高效液相色谱 串联质谱法测定牛羊组织中苯酚类和水杨酸苯胺类抗蠕虫药多残留-文档资料
高效液相色谱法检测液态奶中的苯酚类和水杨酸苯胺类抗蠕虫药

1 0 mi n 。上清 液转 移 至另 一 5 0 mL 离 心 管 中 待
净化 。
1 实 验 部 分
Hale Waihona Puke 1 . 1 仪 器 与 试 剂 MA X 固相萃取 柱 预先用 3 mL 2 5 % 氨化 乙腈溶 液活 化 , 再 将前 述 上清 液 装 载 人柱 , 以约 1 mL / mi n 的流 速过 柱 。待 上 清 液 流 出后 , 依次用 3 mL甲 醇
1 . 4 样 品 的 处 理 与 净 化
了高效 液 相色 谱一 串联 质 谱 法 引用 于牛 组 织 和 牛奶 中氯 氰碘柳 胺 残 留检 测 的研 究 , 尚无 关 于水 杨 酸 苯
胺 类 药物 在奶 中 的多残 留检 测方 法报 道 。本研 究采 用 HP L C分析 奶 中水 杨 酸 苯 胺类 药 物 的多 残 留 , 并 进 行 了方 法学 评价 。
为 了避 免消 费 者受 到药 物 残 留的 危 害 , 欧 盟 于 2 0 1 2年 公 布 了最 新 的 关 于 硝 碘 酚 腈 和 氯 氰碘 柳 胺
匀 。混 合标 准 中间 工作 液 : 准 确量 取 标 准储 备 液 适 量, 用 流动 相稀 释成 含 硝 碘 酚 腈 、 氯 羟柳 胺 、 氯 氰 碘 柳胺、 碘醚 柳 胺 分 别 为 2 、 1 、 4 . 5 、 1 mg / L的混 合 标
在 牛奶 、 羊 奶 中 的 最 高 残 留 限量 ( MR L) 。分 别 如 下: 硝 碘 酚腈 , 牛( 羊) 奶2 0 ̄ g / k g ; 氯 氰 碘 柳胺 , 牛
( 羊) 奶 4 5 ̄ g / k g ; 氯 羟柳 胺 ( 牛奶 ) l 0 ̄ g / k g ; 还尚 未公 布 碘 醚 柳 胺 在 奶 中 的 MR L 。 我 国农 业 部 2 3 5 号公 告 仅 规 定 了牛 、 羊 组 织 中硝 碘 酚 腈 、 氯 氰 碘
牛羊组织中β-受体激动剂残留检测 超高效液相色谱-串联质谱法

DB 21/3004—2013
3.2.15 标准储备液(100 μg/mL):准确称取适量的特布他林、西马特罗、沙丁胺醇、非诺特罗、氯 丙那林、莱克多巴胺、克伦特罗、妥布特罗和喷布特罗对照品,用甲醇分别配制成 100 μg/mL 的标准 储备液,2~8 ℃冰箱中保存,有效期为 3 个月。 3.2.16 混合标准中间液(1 μg/mL):分别准确吸取 1.0 mL 的特布他林、西马特罗、沙丁胺醇、非 诺特罗、氯丙那林、莱克多巴胺、克伦特罗、妥布特罗和喷布特罗标准储备液至 100 mL 容量瓶中, 用甲醇稀释至刻度,2~8 ℃冰箱中保存,有效期为 1 个月。 3.2.17 同位素内标储备液(100 μg/mL):分别取三种同位素内标,各自用甲醇配成浓度约为 100 μg/mL 的储备液,2~8 ℃冰箱中保存,有效期为 3 个月。 3.2.18 混合同位素内标工作液(100 μg/L):将上述三种同位素内标储备液,用 0.2%甲酸水溶液稀 释成约 100 μg/L 的混合工作液,2~8 ℃冰箱中保存,有效期为 1 个月。 3.2.19 混合标准及内标工作液(10 μg/L):准确吸取 1.0 mL 的混合标准中间液(3.2.16)和 10.0 mL 混合同位素内标工作液(3.2.18)至 100 mL 容量瓶中,用 0.2%甲酸水溶液稀释至刻度,2~8 ℃冰箱中 保存,有效期为 1 周。 3.3 仪器和设备 3.3.1 超高效液相色谱-串联质谱仪(配电喷雾离子源)。 3.3.2 涡旋混合器。 3.3.3 冷冻离心机。 3.3.4 恒温定时振荡器。 3.3.5 旋转蒸发仪。 3.3.6 固相萃取装置。 3.3.7 MCX 固相萃取柱(CNW ,for β-agonists):60 mg/3 mL。 3.3.8 氮吹仪。 3.4 测定步骤 3.4.1 样品处理
超高效液相色谱-串联质谱同位素内标法测定羊肉中33种兽药残留

超高效液相色谱-串联质谱同位素内标法测定羊肉中33种兽药残留作者:陈旭晋曹佳陆宇阳苑华宁章雪明张小刚来源:《安徽农业科学》2024年第03期摘要[目的]建立超高效液相色谱-串联质谱同时测定羊肉中33种兽药残留的方法。
[方法]样品用90%乙腈水(含2.0%甲酸)提取,通过Oasis Prime HLB净化,浓缩后用流动相定容。
采用Waters ACQUITY UPLC HSS T3 柱(2.1 mm×100 mm,1.8 μm)分离。
以甲醇和0.1%甲酸水为流动相梯度洗脱,在电喷雾离子源下正、负离子切换扫描,多反应监测(MRM)模式下,结合保留时间和特征离子信息,利用同位素内标法定量。
[结果]33种兽药在0.5~40.0 ng/mL线性关系良好(r>0.99),最低检出浓度在0.08~0.10 ng/mL。
加标回收率为83.7%~115.0%,RSD为0.58%~11.49%。
[结论]该方法具有处理简单、灵敏度高、重复性好等优点,满足羊肉中33种兽药的定量检测。
关键词超高效液相色谱-串联质谱;同位素内标;羊肉;兽药残留中图分类号 S851.34+7 文献标识码 A 文章编号 0517-6611(2024)03-0195-07doi:10.3969/j.issn.0517-6611.2024.03.047Determination of 33 Veterinary Drug Residues in Mutton by Ultra High Performance Liquid Chromatography-Tandem Mass Spectrometry with Isotope Internal Standard MethodAbstract [Objective]To establish a method for the simultaneous determination of 33 kinds of veterinary drug residues in mutton by ultra high performance liquid chromatography-tandem mass spectrometry. [Method]The sample was extracted with 90% acetonitrile water (containing 2.0% formic acid), purified by Oasis Prime HLB, concentrated, and diluted with mobile phase. Then separated by Waters ACQUITY UPLC HSS T3 column(2.1 mm×100 mm,1.8 μm),using 0.1% formic acid and acetonitrile as mobile phase.Under electrospray ionization source, switching between positive and negative ion modes, in Multiple Reaction Monitoring (MRM) mode,utilizing the isotope internal standard method for quantification by combining retention time and characteristic ion information. [Result] The linear relationship of the 33 kinds of veterinary drugs was good in the concentration range of 0.5-40.0 ng/mL(r>0.99),and the lowest detection concentration was 0.08-0.10 ng/mL.Adding standard recovery rates was 83.7%-115.0%,the RSD was 0.58%-11.49%. [Conclusion]The method has the simple processing,high sensitivty and good reproducibility,and is suitable for the quantitative detection of 33 kinds of veterinary drug residues in mutton.Key words UPLC-MS/MS;Isotope internal standard;Mutton;Veterinary drug residues近年來,我国动物生产和兽药使用量的增加已引起人们对兽药残留问题的广泛关注。
高效液相色谱-串联质谱法测定动物组织中的16种喹诺酮类药物残留

高效液相色谱-串联质谱法测定动物组织中的16种喹诺酮类药物残留高效液相色谱-串联质谱法测定动物组织中的16种喹诺酮类药物残留建立了动物组织样品中萘啶酸、恶喹酸、氟甲喹、诺氟沙星、依诺沙星、环丙沙星、洛美沙星、丹诺沙星、恩诺沙星、氧氟沙星、沙拉沙星、二氟沙星、麻保沙星、培氟沙星、司帕沙星、奥比沙星等16种喹诺酮类兽药多残留量的高效液相色谱-串联质谱测定方法.用酸性乙腈萃取样品中的16种喹诺酮类药物残留,然后用正己烷脱脂,旋转蒸发浓缩,以Inertsil C8-3色谱柱分离,在正离子模式下以电喷雾电离串联质谱进行测定.在10,50,100 μg/kg 3个加标水平下进行了验证试验,方法的线性范围为10~100 μg/kg,平均回收率为62.4%~102%,相对标准偏差为1.4%~11.9%.该方法简便、快速、准确,各项技术指标满足国内外法规的要求,可用于鸡肉、鸡肝和鱼肉等动物组织样品中喹诺酮类药物多残留的确证检测.作者:岳振峰林秀云唐少冰陈小霞吉彩霓华红慧刘昱YUE Zhenfeng LIN Xiuyun TANG Shaobing CHEN Xiaoxia JI Caini HUA Honghui LIU Yu 作者单位:岳振峰,林秀云,唐少冰,吉彩霓,华红慧,刘昱,YUE Zhenfeng,LIN Xiuyun,TANG Shaobing,JI Caini,HUA Honghui,LIU Yu(深圳出入境检验检疫局食品检验检疫技术中心,广东,深圳,518067)陈小霞,CHEN Xiaoxia(深圳大学教务处,广东,深圳,518060)刊名:色谱ISTIC PKU英文刊名:CHINESE JOURNAL OF CHROMATOGRAPHY 年,卷(期):2007 25(4) 分类号:O658 关键词:高效液相色谱-串联质谱法喹诺酮类药物残留动物组织。
高效液相色谱-串联质谱法同时测定动物源食品中3种抗菌类药物残留

高效液相色谱-串联质谱法同时测定动物源食品中3种抗菌类药物残留朱晓明;杨涛;陈君慧;江英【摘要】利用高效液相色谱-电喷雾串联质谱(HPLC-ESI-MS/MS)技术,建立了同时测定动物源食品中3种抗菌类药物残留量的分析检测方法。
对色谱、质谱条件进行优化,样品采用环己烷-乙酸乙酯(V/V=1:1)提取,无水硫酸钠除水分,正己烷去脂肪,经GPC富集浓缩目标化合物,旋转蒸发后定容。
以0.1%甲酸-甲醇溶液为流动相,经Hypersil GOLD aQ色谱柱分离后,LC-MS/MS多反应监测模式分析,外标法定量,结果表明:3种目标化合物在100.00~1000.00 ng/mL 浓度下线性良好(R2≥0.9900),添加低、中、高三个水平浓度时,目标化合物的平均回收率为70.07%~85.36%,相对标准偏差小于9.5%。
方法检出限为0.15~0.3μg/kg,定量下限为0.5~1.5μg/kg。
【期刊名称】《食品安全导刊》【年(卷),期】2014(000)026【总页数】4页(P66-69)【关键词】高效液相色谱-质谱联用;动物源食品;乙酰甲喹;卡巴氧;恩诺沙星【作者】朱晓明;杨涛;陈君慧;江英【作者单位】石河子大学食品学院; 新疆乌鲁木齐米东新区质量与计量检测所;新疆产品质量监督检验研究院;石河子大学食品学院;石河子大学食品学院【正文语种】中文新疆是我国最大的畜牧业生产大区之一。
为提高动物源食品的产量和质量,常以低治疗剂量的药物(多种抗菌素、抗寄生虫药物、生物激素等)作为饲料添加剂,防治动物传染性疾病和促进动物的生长。
但因存在违规用药、用药过量或用药后未到达规定休药期即提前屠宰(药物未完全代谢排出体外)的现象,导致肉、蛋、奶、鱼及蜂蜜等食品中药物残留量超过国家标准规定的限量,不但影响人们的健康,还造成肉类、水产品、蜂产品的出口困难,阻碍了食品工业和养殖业的健康发展。
目前对于乙酰甲喹、卡巴氧、恩诺沙星这三种药物的单独测定的研究报道较多,多采用固相萃取法[1,7-10]进行前处理,高效液相色谱法[2-6]、超高效液相色谱-串联质谱法[7-10]等作为分析检测方法,但是国内还未见有乙酰甲喹、卡巴氧、恩诺沙星这3种兽药同时检测的报道,而本研究着力于建立一种可靠、高效、简便易行的液相色谱串联质谱法测定动物源食品中兽药的分析方法。
高效液相色谱—串联质谱法同时测定动物源食中3种抗菌类药物残留

高效液相色谱—串联质谱法同时测定动物源食中3种抗菌类药物残留高效液相色谱—串联质谱法同时测定动物源食中3种抗菌类药物残留摘要:随着养殖业的发展和人们对食品质量安全的日益关注,对动物源食品中抗菌类药物残留的检测变得越来越重要。
本研究采用高效液相色谱—串联质谱法同时测定动物源食中3种常用抗菌类药物残留。
通过优化实验条件,得到了良好的分离和定量效果。
结果表明,该方法具有较高的精密度和准确度,适用于动物源食品中抗菌类药物残留的快速检测和定量分析。
关键词:高效液相色谱,串联质谱,抗菌类药物,残留,定量分析引言:抗菌类药物被广泛用于养殖业中预防和治疗动物疾病,然而过度使用抗菌类药物会导致残留物在动物源食品中超标,对人体健康造成潜在风险。
因此,对动物源食品中抗菌类药物残留进行快速、准确的检测成为保证食品质量安全的重要环节。
实验方法:1. 仪器和试剂本实验使用Agilent 1200高效液相色谱仪和Agilent 6400串联质谱仪,色谱柱为Agilent Zorbax Eclipse XDB C18柱。
试剂为甲醇、乙腈和水(所有试剂均为高纯度试剂)。
2. 样品制备将不同来源的动物源食品样品取一定重量,加入适量的甲醇溶液中。
使用超声波浸提30分钟,离心10分钟,取上层溶液过滤。
取10 mL上清液,质量体积稀释至50 mL。
3. 标准曲线的制备准备不同浓度的抗菌类药物标准溶液,使用同样的萃取方法进行提取和稀释。
4. 色谱条件流动相为甲醇和水的混合溶液,梯度洗脱。
流速为0.3mL/min,柱温为40℃。
串联质谱条件为电喷雾离子源,多反应监测模式。
结果与讨论:通过对动物源食品中抗菌类药物残留的测定,得到了较好的结果。
抗菌类药物的线性范围为10-1000 ng/mL,相关系数大于0.99。
方法的平均回收率在80-110%之间,相对标准偏差小于5%。
结论:本研究成功开发了一种高效液相色谱—串联质谱法,用于同时测定动物源食品中3种常用抗菌类药物残留。
高效液相色谱-串联质谱法同时测定水产品中8类38种兽药残留

高效液相色谱-串联质谱法同时测定水产品中8类38种兽药残留摘要:在养殖过程中,滥用抗生素、大量使用合成等药物的现象越来越严重,兽药具有致癌、至畸、至突变等不良效用,危害人类健康。
近年来由于滥用兽药引起的食品安全问题频发已逐渐成为人们普遍关注的一个社会热点问题。
一些化学合成药物在畜牧水产养殖业中使用较多,随便使用这些药物是很容易使水产品受到污染的。
在这种情况下,人们吃了这种受到污染的肉类食物,就会使得这些毒素在人体内积累,会使人体产生一定的抗药性。
随着兽药在我国实际生产中的广泛应用,人们对其危害有了更多的认识,这些肉类食品中所残留的兽药也是目前食品安全中所关注的重要内容。
因此,对畜牧水产养殖业的相关产品进行严格的检测,能够保证肉类食品的质量。
本文建立了高效液相色谱-串联质谱HPLC-MS/MS同时测定水产品中喹诺酮类、大环内酯类、磺胺类、磺胺增效剂、林可胺类、硝基咪唑类、类和多肽类8类共38种限用兽药残留的检测方法。
关键词:高效液相色谱;串联质谱;兽药残留本试验同时测定鳗鱼、鳙鱼和小龙虾中的类药物残留,在检测的过程中,检测的药物种类虽多,但前处理方法简单并不需要特殊的实验装置来辅助实验,检测周期短且成本低,从而为水产品的大量检测,提供了更加简便的方法。
一、实验部分1.1实验所用的仪器和试剂实验仪器:质谱仪、高效液相色谱仪、Waters C3色谱柱、IKAT25数显型高速分散机、低温高速离心机和氮吹浓缩仪。
实验试剂:甲醇、正已烷、甲酸、冰乙酸、无水硫酸钠,以上试剂均为分析纯,乙腈、甲酸为色谱纯。
1.2样品前处理样品的提取及净化:称5.00g样品置于50mL聚丙烯离心管中,加入约5g无水硫酸钠及15mL1%的乙酸乙腈溶液,用高速分散机进行提取,离心(5000r/min,5min),收集上清液。
沉淀再用20mL1%的乙酸乙腈溶液按照上述步骤重复提取一次,离心,合并上清液,用提取液将其定容到25mL,混合均匀,将其放到-80℃的冷冻机中冷冻1小时。
超高效液相色谱-串联质谱法同时检测牛奶中7种杀虫剂残留
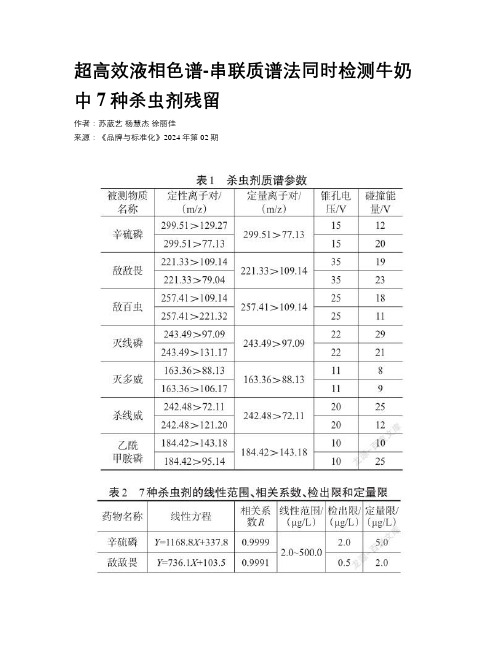
超高效液相色谱-串联质谱法同时检测牛奶中7种杀虫剂残留作者:苏葳艺杨慧杰徐丽佳来源:《品牌与标准化》2024年第02期【摘要】本研究以固相萃取技術作为样品前处理方法,通过超高效液相色谱-串联质谱对处理完的样品进行定性、定量检测,建立了同时测定牛奶中辛硫磷、敌敌畏、敌百虫、灭线磷、灭多威、杀线威和乙酰甲胺磷7种杀虫剂残留量的检测方法。
结果表明,7种杀虫剂辛硫磷的检出限为2.0μg/L,定量限为5.0μg/L;杀线威的检出限为1.0μg/L,定量限为3.0μg/L;敌敌畏、敌百虫、灭线磷、灭多威和乙酰甲胺磷的检出限为0.5μg/L,定量限为2.0μg/L;在2.0~500.0μg/kg浓度范围内具有良好线性关系,R>0.999,平均回收率为70%~95%(RSD【关键词】牛奶;杀虫剂;超高效液相色谱-串联质谱法;检测【DOI编码】10.3969/j.issn.1674-4977.2024.02.001Simultaneous Determination of 7 Insecticides in Milk by Ultra-High Performance Liquid Chromatography Tandem Mass SpectrometrySU Weiyi, YANG Huijie, XU Lijia(Liaoning Inspection,Examination & Certification Centre〔Liaoning Lnstitute forAgro-product Veterinary Drugs and Feed Control〕, Shenyang 110036, China)Abstract: Study by using solid phase extraction technique as a pretreatment method, a tandem mass spectrometry on processed samples were detected qualitatively and quantitatively by ultra-high performance liquid chromatography analysis method to set up the milk of Phoxim, dichlorvos,trichlorfon, ethoprophos, methomyl, oxamyl and acephate.The detection limit of phoxim is2.0μg/L,the quantitative limit of g/Lis 5.0μg/L,the detection limit oxamyl is 1.0μg/L, the quantitative limit of g/Lis3.0μg/L, the detection limit for dichlorvos, trichlorfon, ethoprophos,methomyl,and acephate is 0.5μg/L,the quantitative limit of g/L is 2.0μg/L in the concentration of 2.0~500.0μg/L range and peak area were showed good linear relationship, R>0.999, the average recovery rate of 70%~95% (RSDKeywords: milk; insecticide; UPLC-MS/MS; detection杀虫剂污染是影响奶源安全性的因素之一。
超高效液相色谱——串联质谱法测定畜禽肉中多种磺胺类药物残留

-1
※农业科学
农业与技术 2020ꎬVol 40ꎬNo 16 1 5
瓶内放入适量的上述储备液ꎬ 用甲醇稀释后ꎬ 保存至
离子碰撞能量进行优化ꎬ 通过调节子离子质谱信号丰
方法具有良好的线性关系ꎬ 检出限和定量限分别为 2μgkg-1 、 5μgkg-1 ꎮ 8 种磺胺类药物添加水平为 10μgkg-1 、
20μgkg -1 、 100μgkg -1 时ꎬ 平均回收率 84 0% ~ 114 2%ꎬ 相对标准偏差 0 73% ~ 7 96%ꎮ 该方法操作简易ꎬ 具
2 1 标准溶液配制
(SMZ) 、 磺 胺 噻 唑 ( ST ) 、 磺 胺 甲 基 嘧 啶 ( SMR )
瓶内分别放入精确量取的 8 种 SAs 标准品各 10 00mgꎬ 用
己烷、 乙腈、 乙酸铵 ( 色谱纯) ꎻ 650℃ 环境下灼烧无
配制 1μgmL 的混合标准工作液: 在同一容量
哒嗪 ( SCP ) 、 磺 胺 嘧 啶 ( SDZ ) 、 磺 胺 甲 基 异 恶 唑
冷冻离心机ꎻ Milli - Q 超纯水系统
[1]
ꎮ
8 种磺胺类药物标准品: 磺胺醋酰 ( STD) 、 磺胺
脱溶剂气温度 350℃ ꎻ 雾化气ꎬ 氮气 600Lmin ꎻ 碰
-1
撞气ꎬ 氩气ꎻ 扫描模式ꎬ 多反应监测模式ꎮ
2 实验方法
甲噻二唑 ( STZ) 、 磺胺二甲异恶唑 ( SIX) 、 磺胺氯
1 4 2020ꎬVol 40ꎬNo 16
农业与技术 ※农业科学
超高效液相色谱———串联质谱法
测定畜禽肉中多种磺胺类药物残留
向 俊
1 试验材料
Bꎬ 10min—95%Bꎮ
高效液相色谱-串联质谱在兽药残留分析中的应用

T logy科技食品科技HPLC-MS/MS全称为种高效液相色谱-串联质谱技术,该技术可对多组分进行定性、定量综合分析,在应用中可以对高沸点、非挥发性等进行准确的分离鉴定。
在分离检测的过程中主要利用电喷雾电离和大气压化学电离技术将待测物中的成分分离出来,在送入质谱检测系统中进行检测,便可以较为精准地测量出母离子的特征碎片。
1 动物药物残留分析简介常用的兽药残留量检测方法有微生物法和色谱法。
前者为筛选方法,该方法的原理是抗原抗体反应,在对动物药物残留进行测定的过程中难以对同类型的药物进行区分。
对于禁用兽药(A类),如硝基呋喃等,残留限度(PED)在4%以下;而禁用兽药 (B类),如磺胺类,残留限度(PED)在3%以下。
质谱分析技术可以准确检测食品中的动物药物残留,进而为解决兽药残留问题提供有效的解决 途径。
2 液相色谱质谱联用技术在兽药残留检测中的应用β-内酰胺类在动物医疗中被广泛应用,这种抗生素可以抑制动物细菌性感染,同时还可以对动物体内的细菌细胞合成、抗革兰氏阳性菌等进行阻断隔离,其主要的代表化合物有青霉素、氨苄青霉素。
徐伟、耿士伟等利用电喷雾离子阱技术,对牛奶中7种β-内酰胺类抗生素进行了检测,用乙腈提取和沉淀蛋白质,经C18柱净化浓缩后供LC-MS/MS分析,再利用正离子模式监测,多级离子捕捉器可以提供更多的碎片离子结构信息,获得高灵敏度。
使用LC-MS/MS测量牛奶中的阿莫西林、邻氯青霉素、青霉素G等,并使用内标物d7-青霉素G,样品经高速离心脱脂后,样品过C18柱(pH值过柱时大于6),每一种药物选择3个离子来提高检测的灵敏度。
然后他们将这一方法运用到生奶检测中,以青霉素V为内标,测定了10种牛乳中β-内酰胺类抗生素的残留检测方法[1]。
郭盈岑教授在负离子扫描模式下监测牛肝、肾和肌肉中的6种青霉素含量,用LC-MS/MS测定,定量限为50 μg/kg。
通过LC-MS/MS法测定β-内酰胺、皮质激素、氯霉素等药物的实验研究发现,认为液质结合技术是解决兽药残留分析的有效手段[2]。
高效液相色谱—串联质谱法测定羊肉中6种磺胺类药物残留

N o g ,Z uT o a iceg i nf Y u h a ,Y nQuh n ,WagY n ,Z uLpn n og h ii g ( ’n nr—x npco n u rni ueu iig h n og2 20 ,C i ) jn g E t eiIsetnadQ aat eB ra ,jnn ,S ad n 7 0 0 hn ii y t i n ’ a
A src: A meh d o te d tr n t n o sx up o a d s fufq i0aie, sl daie Sl dmeh xn b tat to fr h eemiai f i s lh n mie s l un x l uf izn , Uf i to ie, o a n a a
第 8期 ( 总第 刊
Acd mi P r dc l f am P o u t P o es g a e c ei i r rd c rc si o ao F s n
No 8 .
Aug .
文 章 编 号 : 17 — 6 6 (0 0 — 0 0 0 6 19 4 2 1) 8 0 9 — 3 1
a eo i i n ee td b g e oma c i ud C r mao r p y t n e ma s s e to tr h e ut h w t a h c tn t l a d d tce y Hih P r r n e L q i h o tg a h — a d m s p cr mee .T e r s l s o h tt e re f s c reain c ef in s o i e r c l r t n u v s R 0 9 8 o rl t o f ce l f l a ai ai c r e = ,9 1~0 9 9 8 w ti h up o a d s c n e tain r n e o o i n b o .9 i n t e s l h n mi e o c nr t a g f h o 1 0~5 0 g L,t e d t cin l t o i t o r 0 gk n h e o ey r tsa e 7 % ~1 5 0 / h ee t i s ft sme h d ae 1 / g a d t e r c v r ae r 8 o mi h 0 %. Ke r s s l h n n d s r s u n l s ; l u d c r mao r p y tn e ma s s e to t r mu tn t s e y wo d : u p o a f e ; e i e a a y i i d s i i h o t g a h — a d m s p cr mee ; q t i u o s
高效液相色谱-串联质谱法检测肉类原料中的药物多残留

肉,菇业MEAT INDUSTRY 2020年第12期总第476期❖由醃妥全与检测❖高效液相色谱-串联质谱法检测肉类原料中的药物多残留沈春华厦门古龙食品有限公司技术中心实验室福建厦门361000摘要建立了高效液相色谱-串联质谱技术,同时检测肉类原料中磺胺类(磺胺二甲囉味、磺胺间二甲氧疇啜、磺胺间甲氧嗨呢、磺胺甲基异噁哇、礦胺囉睫、礦胺二甲异噁哇、磺胺甲氧哒嗪)和喳诺飼类(恩诺沙星)8种药物残留量的方法。
前处理采用1%甲酸-乙睛提取,浓缩后用0.1%甲酸-乙睛定容,正己烷去脂,用高效液相-串联质谱进行定性及定量分析。
所涉8种药物在1~100|xg/L范围内线性良好,相关系数为0.9956~0.9998,方法最低定量限为1jig/kg。
在3个添加水平下,加标回收率为67.8%-115%,相对标准偏差为1.2%~9.1%。
$匕方法可满足肉类原料中,磺胺类和喳•诺飼类多种药物残留的检测与验证。
关键词液相色谱-串联质谱药物残留肉类检测Detection of drugs residue in meat raw materials by high performance liquidchromatography-tandem mass spectrometry methodSHEN ChunhuaAbstract A high performance liquid chromatography-tandem mass spectrometry method was established,and8kinds of drugs residue including sulfonamides(sulfamethazine,sulfadimethoxine, sulfamo nomethoxine,sulfamethoxazole,sulfadiazine,sulfisoxazole and sulfamethoxypyridazine)and quinolones(enrofloxacin)in meat raw materials could be detected simultaneously.1%acidic acetonitrile was adopted to extract in pretreatment.After concentration,the volume was fixed with0.1%formic acid acetonitrile,and the fat was removed with n一Hexane,high performance liquid chromatography-tandem mass spectrometry method was used for qualitative and quantitation analysis・The8kinds of drugs involved had good linearity in the range of1~100|xg/L,and the correlation coefficient was0.9956〜0.9998,and the minimum limit of quantitation was1|xg/kg・Under the three addition levels,the recovery of standard addition was67.8%~115%,and the relative standard deviations was1.2%to9.1%.This method could meet the needs of detection and validation of sulfonamides and quinolones residues in meat raw materials.Key words high performance liquid chromatography一tandem mass spectrometry method;drugs residue;meat;detection肉类作为食品加工行业的一大类原料,随着食品安全问题备受社会日益关注的同时,肉类原料是否安全可靠,已引起了国内食品安全监管机构甚至国际食品法典委员会(CAC)的高度重视⑷。
高效液相色谱-串联质谱法同时测定牛奶和奶粉中的青霉素类药物及其主要酶解代谢产物

高效液相色谱-串联质谱法同时测定牛奶和奶粉中的青霉素类药物及其主要酶解代谢产物李玮;艾连峰;郭春海;马育松;窦彩云【摘要】建立了液相色谱-串联质谱法同时测定牛奶和奶粉中4种青霉素(青霉素G、青霉素V、阿莫西林、氨苄西林)及其4种β-内酰胺酶酶解产物(青霉素G脱羧噻唑酸、青霉素V脱羧噻唑酸、阿莫西林脱羧噻唑酸、氨苄西林脱羧噻唑酸)残留的方法.样品采用乙腈-水提取,浓缩后经HLB柱净化,用液相色谱-串联质谱检测,外标法定量.结果表明,青霉素原药在4 ~200 μg/L,酶解产物在10 ~ 500 μg/L范围呈良好线性,线性相关系数均大于0.99;样品检出限为5 ~50 μg/kg(S/N≥3),定量限为8~100 μg/kg(S/N≥10);对牛奶和奶粉样品分别进行3个水平的加标回收实验(n=6),牛奶中青霉素及其酶解产物的平均回收率为83.48%~96.97%,相对标准偏差为3.86%~10.87%;奶粉中青霉素及其酶解产物的平均回收率为82.70%~95.14%,相对标准偏差为3.02%~ 9.81%.该方法稳定、可靠,适用于牛奶和奶粉中青霉素类药物及其酶解代谢产物的测定.【期刊名称】《色谱》【年(卷),期】2013(031)010【总页数】8页(P946-953)【关键词】高效液相色谱-串联质谱;青霉素;酶解代谢产物;牛奶;奶粉【作者】李玮;艾连峰;郭春海;马育松;窦彩云【作者单位】河北出入境检验检疫局,河北石家庄050051;河北出入境检验检疫局,河北石家庄050051;河北出入境检验检疫局,河北石家庄050051;河北出入境检验检疫局,河北石家庄050051;河北出入境检验检疫局,河北石家庄050051【正文语种】中文【中图分类】O658青霉素是目前治疗牛乳腺炎的首选药物,同时也是牛奶中最常见的残留抗生素,这一问题已受到国内外的广泛重视。
我国农业部2008年颁布实施了《无公害食品生鲜牛乳行业标准》(NY/T 5045-2008)明确了对新鲜牛乳的卫生指标即“抗生素不得检出”,欧美国家也已明文禁止抗生素残留超标的牛奶上市。
高效液相色谱-串联质谱法测定羊肉中的4种β-受体激动剂

高效液相色谱-串联质谱法测定羊肉中的4种β-受体激动剂王伟;尤翠萍【摘要】建立了高效液相色谱-串联质谱法测定羊肉中的4种β-受体激动剂(克伦特罗、莱克多巴胺、沙丁胺醇、特布他林)含量.样品经酶解、提取后,经MCX小柱净化,以乙腈和体积分数为0.001的甲酸溶液为流动相,选择多反应监测(MRM)模式,并且用内标法定量,用高效液相色谱-串联质谱分析测定.β受体激动剂在0.5 ~10μg/kg范围均有良好线性关系,相关系数均大于0.99,定量限为0.05~0.50μg/kg,加标回收率为79.29%~102.63%,相对标准偏差(RSD)为2.5%~5.6%.该方法灵敏度高、结果准确,可作为检测羊肉中的4种β-受体激动剂的方法.【期刊名称】《河北科技师范学院学报》【年(卷),期】2018(032)004【总页数】4页(P32-35)【关键词】β-受体激动剂;同位素内标;高效液相色谱-串联质谱法【作者】王伟;尤翠萍【作者单位】临沂市食品药品检验检测中心,山东临沂,276000;临沂市人民医院【正文语种】中文【中图分类】TS251.7β-受体激动剂(β-agonists)是一类化学合成的苯乙醇胺类物质,常见的盐酸克伦特罗、沙丁胺醇等被俗称为“瘦肉精”,添加在动物饲料或者饮水中,可以促进动物体内的脂肪分解代谢,增加蛋白质合成,从而提高胴体瘦肉率、饲料转化率等[1,2]。
但是残留在动物体内的β受体激动剂如果随之进入人体,对人的肝、肾等内脏器官均可产生毒副作用,危害人的健康,因此国家已经禁止该类药物用于食用动物中,并且要求在肉品中不得检出。
近年来,瘦肉精事件屡有发生,不仅在猪肉中有检出,甚至在牛、羊等其他肉类中也有检出[3],国家食品安全监督抽检实施细则(2018版)已经将克伦特罗、莱克多巴胺、沙丁胺醇、特布他林列入食用肉类的检验项目。
目前已有的β-受体激动剂检测方法主要有酶联免疫吸附法(ELISA)、胶体金法、高效液相色谱(HPLC)法、气质联用(GC/MS)法和液质联用(LC/MS/MS)法等方法[4~13]。
高效液相色谱-串联质谱法测定牛肉中泰拉霉素代谢物残留量研究

溶液,再精密量取100µl,置10ml容量瓶中,用
甲醇-0.1%甲酸溶液(1:9,v/v)稀释至刻度,配制
成浓度为1µg/ml的溶液。
1.3 前处理条件
1.3.1 提取 根据参考文献[7]方法,称取匀浆后
1
山东畜牧兽医
的样品1g(精确至 置 0.01g) 离 50ml 心管中,加入
2mol/L盐酸4.5ml,涡旋1min,振荡提取10min,
置60℃水浴消化1h,10000r/min离心10min,收集
上清液,下层加入2mol/L盐酸4.5ml,重复提取一
次,合并上清液,加水定容至10ml,精密量取
1ml加水4ml涡旋混匀,备用。
1.3.2 净化 MCX固相萃取柱依次用3ml甲醇和
3ml 水 活 化 , 取 备 用 液 全 部 过 柱 , 再 依 次 用
2019 年第 9 期(总第 266 期)
试验研究
试验研究
高效液相色谱-串联质谱法测定牛肉中泰拉霉素代谢物残留量研究
杨修镇 陈 玲 尹伶灵 张呈军 牛华星
(山东省兽药质量检验所 山东省畜产品质量安全监测与风险评估重点实验室 山东 济南 ) 250022 懂玲玲 吉利伟 (山东舍里乐药业有限公司 山东 济宁)
0.1mol/L HCl、甲醇各3ml洗涤,抽干,用5%氨
化甲醇5ml洗脱。洗脱液于50℃水浴氮气吹干,
残余物中加甲醇-0.1%甲酸溶液(1:9,v/v)1ml涡旋
复容, 离心 ,过 滤 1min
10000r/min 5min 0.22µm
膜,滤液供液相色谱-串联质谱测定。
1.4 仪器条件
1.4.1
20
2 结果与分析
2.1 方法的专属性
超高效液相色谱-串联质谱法同时测定牛肉中3类31种药物残留检测方法的建立

宁夏农林科技,超高效液相色谱-串联质谱法同时测定牛肉中3类31种药物残留检测方法的建立陈娟,李莉,马岩,吴春燕,杨俊华,陈海燕宁夏兽药饲料监察所,宁夏银川750011摘要:采用超高效液相色谱-串联质谱方法(UPLC-MS/MS)同时测定牛肉中3种四环素类药物、11种喹诺酮类药物和17种磺胺类药物的残留。
采用Mcllvaine-Na2EDTA缓冲液和磷酸盐缓冲液作为提取液,经HLB固相萃取柱净化,氮气吹干,复溶后,供UPLC-MS/MS测定。
液相色谱条件为:色谱柱为Waters BEH C18柱(50mm×2.1mm,1.7μm);流动相为乙腈-0.1%甲酸水溶液,梯度洗脱;柱温为40℃;流速为0.3mL/min;进样量:2μL。
质谱条件为:电喷雾离子源(ESI+),多反应监测(MRM)方式采集,外标法定量。
结果表明:在5~500μg/L的系列浓度范围内,31种药物药物的相关系数>0.990;方法检测限为5μg/kg,定量限为10μg/kg;从10、50、100μg/L3个添加浓度检测结果可以看出,方法的平均回收率在60%~110%,批内、批间RSD均小于20%。
该方法可作为牛肉中这3类药物残留的确证方法。
关键词:四环素类药物;磺胺类药物;喹诺酮类药物;牛肉;药物残留;液质联用中图分类号:S859.84文献标识码:A文章编号:1002-204X(2018)01-0006-05 Determination of Thirty-one Veterinary Residues in Beef by UPLC-MS/MSCHEN Juan et al.AbstractKey words1材料与方法1.1仪器1.2试剂1.3标准储备液的配制作者简介:陈娟(1980-),女,宁夏贺兰人,高级兽医师,主要从事畜产品质量安全检验工作。
收稿日期:2017-10-316表1牛肉中3类31种药物残留检测对照品表2色谱梯度洗脱程序1.4方法1.4.1色谱的选择1.4.2质谱条件的优化1.4.3色谱条件1.4.4质谱条件1.4.5样品前处理2结果与分析2.1线性关系2.2检测限和定量限2.3回收率及重复性实验表431种药物的标准曲线及相关系数表331种药物定性、定量离子对及对应的锥孔电压和碰撞能量3结论表5添加不同浓度药物回收率及重复性实验结果图2空白牛肉中添加10μg/kg 的31种药物特征离子质量色谱图参考文献:[1]郭黎明,朱奎,江海洋,等.超高效液相色谱-串联质谱法同时测定鸡肝中残留的四环素类、磺胺类和喹诺酮类药物[J].色谱,2009,(4):412-416.[2]班付国,胡兴娟,吴宁鹏,等.超高效液相色说-串联质谱法测定猪肉66种兽药残留的研究[J].中国兽药杂志,2014,48(6):40-51.[3]黄子敬,王晓玲,杨钦沾,等.超高效液相色谱-串联质谱法同时测定畜禽肉中磺胺类、喹诺酮类、硝基咪唑类兽药残留[J].分析实验室,2014,33(10):1184-1188.责任编辑:王银惠。
高效液相色谱-串联质谱法测定牛尿液中14种β-兴奋剂残留

i
shake
r MS2 型);台 式 冷
冻离心机(
BECKMAN ALLEGRA25R 型);
pH 计
(METTLER);恒 温 制 冷 摇 床 (
The
rmo);超 声 波 仪
(
;
/T
;
吹氮浓缩仪(
BRANSON5
5
1
0)
B
i
o
t
a
e
u
r
bo Va
g
p)
移液枪(
1
0~1
0
0μL、
2
0~2
0
0μL、
G兴奋剂类药
物多残留的液质联用确证方法.牛尿液样品经β
G葡萄 糖 醛 苷 酶 水 解、
2 的 乙 酸 Leabharlann 溶 液 提 取、pH5.
固相萃取柱净化富集后,在正离子模式下,采用多反应监测模式 对β
G兴 奋 剂 进 行 定 性 和 定 量 分 析.
该方法 的 定 量 限 为 0.
5 mg/L,在 3 个 浓 度 水 平 进 行 验 证 试 验,结 果 平 均 回 收 率 为 78.
动 物 体 内 脂 肪 代 谢 增 强 ,蛋 白 质 合 成 增 加 ,可 以 明
显 提 高 胴 体 瘦 肉 率 和 饲 料 利 用 率 [1],因 而 被 非 法
作 为 饲 料 添 加 剂 用 于 畜 产 品 的 生 产 ,以 促 进 动 物
生 长 和 改 善 肉 质 . 但 是 ,该 类 药 物 残 留 会 聚 集 在
3% ~
117.
3% ,相对标准偏差为 2.
04%~13.
70% .该方法简便、准确,各项技术指标满足国内外法规的
高效液相色谱-串联质谱法检测牛组织和奶中咪多卡残留的研究

高效液相色谱-串联质谱法检测牛组织和奶中咪多卡残留的研究白玉惠;孙红洋;张骊;朱馨乐;沈昕;黄耀凌【期刊名称】《中国兽药杂志》【年(卷),期】2024(58)4【摘要】建立了一种检测牛组织和牛奶中咪多卡残留检测的高效液相色谱-串联质谱法。
牛组织(肌肉、肝脏、肾脏、脂肪)和奶在NaAc缓冲体系中酶解,经HCl溶液提取,WCX固相萃取柱净化,以0.3%甲酸水溶液(含20 mM甲酸铵)和0.3%甲酸乙腈为流动相进行梯度洗脱,在HILIC色谱柱上分离,在电喷雾正离子(ESI+)模式下,用多反应监测(MRM)模式检测,同位素内标法定量。
结果表明:咪多卡在2.5~1000 ng/mL的浓度范围内呈现良好线性关系,相关系数(R^(2))大于0.99;咪多卡在牛组织和奶中的检测限均为10μg/kg,定量限均为20μg/kg;咪多卡在牛组织和奶中20~4000μg/kg添加浓度水平上的回收率在70.9%~109%范围内;批内RSD在0.55%~9.59%之间,批间RSD在2.21%~12.1%之间。
该方法具有灵敏度高、定量准确,重复性好等特点,可以满足牛组织和奶中咪多卡残留检测的要求。
【总页数】7页(P69-75)【作者】白玉惠;孙红洋;张骊;朱馨乐;沈昕;黄耀凌【作者单位】中国兽医药品监察所(农业农村部兽药评审中心)国家兽药残留基准实验室【正文语种】中文【中图分类】S857.84【相关文献】1.高效液相色谱-串联质谱法测定牛(羊)奶中阿莫西林的残留2.高效液相色谱-串联质谱法检测牛组织中双氯芬酸钠残留的研究3.超高效液相色谱-串联质谱法检测牛可食性组织中氟佐隆残留4.气相色谱-串联质谱法和高效液相色谱-串联质谱法检测大枣中33种禁用农药残留量5.QuEChERS-气相色谱-串联质谱法和高效液相色谱-串联质谱法快速检测蔬菜中267种香港规例中的农药残留量因版权原因,仅展示原文概要,查看原文内容请购买。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
高效液相色谱串联质谱法测定牛羊组织中苯酚类和水杨酸苯胺类抗蠕虫药多残留1 引言氯羟柳胺(OXY)、硝碘酚腈(NIT)、氯氰碘柳胺(CLO)、碘醚柳胺(RAF)是一类苯酚类和水杨酸苯胺类抗蠕虫药,作用机理是通过阻断肝片吸虫的氧化磷酸化过程,使虫体麻痹、死亡,用于驱杀牛、羊的肝片吸虫、蛔虫和绦虫,对血毛线虫、巨片吸虫和羊蝇蛆亦有较好作用。
随着国内驱虫药的广泛应用和研究,发现碘醚柳胺有明显胚胎毒性[1];氯氰碘柳胺对肝、肾具有一定的毒、副作用[2];高剂量硝碘酚腈可引起心脏、神经、肌肉和呼吸的损害[3]。
欧洲医药评价署、澳大利亚以及食品法典委员会均已制定了上述药物的最大残留限量(Maximum residue limit,MRL),残留标志物均为原形药物,残留监测的动物是牛羊。
靶组织MRL分别如下:硝碘酚腈:牛、羊,肌肉400 μg/kg、脂肪200 μg/kg、肝20 μg/kg、肾400 μg/kg。
碘醚柳胺:牛,肌肉30 μg/kg、脂肪30 μg/kg、肝10 μg/kg、肾40 μg/kg;羊,肌肉100 μg/kg、脂肪250 μg/kg、肝150 μg/kg、肾150 μg/kg。
氯氰碘柳胺:牛,肌肉1000 μg/kg、脂肪3000 μg/kg、肝1000 μg/kg、肾3000 μg/kg;羊,肌肉1500 μg/kg、脂肪2000 μg/kg、肝1500 μg/kg、肾5000 μg/kg。
氯羟柳胺:牛、绵羊,肌肉20 μg/kg、脂肪20 μg/kg、肝500 μg/kg、肾100 μg/kg。
我国农业部(2003)235号公告[4]已经颁布硝碘酚腈、碘醚柳胺、氯氰碘柳胺的最高残留限量,氯羟柳胺的最高残留限量正在制定中。
目前,关于苯酚类和水杨酸苯胺类抗蠕虫药在组织中残留的检测方法主要有高效液相色谱与紫外检测器[5~8]、荧光检测器和质谱检测器联用[9,10]、高效液相色谱串联质谱法[11~16]。
国内仅报道高效液相色谱串联质谱法[9]检测氯氰碘柳胺在牛组织和牛奶中残留的研究,国外对于苯酚类和水杨酸苯胺类抗蠕虫药在组织中残留分析方法的报道主要局限于某种组织或者是单残留和两种药物的残留检测方法,尚未报道关于4种苯酚类和水杨酸苯胺类抗蠕虫药在牛、羊组织中的多残留检测方法。
本研究采用HPLC MS/MS分析牛、羊组织中4种苯酚类和水杨酸苯胺类抗蠕虫药的多残留,方法前处理简便易行,灵敏度高,精密度及重复性好,适用于牛、羊组织中苯酚类和水杨酸苯胺类抗蠕虫药的多残留检测。
2 实验部分2.1 仪器与试剂Agilent 1200型液相色谱仪(美国安捷伦公司); API4000电喷雾串联四极杆质谱仪,配Analyst 4.1.5 软件(美国应用生物系统公司); C18 色谱柱(150 mm×2.1 mm, 3.5 μm,美国Waters公司); Centrifuge5804台式高速离心机(美国Eppendorf公司); D10 24型氮气浓缩仪(杭州奥盛仪器XX公司); Milli Q纯水机(美国Millipore公司); CNWBOND MAX 固相萃取小柱(60 mg/3 mL,德国CNW科技公司)。
硝碘酚腈(Nitroxinil,NIT)精制对照品(纯度98.5%,批号 091205),氯羟柳胺(Oxyclozanide,OXY)精制对照品(纯度98.5%,批号1670811),氯氰碘柳胺钠(Closantel,CLO)精制对照品(纯度94.1%,批号 67111001),碘醚柳胺(Rafoxanide,RAF)精制对照品(纯度99.5%,批号 0505028),均购自河北远征药业XX公司;乙腈、甲醇、甲酸(色谱纯,德国CNW科技公司);乙酸铵(色谱纯,美国TEDIA公司);氨水、冰醋酸(分析纯,江苏强盛功能化学XX公司);实验用水为超纯水。
2.2 溶液的配制标准储备液:准确称取适量的硝碘酚腈、氯羟柳胺、氯氰碘柳胺、碘醚柳胺对照品,用乙腈分别配制成1 g/L的单一标准储备液及100 μg/mL的混合标准溶液。
1%三乙胺乙腈溶液:取1.0 mL三乙胺,用乙腈定容至100 mL,混匀。
25% 氨化乙腈溶液:取25.0 mL氨水,用乙腈定容至100 mL,混匀。
5%甲酸乙腈溶液;0.1%甲酸溶液。
2.3 色谱质谱条件2.3.1 色谱条件 C18色谱柱(150 mm×2.1mm,3.5 μm);流速: 0.25 mL/min;柱温:35 ℃;进样量: 5 μL;流动相:甲醇(A),乙腈(B), 0.1%甲酸溶液(C)。
梯度洗脱程序: 0.0 min, 15%A, 25%B; 0.0~1.0 min, 15%~30%A,25%~70%B; 1.0~6.0 min, 30%A, 70%B; 6.0~6.5 min, 30%~15%A, 70%~25%B; 6.5~13.0 min, 15%A, 25%B。
2.3.2 质谱条件用多反应监测(MRM)扫描模式;电喷雾离子源(ESI);负离子扫描;电喷雾电压Symbolm 3500 V,离子源温度650 ℃,气帘气压力15 kPa,雾化气压力60 kPa,辅助气压力 60 kPa。
射入电压、碰撞能量、去簇电压、碰撞室射出电压及定性离子对、定量离子对等参数见表1。
2.4 样品的处理与净化(1)肌肉、肝脏、肾脏组织样品已匀浆组织样品在室温下自然解冻,准确称取(2.00±0.02)g,置于50 mL离心管中,加入乙腈丙酮混合液(60∶40,V/V)15 mL,漩涡混悬15 s,超声提取10 min,300 r/min振摇10 min,8000 r/min 离心10 min 后收集上清液;上清液中加入1 mL 5%氨水待净化。
(2)脂肪组织样品已匀浆组织样品在室温下自然解冻,准确称取(2.00±0.02)g,置于50 mL离心管中,加入1%三乙胺乙腈溶液15 mL,漩涡混悬15 s,超声提取10 min,300 r/min振摇10 min,8000 r/min 离心10 min后收集上清液;上清液中加入2 mL 5%氨水后,置于Symbolm 20 ℃冷藏1 h,取出后在4 ℃下8000 r/min离心10 min。
上清液转移至另一个50 mL离心管中,待净化。
MAX固相萃取柱预先用25%氨化乙腈溶液3 mL活化,再将前述上清液装载入柱,以1.0 mL/min流速过柱。
待上清液流出后,依次用3 mL水和3 mL甲醇分别淋洗,负压抽干后用5%甲酸乙腈溶液3 mL洗脱,收集洗脱液,在40 ℃下氮气吹干,残渣用0.5 mL流动相溶解,转移至2 mL离心管,加入乙腈饱和的正己烷0.5 mL,涡旋,于4 ℃下15000 r/min离心10 min,上清液过0.22 μm滤膜,待测。
3 结果与讨论3.1 质谱参数的确定用针泵连续直接进样(单一标准溶液1 mg/L),发现ESI 准分子离子峰。
优化各质谱参数,进行相应的子离子全扫描,以确定MRM特征诊断离子。
根据欧盟 2002/657/EC[20]制定的分析方法确认的标准和程序规定,各药物的母离子、子离子及部分质谱参数见表1。
3. 2 色谱条件的优化3.2.1 色谱柱的选择由于硝碘酚腈、氯羟柳胺、氯氰碘柳胺、碘醚柳胺的脂溶性较高、极性小,所选色谱柱的固定相应为直链烷烃键合的硅胶,其中最常见的为十八烷基键合硅胶(即C18)。
本研究考察了菲罗门Gemini C18(150 mm×2.0 mm, 5 μm)、CNW C18(250 mm×4.6 mm,5和Waters C18 (150 mm×2.1 mm,3.5 μm)填料的色谱柱。
氯氰碘柳胺和碘醚柳胺在Gemini C18色谱柱上峰形较宽,在样品检测时基质杂音较大;在CNW C18色谱柱上,由于氯氰碘柳胺和碘醚柳胺极性较弱,当加大乙腈的比例后,这两者的分离效果不理想,而且纯标液的基线在后,两种药物出峰之前容易漂移; Waters C18色谱柱对4种药物有良好的保留性质,各待测药物均可得到优异的峰型。
因此采用Waters C18 色谱柱。
3.2.2 流动相的选择比较了甲醇或乙腈(有机相)与乙酸铵缓冲溶液、甲酸水溶液、超纯水(水相)组合对峰型、灵敏度及分辨率的影响。
研究表明,选择甲醇或乙腈作为有机相,均可对4种药物进行有效的分离,但是在甲醇为流动相时,硝碘酚腈的出峰时间在10.26 min,以乙腈为流动相时,硝碘酚腈的出峰时间在3.72 min。
为了缩短药物的出峰时间和优化4种药物的峰形,选择乙腈和甲醇作为流动相。
考察了甲醇或乙腈(有机相)与5 mmol/L乙酸铵、10 mmol/L乙酸铵和0.1%甲酸溶液在不同梯度条件下对药物峰的影响,发现乙酸铵的加入,未能改善氯氰碘柳胺和碘醚柳胺的峰形,峰形较宽,基质杂音较大。
当选择乙腈和0.1%甲酸溶液时,需要加大有机相的比例才能对将氯氰碘柳胺和碘醚柳胺洗脱。
当选择0.1%甲酸乙腈甲醇作为流动相时,采用2.3.1节所示方法进行梯度洗脱,4种药物均可得到良好的峰型。
3.3 样品前处理方法的研究根据4种药物的理化性质以及文献\[12,18,19],考察了乙腈、1%三乙胺乙腈和1%醋酸丙酮、乙腈丙酮、三氯甲烷、乙酸乙酯、甲醇等的提取效果。
发现甲醇、三氯甲烷、乙酸乙酯不能有效地提取这4种药物,回收率在40%~70%;用乙腈或1%三乙胺乙腈时,提取回收率均能达到80%以上;用1%醋酸丙酮提取后,加入5 mL氨水,出现较多杂质,且回收率不稳定(60%~75%);乙腈丙酮混合液提取时,回收率均能达到80%以上,且乙腈丙酮(60∶40,V/V)回收率偏高于乙腈丙酮(80∶20,V/V)。
乙腈具有较强的除蛋白和脱脂作用,对脂肪溶解性差,经过对比发现1%三乙胺乙腈作为脂肪组织的提取剂回收率较好,乙腈丙酮(60∶40,V/V)对肌肉、肝脏、肾脏均有较好的提取效果。
由于4种药物为弱酸性药物,且乙腈和丙酮的极性较强,过柱后,硝碘酚腈的回收率在65%~70%,5%氨水溶液为碱性环境,通过加入5%氨水可以使4种药物给出质子,呈阴离子态,有利于其在阴离子交换柱上被保留。
实验过程中,对比加入1、2和4 mL 5%氨水溶液,发现在肌肉、肝脏、肾脏提取液中加入1 mL 5%氨水溶液,在脂肪提取液中加入2 mL 5%氨水溶液,4种药物的回收率在80%~110%之间。
净化过程中,根据苯酚类和水杨酸苯胺类抗蠕虫药的理化性质,直接比较了菲罗门 Strata X 柱、MAX柱的净化效果,发现硝碘酚腈和碘醚柳胺有很好的保留,回收率均在80%以上,而氯羟柳胺和氯氰碘柳胺在Strata X柱上的回收率较差,分别约为55%和32%。