基,因此需要自己将miRNA中
的T转为U,然后再用于结合情况预测。
3.最后作图时,需要结合实际情况,看是否需要再将T转换回U。