四国药典细菌内毒素对比
各国药典细菌内毒素检查法的比较

各国药典细菌内毒素检查法的比较1968年美国科学家Dr. Levin和Bang建立了鲎试验法。
此后世界各国对鲎试剂的生产和应用得以迅速发展。
1980年美国药典20版首先收载了细菌内毒素检查法。
随后,英国药典、日本药局方、中国药典等也相继收载了该方法。
本文主要对中国药典1995年版与英国药典1995年增补本、美国药典23版和日本药局方13版等的细菌内毒素检查法进行了比较。
1 检验品种的比较中国药典1995年版二部共收载细菌内毒素检查品种12个,2000年版二部增至47个,而美国药典23版收载了471个品种进行细菌内毒素检查。
因此,我们需加紧对更多的品种进行细菌内毒素检查方法的研究,加快该法对药品检验的普及和推广工作,提高我国药品标准和检验水平。
2 检验方法的比较细菌内毒素试验的方法有凝胶法、浊度法、显色基质法、免疫学法等。
其中凝胶法多为限量检查法,浊度法和显色基质法为定量检查法,且后两种方法都属于分光光度法。
浊度法还可分为终点浊度法和动态浊度法;显色基质法也可分为终点显色基质法和动态显色基质法。
凝胶法是较为经典的方法,各国药典都有收载,而收载了细菌内毒素定量测定法的有美国药典、英国药典、欧洲药典和日本药局方等。
中国药典1995年版只收载了凝胶法,2000年版才涉及到定量测定法。
因此,我们还需要对细菌内毒素定量测定进行研究,需要有供定量测定用的标准品、仪器和试剂,以后逐步建立起我国的细菌内毒素定量测定方法。
3 细菌内毒素标准品各国药典均设置了细菌内毒素的标准品,其中中国药典、英国药典和美国药典还可采用根据标准品为基准进行标定的工作标准品或对照标准品。
除英国药典采用U为内毒素的单位外,其余各国药典都用EU为内毒素的单位。
美国药典和中国药典对内毒素标准品每一步稀释时的混匀时间作出了明确规定。
除中国药典外,其余各国药典规定了内毒素标准贮备液在冰箱里保存不得超过14d,且用前应在旋涡混合器上充分混合至少3min。
中美药典细菌内毒素比较(word文档良心出品)

------------USP:在复试中,如果溶液A的两支平行管均为阴性,供试品溶液符合规定
USP:如果供试品溶液以不超过MVD的某个稀释倍数稀释,检测结果为阳性时,可进一步稀释供试品溶液但不能超过MVD,再重新进行试验。------CP;无规定。
USP:与鲎试剂在限定的灵敏度下不发生反应的灭菌注射用水或其它水。
CP:用于细菌内毒素定量测定用的细菌内毒素检查用水,内毒素含量应小于0.005EU/ml。
USP: 未提到。
3、实验用具的准备
CP:实验所用器皿需经处理,除去可能存在的外源性内毒素,常用的方法是250℃下干烤至少1小时,也可用其它确证不干扰定义,等于K/M
除了鞘内给药以外的任何给药途径,K均为5 USP-EU/kg,鞘内给药时,K为0.2 USP-EU/kg,对于非鞘内给药放射性药品,内毒素限值的计算为175/V,V为以mL为单位的最大推荐剂量,对于鞘内给药的放射性药品,其内毒素限值为14/V,对于按以每平方体表面积计算的给药剂量(通常是抗肿瘤药品),计算公式为K/M,其中K=5EU/kg,M为(最大剂量/m2/小时×1.80m2)/70kg。
5、供试品溶液的制备
CP:|某些供试品需进行复溶、稀释或在水性溶液中浸提 用LRW复溶或稀释药品或抽提医疗器械,某些物质可能更适于用其它水性溶液来溶解、稀释或抽提。
对于过酸、过碱或本身有缓冲能力的供试品需调节被测溶液(或其稀释液)的pH值,一般要求供试品溶液的pH值在6.0-8.0的范围内。
USP:用LRW复溶或稀释药品或抽提医疗器械,某些物质可能更适于用其它水性溶液来溶解、稀释或抽提。
中国药典和美国药典中的细菌内毒素检测法的不同点比较
药典三部版通则细菌内毒素检查法

药典三部版通则细菌内毒素检查法标准化工作室编码[XX968T-XX89628-XJ668-XT689N]1143 细菌内毒素检查法本法系利用鲎试剂来检测或量化由革兰阴性菌产生的细菌内毒素,以判断供试品中细菌内毒素的限量是否符合规定的一种方法。
细菌内毒素检查包括两种方法,即凝胶法和光度测定法,后者包括浊度法和显色基质法。
供试品检测时,可使用其中任何一种方法进行试验。
当测定结果有争议时,除另有规定外,以凝胶限度试验结果为准。
本试验操作过程应防止内毒素的污染。
细菌内毒素的量用内毒素单位(EU)表示,1EU与1个内毒素国际单位(IU)相当。
细菌内毒素国家标准品系自大肠埃希菌提取精制而成,用于标定、复核、仲裁鲎试剂灵敏度、标定细菌内毒素工作标准品的效价,干扰试验及检查法中编号B和C溶液的制备、凝胶法中鲎试剂灵敏度复核试验、光度测定法中标准曲线可靠性试验。
细菌内毒素工作标准品系以细菌内毒素国家标准品为基准标定其效价,用于干扰试验及检查法中编号B和C溶液的制备、凝胶法中鲎试剂灵敏度复核试验、光度测定法中标准曲线可靠性试验。
细菌内毒素检查用水应符合灭菌注射用水标准,其内毒素含量小于0.015EU/ml(用于凝胶法)或0.005EU/ml(用于光度测定法),且对内毒素试验无干扰作用。
试验所用的器皿需经处理,以去除可能存在的外源性内毒素。
耐热器皿常用干热灭菌法(250℃、30分钟以上)去除,也可采用其他确证不干扰细菌内毒素检查的适宜方法。
若使用塑料器皿,如微孔板和与微量加样器配套的吸头等,应选用标明无内毒素并且对试验无干扰的器具。
供试品溶液的制备某些供试品需进行复溶、稀释或在水性溶液中浸提制成供试品溶液。
必要时,可调节被测溶液(或其稀释液)的pH值,一般供试品溶液和鲎试剂混合后溶液的pH值在6.0~8.0的范围内为宜,可使用适宜的酸、碱溶液或缓冲溶液调节pH值。
酸或碱溶液须用细菌内毒素检查用水在已去除内毒素的容器中配制。
中国药典和美国药典的内毒素检测要求

中国药典和美国药典的内毒素检测要求鲎试剂-LAL,中国药典和美国药典的内毒素检测要求鲎试剂是从栖⽣于海洋的节肢动物“鲎”的兰⾊⾎液中提取变形细胞溶解物,经低温冷冻⼲燥⽽成的⽣物试剂,专⽤于细菌内毒素检测。
鲎试验法是国际上⾄今为⽌检测内毒素最好的⽅法,它简单﹑快速﹑灵敏﹑准确,因⽽被欧美药典及我国药典定为法定内毒素检查法,并已被世界各国所采⽤。
⽬前国际上销售的鲎试剂有两种,⼀种称美洲鲎试剂(Limulus Amebocyte Lysate),缩写为LAL,由美国⽣产;另⼀种称东⽅鲎鲎试剂(Tachypleus Amebocyte Lysate),缩写为TAL。
实验证明:美洲鲎试剂-LAL⽐东⽅鲎试剂LAL更纯洁,检测效果更好。
TAL检查内毒素有很多⽅法,⽬前应⽤最⼴泛的是凝胶法,此外还有动态浊度法﹑显⾊基质法,⽐⾊法等。
其⽤途有以下⼏⽅⾯:1. 药检:⽤于注射药品﹑放射性药物﹑⽣物制品﹑注射器及⽣产⼯艺流程中的内毒素检测;2. 临床:⽤于检测病⼈各种体液中的内毒素含量;3. 其他:⽤于检测⽔或⾷品中的内毒素含量。
2005年版中国药典《细菌内毒素检查法-鲎试剂法》(鲎试剂-LAL法)本法系利⽤鲎试剂来检测或量化由⾰兰阴性菌产⽣的细菌内毒素,以判断供试品中细菌内毒素的限量是否符合规定的⼀种⽅法。
细菌内毒素检查包括两种⽅法,即凝胶法和光度测定法,后者包括浊度法和显⾊基质法。
供试品检测时,可使⽤其中任何⼀种⽅法进⾏试验。
当测定结果有争议时,除另有规定外,以凝胶法结果为准。
细菌内毒素的量⽤内毒素单位(EU)表⽰。
细菌内毒素国家标准品系⾃⼤肠杆菌提取精制⽽成,⽤于标定、复核、仲裁鲎试剂灵敏度和标定细菌内毒素⼯作标准品的效价。
细菌内毒素⼯作标准品系以细菌内毒素国家标准品为基准标定其效价,⽤于试验中鲎试剂灵敏度复核、⼲扰试验及设置的各种阳性对照。
凝胶法细菌内毒素检查⽤⽔系指内毒素含量⼩于0.015EU/ml的灭菌注射⽤⽔。
细菌内毒素检测试验注意事项与误差分析

➢内毒素检测人员应经过严格的培训,并应配备相 应的防护设施(如穿防护服、戴口罩、头套等)。
——2010年版药品GMP实施指南
7 样品
➢β-葡聚糖样物质的影响 ➢PH值的影响 ➢金属离子和弱酸阴离子的影响 ➢药物浓度的影响
1) β-葡聚糖样物质的影响
常见现象:假阳性。
➢在进行鲎试剂灵敏度复核、干扰实验和供试 品细菌内毒素检查时,各个实验中要求的对照 应同时进行,并在实验有效的情况才能进行计 算和判断。
二、影响试验结果的因素分析
鲎试剂 细菌内毒素工作标准品 细菌内毒素检查用水 环境 实验器皿 实验人员 样品 试验
1 鲎试剂
➢质量标准:WS1-363(B-123)-91 性状、鉴别、检查、灵敏度测定、 用途、规格、贮藏
鲎试剂的特异性系指在药品、生物制品和血液制 品中鲎试剂能准确测出被测物质内毒素的能力, 既不能造成样品内毒素的漏检,也不能将其它物 质错判为内毒素。
目前我公司研究证明,已知能与鲎试剂发生反应的 物质有细菌内毒素和β-葡聚糖,而内毒素反应曲 线为S型特征曲线,β-葡聚糖反应曲线为经过0点 的斜线。鲎试剂的特异性差的另一种表现,是将其 它物质(如β-葡聚糖)判定为内毒素,错误将合 格药品判为不合格,造成制药厂直接经济损失。
2) 湿度 ➢ 吸潮 ➢ 灵敏度增高 ➢ 挥发
5 实验器皿
➢器皿的计量管理(校正) ➢器皿的选择标准 ➢器皿的正确处理
1) 器皿的选择标准
鲎试验对器皿的要求:刻度准确;内表面光 滑;易于清理。细菌内毒素检查实验不宜选用 注射器,最好使用经过校正的玻璃刻度移液管 或精密移液器。注射器刻度不精确,内壁磨沙 面吸附内毒素,而且要特别注意注射器针头引 入的铁离子对试验结果的干扰,故不宜选用注 射器。
中国药典和美国药典中的细菌内毒素检测法的不同点比较

灵敏度复核实验
未说明
至少用每批鲎试剂中的1支试剂进行灵敏度复核实验
当最大浓度2.0λ管为阳性,最低浓度0.25λ管均为阴性,阴性对照管为阴性时,验方为有效
浓度最低的标准品溶液的所有重复管均为阴性,实验方为有效
按下式计算反应终点浓度的几何平均值,即为鲎试剂灵敏度的测定值(λc):
λc=lg-1(ΣX/4)
供试品溶液的制备
某些供试品需进行复溶、稀释或在水性溶液中浸提
用LRW复溶或稀释药品或抽提医疗器械,某些物质可能更适于用其它水性溶液来溶解、稀释或抽提
对于过酸、过碱或本身有缓冲能力的供试品需调节被测溶液(或其稀释液)的pH值,一般要求供试品溶液的pH值在6.0-8.0的范围内
如果需要,可调节待测溶液(或其稀释液)的pH值,以使鲎试剂和样品的混合物的pH范围落在鲎试剂生产商指定的范围内,这通常适用于pH值在6.0-8.0范围内的产品
与鲎试剂在限定的灵敏度下不发生反应的灭菌注射用水或其它水
用于细菌内毒素定量测定用的细菌内毒素检查用水,内毒素含量应小于0.005EU/ml
未提到
实验用具的准备
实验所用器皿需经处理,除去可能存在的外源性内毒素,常用的方法是250℃下干烤至少1小时,也可用其它确证不干扰细菌内毒素检查的适宜方法。
使用经验证的除热原程序对所有玻璃器皿和遇热稳定的材料在热空气烘箱中进行除热原,常用的最低温度和最少的时间是250℃下30分钟
中国药典和美国药典中的细菌内毒素检测法的不同点比较requirementscpusp细菌内毒素工作标准品cse对cse的用途及效价进行定义细菌内毒素工作标准品中每1ng细菌内毒素的效价应不小于2eu不大于50eu未提到cse细菌内毒素检查用水与灵敏度为003euml或更高灵敏度的鲎试剂在371条件下24小时不产生凝集反应的灭菌注射用水与鲎试剂在限定的灵敏度下不发生反应的灭菌注射用水或其它水用于细菌内毒素定量测定用的细菌内毒素检查用水内毒素含量应小于0005euml未提到实验用具的准备实验所用器皿需经处理除去可能存在的外源性内毒素常用的方法是250下干烤至少1小时也可用其它确证不干扰细菌内毒素检查的适宜方使用经验证的除热原程序对所有玻璃器皿和遇热稳定的材料在热空气烘箱中进行除热原常用的最低温度和最少的时间是250下30分钟试验操作过程应防止微生物污染未提到内毒素标准品贮液的制备未提到usp的rse的效价定为10000uspeu支用5ml鲎试剂检查用水复溶部内容物用漩涡混合器间歇混合30分钟并用此原液作系列稀释将原液置于冰柜中保存不超过14天作以后的稀释之用在使用前用漩涡混合器强力混合不少于钟在作下一步稀释前需对前面的稀释液混合不少于30秒不要贮存稀释液因为没有数据能证明其不会因为吸附作用而失去活性
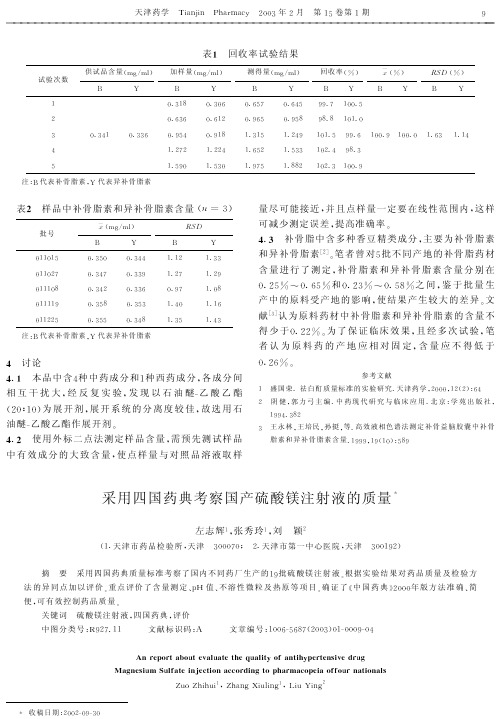
四国药典考察国产硫酸镁注射液的质量
R ST U V W T X Y Z W [ X U \ Y ] [ Y X UX ^ U_ [ Y ] ‘ X aW bY S X ‘ ^ a V U T X U S c ‘ \ Ud T [ e fY e S U c ‘ [ gh [ ] b Y X U‘ S i U j X ‘ W SY j j W T d ‘ S eX WV ^ Y T gY j W V U ‘ YW b b W [ TS Y X ‘ W S Y ] c <k k l mk n o n l o n p q $r o l & o q $<s o l)o q $
表 [ 抗高血压药硫酸镁注射液 ! 规格 9 的评价 b c, d " 6H0 ?
生产 6 h 厂 报告书 编号 检验项目 ! 参见 ] 中国药典 ^ 年版 > 页? 9 6 6 6 6 " 生产批号 批准文号 性状 鉴别 = <值 规定 c ! b 6 e7 # 6 ? 热原 符合 均为无色 的澄明液 体 镁盐与硫 酸盐均呈 正反应 符合 i b 9 规定 符合 9 6 6 " 9 j 6 > 6 " 6 ‘ 9 ‘ ‘ i b 9 规定 符合 6 6 " 9 j " 6 6 " 6 ‘ 9 c " 无锡市第 9 七制药有 限公司 9 6 6 " 5 ‘ 9 > 6 " 6 ‘ 9 c ‘ 苏卫药准 字! " > j > ? 号 9 > 7 7 6 5 均为无色 的澄明液 体 镁盐与硫 酸盐均呈 正反应 符合 c b > 规定 符合 9 6 6 " 5 ‘ 5 6 6 " 6 9 " c 5 c b 7 规定 符合 9 6 6 " 9 j " " 上海旭东 海普药业 9 6 6 " 9 j " 9 有限公司 9 6 6 " 9 j " 5 6 " 6 i 6 5 6 " 6 i 6 9 6 " 6 i 6 " 沪卫药准 字! " > > c ? 第6 号 " 5 6 9 均为无色 的澄明液 体 镁盐与硫 酸盐均呈 正反应 i b " c b > i b 6 规定 符合 规定 符合 规定 符合 i b 6 苏卫药准 常州第二 9 6 6 " 9 j " c 6 " 6 5 " ‘ 9 制药厂 9 6 6 " 9 j " i 6 " 6 5 " c " ! " > > " ?第 9 > 7 7k 6 ‘ 号 均为无色 的澄明液 体 镁盐与硫 酸盐均呈 正反应 i b " c b > 规定 符合 规定 符合 规定 符合 规定 符合 规定 符合 规定 符合 规定 符合 规定 符合 规定 符合 规定 符合 规定 6 b c 6 b c 6 " b c 6 " b 6 " b 6 5 b c 符合 规定 符合 规定 符合 规定 符合 规定 符合 规定 符合 规定 符合 规定 符合 规定 > > b c > > b c " 6 6 b c " 6 6 b 9 > > b 6 " 6 6 b 5 > > b 9 > 7 b 9 i b 9 规定 符合 规定 符合 规定 符合 规定 6 6 6 符合 规定 符合 规定 符合 规定 > 7 b 6 > j b 9 > > b 7 i b 9 规定 装量 符合 规定 " b c 澄明度 规定 f7 ! b c g? 无菌 符合 规定 > j b c 含量测定 ! 规定 > c b 6 ge" 6 c # 6 g?
中国药典和美国药典中的细菌内毒素检测法的不同点比较(word文档良心出品)

某些供试品需进行复溶、稀释或在水性溶液中浸提
用LRW复溶或稀释药品或抽提医疗器械,某些物质可能更适于用其它水性溶液来溶解、稀释或抽提
对于过酸、过碱或本身有缓冲能力的供试品需调节被测溶液(或其稀释液)的pH值,一般要求供试品溶液的pH值在6.0-8.0的范围内
如果需要,可调节待测溶液(或其稀释液)的pH值,以使鲎试剂和样品的混合物的pH范围落在鲎试剂生产商指定的范围内,这通常适用于pH值在6.0-8.0范围内的产品
阴性对照系列D的结果不超出所用鲎试剂所说明的空白对照限值
用下式计算终点浓度的对数值的平均值,然后再计算该平均值的反对数:
终点浓度的几何平均值= antilog(Σe/f)
Σe是稀释系列的终点浓度的对数值之和,f为重复管数,反应终点浓度的几何平均值即为鲎试剂灵敏度的测定值
凝胶法限量实验
溶液制备中的表添加了内毒素的溶液为“供试品溶液”
为“经稀释的供试品溶液”
灵敏度复核实验
未说明
至少用每批鲎试剂中的1支试剂进行灵敏度复核实验
当最大浓度2.0λ管为阳性,最低浓度0.25λ管均为阴性,阴性对照管为阴性时,实验方为有效
浓度最低的标准品溶液的所有重复管均为阴性,实验方为有效
按下式计算反应终点浓度的几何平均值,即为鲎试剂灵敏度的测定值(λc):
λc=lg-1(ΣX/4)
中国药典和美国药典中的细菌内毒素检测法的不同点比较
Requirements
CP
USP
细菌内毒素工作标准品(CSE)
对CSE的用途及效价进行定义“细菌内毒素工作标准品中每1ng细菌内毒素的效价应不小于2EU,不大于50EU”
未提到CSE
细菌内毒素检查用水
与灵敏度为0.03EU/ml或更高灵敏度的鲎试剂在37±1℃条件下24小时不产生凝集反应的灭菌注射用水
四国药典细菌内毒素对比

CP(2015 版)1143 EP (8.0) 2.6.14 USP( 36) 85JP( 16) 6.06仅在最低浓度的标准溶液的所有平行管的检查结果均为阴性的情况下,试验方为有效。
反应终点浓度指系列递减的内毒素浓度中最后一个呈阳性结果的浓度。
将终点浓度取对数,计算它们的平均值,再将平均值的结果取反对数,最后的表达式如下:终点浓度的几何平均值=lg-1(工e/f)工e =所用系列溶液的终点浓度的对数值的和f=平行管的数量反应终点的浓度的几何平均值即为鲎试剂灵敏度的测量值(IU/ml )。
当终点浓度的几何平均值在0.5入至2.0入之间时,可判定受试鲎试剂的标示灵敏度为入,可用于内毒素的检查。
(ii )干扰因素试验开展该项实验的目的是检查供试品溶液中反应的增强因素或阻抑因素的存在。
按表2.6.14.-1 制备溶液A B、C D。
供试品的稀释度不得超过MVD且供试品溶液不能检查出内毒素,具体操作见(1)预备试验的(i )鲎试剂标示灵敏度的复核试验项。
表 2614-1人=经检查无内毒素的溶液工e为所用系列溶液的终点浓度的对数值的和;f为平行管的数量。
反应终点的几何平均值即为LAL试剂的标示灵敏度(单位为EU/mL。
当终点浓度的几何平均值在0.5入至2.0入之间,可判定受试 LAL试剂的标示灵敏度为入。
凝胶法的干扰因素试验一一按表1制备溶液A B C D,使用的供试品溶液应为未检验出内毒素且不超过MVD勺溶液,按LAL试剂灵敏度复核试验项下操作。
用检查法给出的公式计算溶液B、C的反应终点的几何平均值。
示灵敏度范围内时,试验方有效。
计算溶液B的鲎试剂灵敏度,如果值在 [0.5入,2.0入]间,可判定供试品溶液在该浓度下无干扰作用;反之则判定供试品溶液对试验能形成干扰。
若供试品溶液在小于 MVD的稀释倍数下对试验有干扰,应将供试品溶液进行不超过MVD勺进一步稀释,再重复干扰试验。
使用灵敏度更高如溶液的平行管的检查结果均为阳性时:- 如供试品的稀释倍数为 MVD供试品不符合规定。
四国药典细菌内毒素对比

CP(2015版)1143 EP(8.0)2.6.14 USP(36)85
JP(16)6.06
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
文案大全
小结:
细菌内毒素检测法,CP、EP、USP、JP中所描述的检测方法一致,包括种类、操作基本相同。
EP、USP、JP中有关于鲎试剂的介绍,CP中没有对这方面进行描述。
检测方法EP、USP、JP比CP描述的更加详细,例如鲎试剂的反应原理、供试品溶液表示方法等。
文案大全。
细菌内毒素检测

2006年6月
山东省药品检验所
21
二、信息的收集 在建立细菌内毒素检查法之前,应尽可能的收 集有关该药品的基本信息,例如:有关样品的 溶解性信息,推荐的稀释液,在水中的溶解度, 以及最适溶剂等;样品的pH值范围;分子量大 小;产品规格、体积或重量;拟用于临床的用 法和用量;还应了解是否为已知的鳌合剂;是 否具有酶活性(如胰岛素或丝氨酸蛋白酶); 活性组分是否会在70℃水浴中被灭活,是否可 能含有纤维素物质等。
31
大输液品种的限值一般定为0.5EU/ml, 灭菌注射用水则定为0.25EU/ml。 内毒素限值没有考虑到几个药物联合用 药问题,但一般情况下,药品中内毒素 的含量远低于限值,并且药品生产厂家 的内控标准严于法规标准,所以药典内 毒素限值也被称为最大允许内毒素浓度 (Maximum allowable endotoxin concentration)。
2006年6月 山东省药品检验所 11
细菌内毒素检查方法的建立
方法 标准物质 限值的确定 干扰试验 检验
2006年6月
山东省药品检验所
12
一、方法
细菌内毒素检查法包括凝胶法和光度测定法 凝胶法为限量测定方法,光度测定法为定量测 定方法 凝胶法分为限量试验和半定量试验;光度测定 法包括浊度法(动态浊度法、终点浊度法)、 显色法(动态显色法和终点显色法) 两种方法可任选一种进行内毒素的检测 当测定结果有争议时,除有特殊规定外,以凝 胶法结果为准
《中国药典》细菌内毒素检 验及方法建立
1
概述
前言
细菌内毒素
一、前言
热源检查易受试验动物个体差异、方法灵敏度、 药物本身性质的影响,受到一定限制,与家兔 热原法相比,细菌内毒素检查法具有劳动强度 低,实验成本小等特点,还可以通过对原料、 活性成分、灭菌水、容器、赋形剂和终产品的 细菌内毒素检测,对生产过程进行质控,更明 显的特点是可以定量检测药品生产过程中可能 污染的痕量内毒素,为区分干扰作用与内毒素 污染提供了极为重要的技术手段。
中美药典细菌内毒素比较

中国药典和美国药典中的细菌内毒素检测法的不同点比较1、细菌内毒素工作标准品(CSE)CP 对CSE的用途及效价进行定义“细菌内毒素工作标准品中每1ng细菌内毒素的效价应不小于2EU,不大于50EU”USP未提到CSE2、细菌内毒素检查用水CP :与灵敏度为0.03EU/ml或更高灵敏度的鲎试剂在37±1℃条件下24小时不产生凝集反应的灭菌注射用水;USP:与鲎试剂在限定的灵敏度下不发生反应的灭菌注射用水或其它水。
CP:用于细菌内毒素定量测定用的细菌内毒素检查用水,内毒素含量应小于0.005EU/ml。
USP:未提到。
3、实验用具的准备CP:实验所用器皿需经处理,除去可能存在的外源性内毒素,常用的方法是250℃下干烤至少1小时,也可用其它确证不干扰细菌内毒素检查的适宜方法。
USP:使用经验证的除热原程序对所有玻璃器皿和遇热稳定的材料在热空气烘箱中进行除热原,常用的最低温度和最少的时间是250℃下30分钟。
4、内毒素标准品贮备液和标准品溶液的制备CP:未提到USP:RSE的效价定为10,000USP EU/支,用5ml鲎试剂检查用水复溶1支RSE的全部内容物,用漩涡混合器间歇混合30分钟,并用此原液作系列稀释,将原液置于冰柜中保存不超过14天,作以后的稀释之用,在使用前用漩涡混合器强力混合不少于3分钟,在作下一步稀释前需对前面的稀释液混合不少于30秒,不要贮存稀释液,因为没有数据能证明其不会因为吸附作用而失去活性。
5、供试品溶液的制备CP:|某些供试品需进行复溶、稀释或在水性溶液中浸提用LRW复溶或稀释药品或抽提医疗器械,某些物质可能更适于用其它水性溶液来溶解、稀释或抽提。
对于过酸、过碱或本身有缓冲能力的供试品需调节被测溶液(或其稀释液)的pH值,一般要求供试品溶液的pH值在6.0-8.0的范围内。
USP:用LRW复溶或稀释药品或抽提医疗器械,某些物质可能更适于用其它水性溶液来溶解、稀释或抽提。
《美国药典》《欧洲药典》《日本药典》与《中国药典》中中药饮片微生物限度检查及标准的比较研究

《美国药典》《欧洲药典》《日本药典》与《中国药典》中中药饮片微生物限度检查及标准的比较研究作者:范一灵李琼琼秦峰刘浩杨美成来源:《中国药房》2020年第22期摘要目的:比較《美国药典》43版(USP43)、《欧洲药典》10.0版(EP10.0)、《日本药典》17版(JP17)与《中国药典》2020年版(ChP2020)中中药饮片微生物限度检查方法及标准的差异,为我国中药饮片相关微生物标准的修订和完善提供参考。
方法:比较USP43、EP10.0、JP17和ChP2020在中药饮片的微生物计数法(包括抽样与取样、菌种和培养基选择、微生物和耐热菌计数等)、控制菌检查(包括样品前处理、增菌、分离、鉴定等)、微生物相关限度标准等方面的差异。
结果与结论:在中药饮片微生物的检查方法上,USP43、EP10.0、JP17都有各自独立的规定,ChP2020则新增了“通则1108”。
在检验项目上,除需氧菌总数、霉菌和酵母菌总数外,ChP2020与EP10.0规定了3种控制菌(耐胆盐革兰阴性菌、大肠埃希菌、沙门菌)的检查方法;在此基础上,JP17补充了金黄色葡萄球菌的检查方法;USP43增加了梭菌的检查方法,并最早提出不可接受微生物风险评估理念;ChP2020还新增了耐热菌计数方法。
在微生物限度标准上,USP 43对中药饮片的分类最为细致,要求较为严格且高于EP10.0、JP17;ChP2020仍未对中药饮片控制菌检查设立统一的限度标准。
虽然,ChP2020对“中药提取物及中药饮片的微生物限度标准”进行了修订,但相较于美国、欧洲和日本药典的规定还不完善。
建议根据我国中药饮片微生物污染和控制现状,逐步完善药典对中药相关产品的微生物检验和限度标准,合理细化相应产品的微生物限度水平。
关键词中药饮片;药典;微生物检查;限度标准;比较研究中图分类号 R921;R927.1 文献标志码 A 文章编号 1001-0408(2020)22-2695-06DOI 10.6039/j.issn.1001-0408.2020.22.02ABSTRACT; ;OBJECTIVE: To compare the difference of microbiological limit test and criteria of TCM decoction pieces among 43 edition of United States Pharmacopeia; (USP43),10.0 edition of European Pharmacopeia; (EP10.0), 17 edition of Japanese Pharmacopeia;(JP17) and 2020 edition of Chinese Pharmacopeia (ChP2020), and to provide refernce for the revision and improvement of microbiological standards for TCM decoction pieces in China. METHODS: The differences in the microbial enumeration tests method (including sampling and sample preparation, selection of bacteria and culture medium, count of microorganisms and heat-resistant bacteria, etc.), tests for specified microorganisms (including sample pretreatment,enrichment, separation and identification, etc.) and microbial related limit criteria were compared among USP43, EP10.0, JP17 and ChP2020. RESULTS & CONCLUSIONS: In terms of microbiological examination of TCM decoction pieces, USP43, EP10.0, JP17 had their own independent provisions. Chp2020 added “general rule 1108”. In terms of inspection items, in addition to the total aerobic bacteria count and total combined yeasts and molds count, ChP2020 and EP10.0 provided three methods for the inspection of control bacteria (bile-resistant Gram-negative bacteria, Escherichia coli, Salmonella). On the basis, JP17 supplemented Staphylococcus aureus test; However, USP43 added Clostridium test method and put forward the concept of objectionable microorganisms risk assessment; ChP2020 also added a new method for counting heat-resistant bacteria. In terms of microbial limit criteria, USP43 was the most detailed in the classification of TCM decoction pieces, which was more strict than EP10.0 and JP17; ChP2020 had not set up a unified limit for the inspection of control bacteria of TCM decoction pieces. ChP2020 revised the “microbial limit standard for TCM extracts and TCM decoction pieces”, but it was notperfect compared with the Pharmacopoeia of the United States, Europe and Japan. It is suggested that according to the current situation of microbial contamination and control of TCM decoction pieces, the microbial limit test and criteria of TCM related products in Pharmacopoeia should be gradually improved, and the microbial limit level of corresponding products should be reasonably refined.KEYWORDS; ;TCM decoction piece; Pharmacopeia; Microbial limit test; Limit criteria; Comparative study中药饮片是由中药材通过炮制加工而成的可直接用于中医临床的产品,其大多源自天然植物、动物或矿物,通常携带有大量微生物[1-2]。
四国药典有关药品微生物限度标准的比较

四国药典有关药品微生物限度标准的比较微生物限度规定的作用,是为药品生产提供一个标准或指导,以确保药品使用的安全。
各国药典标准分为强制性的和非强制性的可达到的限度标准,这些指标正确、有效地规范了药品生产、检定和监督的程序。
药品要能反映不引起生物降解物和没有药源性污染的微生物存在是必要的,严格控制条件致病菌及致病菌。
一、CP、USP、BP、JP的微生物限度要求的特点及其发展趋势⒈各国药典收载微生物限度检查法的时间不同(见表1)表1 各国药典收载微生物限度检查法的时间各国药典CP USP BP JP收载微生物限度检查法的时间1995版* 1975(19)1973---方法1988---品种**第十三改正版****1978年颁布第一个药品卫生标准;1986年颁布了修改的药品卫生标准;1989年下发药品卫生标准补充规定和说明,1995年版中国药典收载微生物限度检查法(标准仅为少数剂型)**品种98版43个,其中原料药品种38个,制剂品种仅5个。
***仅有6个品种。
⒉品种不断扩大USP版本19(1975)22(1990)23 24(2000)*217种包括原料药品种72个(占1/3);制剂品种(占2/3)。
⒊活菌数要求各有特点⒋控制菌的要求各有特点a 10g或10ml样品不得检出。
b 1g或1ml 样品不得检出。
c 仅为个别品种要求10~103/1g或1ml。
*d 不列在剂型项内而以说明提出,意即不作为常规检查,如有检出,以不合格处理的依据。
二、稀释剂的种类* pH7.0缓冲氯化钠-蛋白胨水:磷酸二氢钾3.56g,磷酸氢二钠4.30g,蛋白胨(肉胨或酪胨1.0g纯水1000ml/L。
溶解以上成份,加入1~10g 吐温-20或吐温-80。
121℃高压蒸汽灭菌30min。
作用:可调节供试液pH至近中性,其中蛋白胨对菌细胞有保护作用,有利于菌数及控制菌的测定。
吐温加入对含油性供试品的助溶具有作用。
** USP、BP微生物限度(污染)检查用稀释剂,除磷酸盐缓冲液、磷酸盐缓冲胨水外,尚采用以上两种培养基,直接稀释供试品并作增菌培养.。
细菌内毒素的简介

4中国药典2005年版细菌内毒素检查法的修订内容
中国药典2005年版较中国药典2000 年版有重大修订,主要修订内容如下:
4.1.定义改变:2005年版药典定义细 菌内毒素检查法为利用鲎试剂来检 测或量化由革兰氏阴性菌产生的细 菌内毒素,以判断供试品中细菌内 毒素的限量是否符合规定。
脂多糖
稳定性
不稳定,60-80℃破坏
160-240℃破坏
毒性作用 抗原性 举例
强,有选择性 强,可制成类毒素 肉毒毒素,白喉外毒素
弱 弱 大肠杆菌内毒素
第六页,共38页。
▪ 内毒素的主要生物活性: 1 发热反应 2 白细胞反应 3 代谢性酸中毒 4 感染性休克 5 弥漫性血管内凝血(DIC)
▪ 2.8细菌内毒素标准品 细菌内毒素标准品分为细菌内毒素国际标准品、
2个以上厂家的鲎试剂
检 供试品至少3个
样品稀释到MVD, 样品稀释到MVD,
查 浸提介质:内毒素 1支NC, 1支PC,1支 2支NC, 2支PC,2支PPC
法 含量<0.05EU/ml PPC
2支供试品。
输液器10ml,输血 2支供试品。
器15ml,反复荡洗
5次后两端密闭, 37 ℃2h后合并
2支NC, 2支PC,
细菌内毒素检查
▪ 本讲座共分三部分 ▪ 一、基础部分 ▪ 二、标准部分 ▪ 三、实验操作部分
第一页,共38页。
一、基础部分
1. 前言
防止输注用药品及器械的热原污染,是临床上十分重要的。 半个多世纪以来,热原检查法对保证注射用药品安全发挥了 重要作用。但随着制药工业的发展,该方法已不完全适合许 多品种的热原检查。为此,人们研究了一种替代热原检查法 的方法,这就是我们今天正在研究和扩大应用的细菌内毒素 检查法。细菌内毒素检查是输注药品及器械质量控制中的一 项重要指标。
四国药典细菌内毒素对比

.CP(2015版)1143EP(8.0)2.6.14USP(36)85JP(16)6.06定义CP(2015) EP(8.0) USP(36) JP(16)本法系利用鲎试剂来检测或量化由革兰阴性菌产生的细菌内素素本法利用鲎试剂(从美洲鲎或中华鲎——阿米巴细胞溶解产物制备而来)检测由革兰氏阴性菌产生的细菌内毒素或对内毒素进行定量检查和定量供试品内毒素的方法。
本法利用鲎(美洲鲎或中华鲎)阿米巴细胞溶解产物制备的用于内毒素检测的鲎试剂( LAL)检定内毒素本法利用鲎试剂(从美洲鲎或中华鲎——阿米巴细胞溶解产物制备而来)检测由革兰氏阴性菌产生的细菌内毒素或对内毒素进行定量方法CP(2015) EP(8.0)一、凝胶法二、光度测定法1、浊度法2、显色基质法方法 A:凝胶法:限度试验方法 B:凝胶法:定量试验方法 C:动态浊度法方法 D:终点显色法方法 F:终点浊度法.USP(36) JP(16)仪器CP(2015) EP(8.0) USP(36) JP(16)一、凝胶法二、光度测定法1、浊度法2、显色法一、凝胶法二、光度测定法1、浊度法2、显色法干热灭菌法(250℃, 30 分钟以上),塑料器具应无内毒素并且对试验无干扰干热灭菌法(250℃, 30 分钟以上),塑料器具应无内毒素并且对试验无干扰干热灭菌法(250℃, 30 分钟以上),塑料器具应无内毒素并且对试验无干扰干热灭菌法(250℃, 30 分钟以上),塑料器具应无内毒素并且对试验无干扰溶液制备CP(2015) EP(8.0)供试品溶液的制备某些供试品需进行复溶、稀释或在水性溶液中浸提制成供试品溶液。
必要时,可调节被测溶液( 或其稀释液)的pH值,一般供试品溶液和鲎试剂混合后溶液的pH 值在 6.0-8.0的范围内为宜,可使用适宜的酸、碱溶液或缓冲液调节pH 值。
酸或碱溶液须用细菌内毒素检査用水在已去除内毒素的容器中配制。
缓冲液必须经过验证不含内毒素和干扰因子。
细菌内毒素检查结果准确性的影响因素

一、试验误差分析及改进措施
鲎试剂质量及灵敏度标示值得的影响品成份及理化性质的影响 实验员的影响 环境的影响
1 鲎试剂质量及灵敏度标示值得的影响
➢鲎试剂的质量:抗干扰能力,稳定性; ➢鲎试剂灵敏度标示值的准确性 。
3 实验器皿的影响
器皿的计量管理(校正) 器皿的选择标准 器皿的正确处理
1) 器皿的选择标准
鲎试验对器皿的要求:刻度准确;内表面光 滑;易于清理。细菌内毒素检查实验不宜选用 注射器,最好使用经过校正的刻度玻璃吸管或 吸嘴。注射器刻度不精确,内壁磨沙面吸附内 毒素,而且要特别注意注射器针头引入的铁离 子对试验结果的干扰,故不宜选用注射器。
谢 谢!
鲎试剂灵敏度的误差 是否用细菌内毒素国家标准品标定? 标示值是否准确?
2 细菌内毒素标准品引起的误差
细菌内毒素标准品种类 细菌内毒素标准品效价误差 细菌内毒素标准品操作误差 细菌内毒素单位表示误差 细菌内毒素标准品的错误使用
1) 细菌内毒素标准品种类 ➢细菌内毒素国际标准品 ➢细菌内毒素国家标准品 ➢细菌内毒素工作标准品
应该特别注意,该规程明确规定最后冲洗玻璃器皿 必须“用蒸馏水冲洗,禁用纯化水冲洗。”有的客 户使用反渗透制备的纯化水冲洗试验用玻璃器皿, 结果试验中所有的阳性对
照都为阴性,复核鲎试剂灵敏度时试验结果误差较 大,这主要是由于纯化水和蒸馏水的质量不同所造 成的。因纯化水中存在的阴阳离子经250℃高温干烤 不能完全被破坏,这时不同的离子就产生对鲎试剂 酶反应抑制或增强的反应,为了保证试验结果的准 确性和检验报告的权威性,我们应该严格遵守操作 规程,用多效蒸馏水冲洗鲎试验用玻璃器皿。
2) PH值对反应速度的影响 常见现象:样品阳性不成立。
2019年热原和细菌内毒素的区别.doc

热原和细菌内毒素一、热原(progon)医院临床在使用药品注射剂时,常有发生冷感、寒战、发热、头痛、恶心、呕吐、肤色灰白、休克、严重时导致死亡,这种症状称为热原反应。
为提高药品质量和用药安全,人们对热原进行了广泛的研究,直到1923年Seibert提出了用家兔检测热原的方法。
在1942年美国药典首先将家兔热原检查项收入药典成为法定方法,中国药典1953年版开始收载该方法,随后的世界各国药典都以动物热原检查法作为药品质量监测的方法之一。
家兔热原检查法的优点,可在规定时间里观察到家兔的体温变化,相应反应了热原质引起哺乳类动物复杂的体温反应过程。
所以,在半个多世纪以来热原检查法,为保障药品质量和用药安全发挥了重要作用。
但随着制药工业的发展和临床用药的要求,该方法的局限性越来越明显。
这种热原检查法,只局限于某种药物进入体内(血循环)是否能引起体温变化或热原反应作为判断药品是否污染热原的方法,已不能满足医药工业发展的需要。
其缺点:①标准化程度低,无法判断检查样品中存在的热原质到底是什么或是哪一种物质。
②由于试验动物家兔是处在被细菌污染的环境中,通过吸入或皮肤感染细菌内毒素而被免疫,导致动物的个体差异较大。
③试验动物受到药品的药理活性干扰,而影响体温变化(如放射性药品、抗生素、生物制品等),实验结果难以判断。
④设备及实验费用昂贵(如建设动物房、水电、动物饲料等耗费),做一种药品需要几百元/次,而鲎试剂仅几十元/次。
综上情况分析,鲎试验法可避免以上动物热原检查法的不足,该技术的成功和应用真可谓是药品质量监控一场大革命。
什么是热原?目前国内外仍未有统一的认识,但从国内外文献报道中,一个共同的意见,都普遍认为:它是指细菌内毒素的脂多糖。
欧洲药典委员会副主席J.Van Noordwijk提出:“严格地讲,不是每一种热原都具有脂多糖的结构,但所有已知的细菌内毒素脂多糖都有热原活性”。
在药品生产质量管理规范(GMP)条件下,药品生产的质量控制一般可以接受的观点是:不存在细菌内毒素意味着不存在热原。
细菌内毒素与热原的区别

热原和细菌内毒素一、热原(progon)医院临床在使用药品注射剂时,常有发生冷感、寒战、发热、头痛、恶心、呕吐、肤色灰白、休克、严重时导致死亡,这种症状称为热原反应。
为提高药品质量和用药安全,人们对热原进行了广泛的研究,直到1923年Seibert提出了用家兔检测热原的方法。
在1942年美国药典首先将家兔热原检查项收入药典成为法定方法,中国药典1953年版开始收载该方法,随后的世界各国药典都以动物热原检查法作为药品质量监测的方法之一。
家兔热原检查法的优点,可在规定时间里观察到家兔的体温变化,相应反应了热原质引起哺乳类动物复杂的体温反应过程。
所以,在半个多世纪以来热原检查法,为保障药品质量和用药安全发挥了重要作用。
但随着制药工业的发展和临床用药的要求,该方法的局限性越来越明显。
这种热原检查法,只局限于某种药物进入体内(血循环)是否能引起体温变化或热原反应作为判断药品是否污染热原的方法,已不能满足医药工业发展的需要。
其缺点:①标准化程度低,无法判断检查样品中存在的热原质到底是什么或是哪一种物质。
②由于试验动物家兔是处在被细菌污染的环境中,通过吸入或皮肤感染细菌内毒素而被免疫,导致动物的个体差异较大。
③试验动物受到药品的药理活性干扰,而影响体温变化(如放射性药品、抗生素、生物制品等),实验结果难以判断。
④设备及实验费用昂贵(如建设动物房、水电、动物饲料等耗费),做一种药品需要几百元/次,而鲎试剂仅几十元/次。
综上情况分析,鲎试验法可避免以上动物热原检查法的不足,该技术的成功和应用真可谓是药品质量监控一场大革命。
什么是热原?目前国内外仍未有统一的认识,但从国内外文献报道中,一个共同的意见,都普遍认为:它是指细菌内毒素的脂多糖。
欧洲药典委员会副主席J.V an Noordwijk提出:“严格地讲,不是每一种热原都具有脂多糖的结构,但所有已知的细菌内毒素脂多糖都有热原活性”。
在药品生产质量管理规范(GMP)条件下,药品生产的质量控制一般可以接受的观点是:不存在细菌内毒素意味着不存在热原。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
EP(8.0)
最大有效稀释倍数(MVD)指可检测出内毒素限值的供试品溶液的最大稀释倍数。MVD按下列公式确定:
MVD = 内毒素限值×供试品溶液浓度/λ
内毒素限值:在剂量的基础上,注射药物的活性成分的内毒素限度为K/M
JP(16)
内毒素储备标准溶液的制备
以BET检查用水溶解内毒素10000对照标准品或内毒素100对照标准品,制取细菌内毒素试验(BET)的内毒素储备标准溶液。内毒素的量用内毒素单位(EU)表示。1EU相当于1个内毒素国际单位(IU)。
内毒素标准溶液的制备
充分混合内毒素储备标准溶液后,用水稀释,即得BET检查用内毒素标准溶液。得到的稀释液应尽快使用,以免因吸附而导致活性损失。
EP(8.0)
内毒素储备标准溶液的制备
用内毒素标准品制备内毒素储备标准溶液;所用的内毒素标准品必须先用国际标准品校准,如内毒素标准BRP。
内毒素以国际单位(IU)表示。IU的换算见国际卫生组织公布的国际标准。
注:一国际单位(IU)内毒素相当于一个内毒素单位(E.U.)。
根据包装说明书上的标准和内毒素储备标准溶液的标签上关于制备和贮存的说明。
EP(8.0)
干热灭菌法(250℃,30分钟以上),塑料器具应无内毒素并且对试验无干扰
USP(36)
干热灭菌法(250℃,30分钟以上),塑料器具应无内毒素并且对试验无干扰
JP(16)
干热灭菌法(250℃,30分钟以上),塑料器具应无内毒素并且对试验无干扰
溶液制备
CP(2015)
供试品溶液的制备
某些供试品需进行复溶、稀释或在水性溶液中浸提制成供试品溶液。必要时,可调节被测溶液(或其稀释液)的pH值,一般供试品溶液和鲎试剂混合后溶液的pH值在6.0-8.0的范围内为宜,可使用适宜的酸、碱溶液或缓冲液调节pH值。酸或碱溶液须用细菌内毒素检査用水在已去除内毒素的容器中配制。缓冲液必须经过验证不含内毒素和干扰因子。
K=人每公斤体重每小时最大可接受的内毒素剂量(EU/kg)
M=人用每公斤体重每小时的最大供试品剂量。
专论中,注射剂的活性成分的内毒素限度的单位有IU/ml、IU/mg、IU/生物活性单位等。
供试品溶液的浓度表示法:
-当内毒素限度以质量(IU/mg)表示时,用mg/ml表示浓度。
-当内毒素限度以(IU/ml)表示时,用ml/ml表示浓度。
表1凝胶法干扰试验溶液的制备
编号
内毒素浓度/被加入内毒素的溶液
稀释用液
稀释倍数
稀释后内毒素的浓度
平行管数
A
无/供试品溶液
—
—
-
2
B
2λ/供试品溶液
供试品溶液
1
2
4
8
2λ
1λ
0.5λ
0.25λ
4
4
4
4
C
2λ/检查用水
检查用水
1
2
4
8
2λቤተ መጻሕፍቲ ባይዱ
1λ
0.5λ
0.25λ
2
2
2
2
D
无/检查用水
-
-
-
2
注:A为供试品溶液;B为干扰试验系列;C为鲎试剂标示灵敏度的对照系列;D为阴性对照。
λc=antilg(∑X/n)
式中X为反应终点浓度的对数值(lg)。反应点浓度是指系列递减的内毒素浓度中最后一个呈阳性结果的浓度;
n为每个浓度的平行管数。
当λc在0.5-2λ(包括0.5λ和2λ)时,方可用于细菌内毒素检査,并以标示灵敏度λ为该批鲎试剂的灵敏度。
干扰试验按表1制备溶液A、B、C和D,使用的供试品溶液应为未检验出内毒素且不超过最大有效稀释倍数(MVD)的溶液,按鲎试剂灵敏度复核试验项下操作。
内毒素限值的确定及最大有效稀释倍数
CP(2015)
药品、生物制品的细菌内毒素限值
( L )一般按以下公式确定:
L=K/M
式中L为供试品的细菌内毒素限值,一般以EU/ml、EU/mg或EU/U (活性单位)表示;
K为人每千克体重每小时最大可接受的内毒素剂量,以EU/(kg•h )表示,注射剂K=5EU/(kg•h),放射性药品注射剂K =2.5EU/(kg•h),鞘内用注射剂K=0.2E U /(kg•h);
将试管从恒温器中轻轻取出,缓缓倒转180°,若管内形成凝胶,并且凝胶不变形、不从管壁滑脱者为阳性;未形成凝胶或形成的凝胶不坚实、变形并从管壁滑脱者为阴性。保温和拿取试管过程应避免受到振动,造成假阴性结果。
当最大浓度2λ管均为阳性,最低浓度0.25λ管均为阴性,阴性对照管为阴性,试验方为有效。按下式计算反应终点浓度的几何平均值,即为鲎试剂灵敏度的测定值(λc)。
JP(16)
本法利用鲎试剂(从美洲鲎或中华鲎——阿米巴细胞溶解产物制备而来)检测由革兰氏阴性菌产生的细菌内毒素或对内毒素进行定量
CP(2015版)1143
EP(8.0)2.6.14
USP(36)85
JP(16)6.06
方法
CP(2015)
一、凝胶法
二、光度测定法
1、浊度法
2、显色基质法
EP(8.0)
供试品溶液的制备
除非另有说明,以BET检查用水溶解或稀释药品来制备供试品溶液。药用容器内盛放的供试品溶液的制备方法见其他具体规程。如有必要,调节待测溶液的pH值,使鲎试剂和供试品溶液的混合物的pH值在所选鲎试剂的使用pH范围内。一般要求供试品溶液的pH值范围为6.0-8.0。试液或调节pH值用溶液应用BET检查用水配制,在去除内毒素的容器中贮存。
M为人用每千克体重每小时的最大供试品剂量,以ml/(kg•h )、mg/(kg•h)或U/(kg•h )表示,人均体重按60kg计算,人体表面积按1.62m2计算。
注射时间若不足1小时,按1小时计算。供试品每平方米体表面积剂量乘以0.027即可转换为每千克体重剂量(M)。
按人用剂量计算限值时,如遇特殊情况,可根据生产和临床用药实际情况做必要调整,但需说明理由。
-当内毒素限度以生物单位(IU/Unit)表示时,用Units/ml表示浓度。
λ=指凝胶法中鲎试剂的标示灵敏度,或指浊度法或显色法使用的标准回归曲线上最低点(IU/ml)的对应值。
USP(36)
最大有效稀释倍数(MVD)指可检测出内毒素限值的供试品溶液的最大稀释倍数。按下列公式确定:
MVD=内毒素限值×供试品溶液浓度/λ
只有当溶液A和阴性对照溶液D的所有平行管都为阴性,并且系列溶液C的结果符合鲎试剂灵敏度复核试验要求时,试验方为有效。当系列溶液B的结果符合鲎试剂灵敏度复核试验要求时,认为供试品在该浓度下无干扰作用。
其他情况则认为供试品在该浓度下存在干扰作用。若供试品溶液在小于MVD的稀释倍数下对试验有干扰,应将供试品溶液进行不超过MVD的进一步稀释,再重复干扰试验。
最大有效稀释倍数是指在试验中供试品溶液被允许达到稀释的最大倍数(1-MVD) ,在不超过此稀释倍数的浓度下进行内毒素限值的检测。用以下公式来确定MVD:
MVD =cL/λ
式中L为供试品的细菌内毒素限值;
c为供试品溶液的浓度,当L以EU/mg或EU/U表示时,c的单位需为mg/ml或U/ml ,当L以EU/ml表示时,则c等于l.0ml/ml。如需计算在MVD时的供试品浓度,即最小有效稀释浓度,可使用公式c=λ/L;
可通过对供试品进行更大倍数的稀释或通过其他适宜的方法(如过滤、中和、透析或加热处理等)排除干扰。为确保所选择的处理方法能有效地排除干扰且不会使内毒素失去活性,要使用预先添加了标准内毒素再经过处理的供试品溶液进行干扰试验。
MVD = 内毒素限值×供试品溶液浓度/λ
内毒素限值:
根据剂量,注射剂的内毒素限度即为K/M的值,其中K为人每公斤体重可接受的内毒素中热原的最小量(EU/kg),M指人用每公斤体重每小时的最大供试品剂量。
供试品溶液的浓度表示法:
当内毒素限度以质量(EU/mg)表示时,用mg/mL表示浓度。
当内毒素限度以(EU/mEq)表示时,用mEq/mL表示浓度。
供试液——供试溶液是通过用细菌内毒素检查用水溶解、稀释药物制备。某些物质或制剂可能在其他水溶液中被更适当地溶解、稀释。如有必要,调节待测溶液的pH值,使鲎试剂和样品的混合物pH在由鲎试剂生产者定的pH范围内。通常使用于pH范围在6.0-8.0的产品。pH的调节可以用鲎试剂生产者推荐的酸、碱或适当的缓冲液。酸和碱可以在已去除内毒素的容器中用浓缩液或固体和细菌内毒素检查用水制备。缓冲剂必须进行验证无内毒素和干扰因子。
鲎试剂灵敏度复核试验在本检査法规定的条件下,使鲎试剂产生凝集的内毒素的最低浓度即为鲎试剂的标示灵敏度,用EU/ml表示。当使用新批号的鲎试剂或试验条件发生了任何可能影响检验结果的改变时,应进行鲎试剂灵敏度复核试验。
根据鲎试剂灵敏度的标示值(A),将细菌内毒素国家标准品或细菌内毒素工作标准品用细菌内毒素检査用水溶解,在旋涡混合器上混匀15分钟,然后制成2λ、λ、0.5λ和0.25λ四个浓度的内毒素标准溶液,每稀释一步均应在旋涡混合器上混匀3 0秒。取分装有0.1ml鲎试剂溶液的10mm×75mm试管或复溶后的0.1ml/支规格的鲎试剂原安瓿18支,其中16管分别加人0.lml不同浓度的内毒素标准溶液,每一个内毒素浓度平行做4管;另外2管加入0.1ml细菌内毒素检査用水作为阴性对照。将试管中溶液轻轻混匀后,封闭管口,垂直放入37±1℃的恒温器中,保温60分钟±2分钟。
内毒素标准溶液的制备
充分混合内毒素储备标准溶液后,用细菌内毒素试验检查用水(BET检查用水)稀释,制成适当的系列稀释液,即得BET检查用内毒素标准溶液。
得到的稀释液应尽快使用,以免因吸附而导致活性损失。
供试品溶液的制备