高斯常见错误及注意地方
高斯错误修正

收敛失败是很正常的事也是很头痛的事,在Gaussian98高级注释一文中提到以下几种做法:我经常用的是3/5/8/12,而且如果你是计算过渡金属,降低收敛标准你就得小心。
1. 在Guess关键字中使用Core,Huckel或Mix选项,试验不同的初始猜测。
2. 对开壳层体系,尝试收敛到同一分子的闭壳层离子,接下来用作开壳层计算的初始猜测。
添加电子可以给出更合理的虚轨道,但是作为普遍的经验规则,阳离子比阴离子更容易收敛。
选项Guess=Read定义初始猜测从Gaussian 计算生成的checkpoint文件中读取。
3. 另一个初始猜测方法是首先用小基组进行计算,由前一个波函得到用于大基组计算的初始猜测(Guess=Read自动进行)。
4. 尝试能级移动(SCF=Vshift)。
5. 如果接近SCF但未达到,收敛标准就会放松或者忽略收敛标准。
这通常用于不是在初始猜测而是在平衡结构收敛的几何优化。
SCF=Sleazy 放松收敛标准,Conver选项给出更多的控制。
6. 一些程序通过减小积分精度加速SCF。
对于使用弥散函数,长程作用或者低能量激发态的体系,必须使用高积分精度:SCF=NoVarAcc。
7. 尝试改变结构。
首先略微减小键长,接下来略微增加键长,接下来再对结构作一点改变。
8. 考虑使用不同的基组。
9. 考虑使用不同理论级别的计算。
这并不总是实用的,但除此之外,增加迭代数量总是使得计算时间和使用更高理论级别差不多。
10. 关闭DIIS外推(SCF=NoDIIS)。
同时进行更多的迭代( SCF=(MaxCycle=N) )。
11. 更多的SCF迭代( SCF(MaxCycle=N),其中N是迭代数)。
这很少有帮助,但值得一试。
12. 使用强制的收敛方法。
SCF=QC通常最佳,但在极少数情况下SCF=DM 更快。
不要忘记给计算额外增加一千个左右的迭代。
应当测试这个方法获得的波函,保证它最小,并且正好不是稳定点(使用Stable关键字)。
高斯常见错误及注意地方

优化1在优化时采用Scf(tight)的选项,增加收敛的标准。
再去计算频率。
如果还有虚频,参见下一步。
2.对称性的影响,很多情况下的虚频是由于分子本身的对称性造成的。
这样,在优化时,如果必要,要将对称性降低,还有,输入文件有时是用内坐标。
建议如果有虚频的话,将内坐标改成直角坐标优化。
3.如果上述方法还有虚频,看一下虚频,找到强度较大的,将在频率中产生的原子的振动坐标加到相应的输入文件中。
这样,重新计算。
直到虚频没有。
4实际上,如果分子柔性较大,很难找到最低点,这是电子结构计算的问题,这种情况下,需要动力学的东西,用构象搜寻的办法解决。
如:模拟退火,最陡下降法,淬火法等。
将得到的能量最低的构象做一般的电子结构计算,这样,应当没有问题。
不要讲你还没有得到最稳定的结构,那么,是你的分子有问题,要么计算错了,要么就是游离出现代计算的范畴1。
低频率振动模式时,力常数很小,必须使用opt=tight以确保适当的收敛和随后任务步骤中频率计算的可靠度。
比如我曾经算过一个体系,第一个频率振动的力常数只有10~20,很不放心,加入tight后,优出来的结构却与前面大相径庭,而且还出现了一个虚频。
2。
做IRC计算的时候,如果步长很小,必须要用verytight。
就曾有人步长设为1,而不加verytight,算出来的反应路径是v字形倒海鸥状的。
3。
使用int指定用于数值积分的积分网格来用于消虚频的时候,必须注意,在比较能量的时候,对所有的计算要使用相同的积分网格。
检查错误1.检查是否有初始文件错误在命令行中加入%kJob L301 or %kJob L302如果通过则一般初始文件ok。
常见初级错误:a.自旋多重度错误b.变量赋值为整数c.变量没有赋值或多重赋值d.键角小于等于0度,大于等于180度e.分子描述后面没有空行f.二面角判断错误,造成两个原子距离过近g.分子描述一行内两次参考同一原子,或参考原子共线2.SCF(自洽场)不收敛则一般是L502错误省却情况做64个cycle迭代(G03缺省128 cycles)a.修改坐标,使之合理b.改变初始猜Guess=Huckel或其他的,看Guess关键词。
Gaussian菜鸟常见问题分析

Gaussian菜鸟常见问题分析1.检查是否有初始文件错误在命令行中加入%kJob L301 or%kJob L302如果通过则一般初始文件ok。
常见初级错误:a.自旋多重度错误b.变量赋值为整数c.变量没有赋值或多重赋值d.键角小于等于0度,大于等于180度e.分子描述后面没有空行f.二面角判断错误,造成两个原子距离过近g.分子描述一行内两次参考同一原子,或参考原子共线2.SCF(自洽场)不收敛则一般是L502错误省却情况做64个cycle迭代(G03缺省128 cycles)a.修改坐标,使之合理b.改变初始猜Guess=Huckel或其他的,看Guess关键词。
c.增加叠代次数SCFCYC=N(对小分子作计算时最好不要增加,很可能结构不合理)d.iop(5/13=1)这样忽略不收敛,继续往下做。
3.分子对称性改变a.修改坐标,强制高对称性或放松对称性b.给出精确的、对称性确定的角度和二面角。
如CH4的角度给到109.47122 c.放松对称性判据Symm=loose d.不做对称性检查iop(2/16=1)(最好加这个选项)iop(2/16=2)则保持新的对称性来计算4.Opt时收敛的问题a.修改坐标,使之合理b.增加叠代次数optcyc=N 5.优化过渡态,若势能面太平缓,则不好找到。
iop(1/8=10)默认30(下一个结构和该结构的差别0.3),可改成10。
如果每一步都要用到小的步长,应该加opt(notrustupdate)6.在CI(组态)方法中如QCISD(T),CCSD(T),CID方法中,省却最大循环50,若出错(L913错误) 解决方法:#P QCISD(maxcyc=N)注:N≤512 7.优化过渡态opt=TS(给出过渡态)opt=qst2(给出反应物和产物)opt=qst3(给出反应物和产物和过渡态)a.用G03时的出错opt=ts必须加FC(force constant)写法:opt=(TS,calcFc)or opt=(TS,calchffc)计算HF力常数,对QCISD,CCSD等方法用;or opt=(TS,modRedundant)(最好写这个)b.如果计算采用QCISD计算(不好计算FC)则写为QCISD opt=(TS,calcHFFC)(用HF计算FC)8.无法写大的Scratch文件RWF a.劈裂RWF文件%rwf=loc1,size1,loc2,size2,….,locN,-1 b.改变计算方法MP2=Direct可以少占硬盘空间c.限制最大硬盘maxdisk=N GB,*MB,有些系统写2GB会出错,可以写2000MB 9.FOPT出错原因是变量数与分子自由度数不相等。
gaussian基组重叠误差

高斯基组重叠误差是指在计算分子结构、能量和性质时,由于采用有限的高斯基组而产生的误差。
高斯基组是一种常用的量子化学计算方法,它是通过高斯函数的线性组合来描述原子和分子的电子轨道,从而计算分子的性质和行为。
然而,由于高斯基组的有限性,会导致在计算中产生一定的误差,其中最常见的误差之一就是重叠误差。
1. 高斯基组的理论基础和应用高斯基组是由密度泛函和哈特里-福克方法等理论演化而来的,它是一种在量子化学计算中被广泛应用的方法。
通过高斯函数的线性组合,高斯基组可以有效地描述电子的轨道分布和排布,并且具有较高的计算精度和效率。
在分子动力学模拟、分子能量计算、反应动力学等领域都有着重要的应用。
2. 高斯基组重叠误差产生原因在采用高斯基组进行量子化学计算时,由于基组的有限性,它无法完美描述真实电子轨道的特性,从而导致了重叠误差的产生。
具体而言,由于高斯基组的函数形式和数量都是有限的,因此在电子轨道相互重叠的区域会产生误差,进而影响到计算结果的精度和准确性。
3. 重叠误差的影响和后果重叠误差在量子化学计算中会对分子结构、键能、反应速率等多个方面产生影响和后果。
它会导致计算出的分子结构与实际情况相差较大,从而影响到后续的性质和行为的计算结果。
重叠误差还会对分子间相互作用能产生一定的影响,尤其是对于含有氢键等弱键的分子体系,重叠误差会使得其计算结果偏离真实值。
重叠误差还会影响到分子内部的电子密度分布和轨道的形状,进而影响到分子的稳定性和反应特性。
4. 减小重叠误差的方法和策略为了尽可能减小高斯基组重叠误差的影响,科研工作者们提出了一系列的优化方法和策略。
可以通过增加高斯基组的数量和扩展基组的范围来改善计算精度,例如采用多级基组计算或者采用diffuse函数来扩展基组。
使用一些先进的密度泛函和校正方法也可以有效减小重叠误差的影响,例如采用长程校正和修正势能项等方法。
还可以通过对计算模型的参数和设定进行合理的调整和优化来减小重叠误差的影响,例如采用高级的优化算法和设定合理的收敛标准。
高斯错误修改总结

A list of error messages and possible solutions -Gaussian calculations can fail with various error messages. Some error messages from .out and .log files - and possible solutions - have been compiled here to facilitate problem solving.-These are divided into:-Syntax and similar errors-语法类错误Memory and similar errors-内存类错误Convergence problems -不收敛错误Errors in solvent calculations -溶剂中的计算错误Errors in log files-错误文件-ERROR MESSAGES IN OUTPUT FILES-Syntax and similar errors:End of ZSymb.-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l101.exe Solution: The blank line after the coordinate section in the .inp missing. (输入文件空行丢失) Unrecognized layer "X".-(不识别层X)Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l101.exeSolution: Error due to syntax error(s) in coordinate section (check carefully). If error is "^M", it is caused by DOS end-of-line characters (e.g. if coordinates were written under Windows). Remove ^M from line ends using e.g. emacs. To process .inp files from command line, use sed -i 's/^M//' (Important: command does not work if ^M is written as characters - generate ^M on command line using ctrl-V ctrl-M).-QPERR --- A SYNTAX ERROR WAS DETECTED IN THE INPUT LINE.-Solution: Check .inp carefully for syntax errors in keywords -RdChkP: Unable to locate IRWF=0 Number= 522.-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l401.exe or-FileIO operation on non-existent file.-[...] Error termination in NtrErr:-NtrErr Called from : Operation on .chk specified (e.g. geom=check, opt=restart),but .chk was not found. Check that:-%chk= was specifed in .inp-.chk has the same name as .inp-.chk is in the same directory as .inp -run script transports .chk to temporary folder upon job start. Run scripts downloaded here should do this. -The combination of multiplicity N and M electrons is impossible.-(多重性)Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l301.exeSolution: Either the charge or the multiplicity of the molecule was not specified correctlyin .inp.-(电荷和多重性指定错误)Memory and similar errors: Out-of-memory error in routine RdGeom-1 (IEnd= 1200001 MxCore= 2500)-Use %mem=N MW to provide the minimum amount of memory required to complete this step-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l101.exe or-Not enough memory to run CalDSu, short by 1000000 words.-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l401.exe or-[...] allocation failure: -(表示配分失败)Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l1502.exe Solution: Specify more memory in .inp (%mem=Nmb). Possibly, also increase pvmem value in run script. Especially solvent calculations can exhibit allocation failures and explicit amounts of memory should be specified.-galloc: could not allocate memory.-(无法分配内存)Solution: The %mem value in .inp is higher than pvmem value in run script. Increase pvmem or decrease %mem. -Probably out of disk space(磁盘空间). Write error in NtrExt1 Solution: /scratch space is most likely full. Delete old files in temporary folder. -Convergence problems: Density matrix is not changing but DIIS error= 1.32D-06 CofLast= 1.18D-02.-(收敛问题)The SCF is confused. Error termination via Lnk1e in/global/apps/gaussian/g03.e01/g03/linda-exe/l502.exel Solution: Problem with DIIS. Turn it off completely, e.g. using SCF=qc, or partly by usingSCF=(maxconventionalcycles=N,xqc), where N is the number of steps DIIS should be used (see SCF keyword). -Convergence criterion not met. SCF Done: E(RHF) = NNNNNNN A.U. after 129 cycles -[...] Convergence failure -- run terminated. Error termination via Lnk1e in/global/apps/gaussian/g03.e01/g03/linda-exe/l502.exe Solution: One SCF cycle has a default of maximum 128 steps, and this was exceeded without convergence achieved. Possible solution: In the route section of input file, specify SCF=(MaxCycle=N), where N is the number of steps per SCF cycles. Alternatively, turn of DIIS (e.g. by SCF=qc) (see SCF keyword).--Problem with the distance matrix.-(距离矩阵)Error termination via Lnk1e in /pkg/gaussian/g03/l202.exe Solution: Try to restart optimization from a different input geometry. -(重新不同几何异构体的输入优化)New curvilinear step not converged(新曲线步骤不收敛). Error imposing constraints-Error termination via Lnk1e in /pkg/gaussian/g03/l103.exe-Solution: Problem with constrained coordinates (e.g. in OPT=modredun calculation). Try to restart optimization from a slightly different input geometry. -(一种稍微不同的输入几何)-Optimization stopped. -- Number of steps exceeded, NStep= N-[..] Error termination request processed by link 9999.-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l9999.exe Solution: Maximum number of optimization steps is twice the number of variables to be optimized. Try increasing the value by specifying OPT=(MaxCycle=N) in .inp file, where N is the number of optimization steps (see OPT keyword). Alternatively, try to start optimization from different geometry.--Errors in solvent calculations: AdVTs1: ISph= 2543 is engulfed by JSph= 2544 but Ae( 2543) is not yet zero!-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l301.exe Solution: Problem is related to building of the cavity in solvent calculations(溶剂效应优化计算错误). One possible solution is to change the cavity(腔) model (default in g03 is UAO, can be changed by adding RADII keyword in section below coordinates in the .inp file, e.g. RADII=UFF, see SCRF keyword).--Hydrogen X has 2 bounds. Keep it explicit at all point on the-potential energy surface to get meaningful results.Solution: In UAO cavity model, spheres are placed on groups of atoms, with hydrogens assigned to the heavy atom, they are bound to. If assignment fails (e.g. because heavy atom-H bond is elongated), cavity building fails. Possible solutions: a) use cavity model that also assigns spheres to hydrogens (e.g. RADII=UFF) or b) Assign a sphere explicity on problematic H atom (use SPHEREONH=N, see SCRF keyword)--ERROR MESSAGES IN LOGFILES =>> PBS: job killed: wall time N exceeded limit M-signal number 15 received. Solution: Job did not finish within specified wall time. Retrieve .out and .chk files from temporary folder /global/work/$USER/$JOB (or $PBS_JOBID) and restart calculation if possible (using e.g. opt=restart or scf=restart). -cp: cannot stat $JOB.inp: No such directory Solution: The .inp not in the directory from where the job was submitted (or its name was misspelled during submission. If error reads: cp: cannot stat $JOB .inp .inp, the .inp submitted with extension).-ntsnet: unable to schedule the minimum N workers Solution: The value of %N proc Linda=N in the .inp higher than the number of nodes asked for during submission. Make sure these values match.Connection refused [...] died without ever signing in-Sign in timed out after 0 worker connections. Did not reach minimum (N), shutting downSolution: Error appears if you run parallel calculations but did not add this your $HOME directory: .tsnet.config containing only the line: Tsnet.Node.lindarsharg: ssh (see also guidelines for submission). -Density matrix is not changing but DIIS error - Suggested solutions1/- SCF=qc will probably solve the problem, albeit at a cost- Change the SCF converger to either SD, Quadratic or Fermi2/- lower the symmetry of optimize with and optimizewith the "nosymm" keywordI solved the problem using a variation on the first suggestion. Normally the scf took less than 80 cycles to converge. So i used scf=(Maxconventionalcycles=100,xqc) which resulted in a good compromise between using scf=qc and optimisation speed.In the case of the DIIS error the scf always took more than 100 cycles before the error,so by adding scf=(Maxconventionalcycles=100,xqc) the scf switched to qc after 100 cycles in the standard DIIS mode.l9999错误是优化圈数不够,把out文件保存成gjf,修改后接着优化。
高斯常见错误

高斯常见错误近来一直在学习高斯,因为不精通常遇到各种错误。
结合自学的东西和查阅的资料总结出来一些错误,希望对和我一样的高斯初学者有所帮助。
1、Q:Error termination in NtrErr: ntran open failure returned to fopen. Segmentation faultE:Can't open a file.2、Q:Internal consistency error detected in FileIO for unit 1I= 4 J=0 I Fail= 1.E:Gaussian is limited to 16 GB of scratch space on the 32-bit nodes.3、Q:Out-of-memory error in routine UFChkP (IEnd= 12292175MxCore= 6291456)Use %Mem=12MW to provide the minimum amount of memory required to complete this step. Error termination via Lnk1e at Thu Feb 2 13:05:32 2006.E efault memory (6 MW, set in $GAUSS_MEMDEF) is too small for unfchk.4、Q:galloc: could not allocate memory.: Resource temporarily unavailableor Out-of-memory error in routine...or End of file in GetChg. Error termination via Lnk1e ...E:Not enough memory.5、Q:IMax=3 JMax=2 DiffMx= 0.00D+00Unable to allocate space to process matrices in G2DrvN:NAtomX= 58 NBasis= 762 NBas6D= 762 MDV1= 6291106 MinMem= 105955841.E:Gaussian has 6 MW free memory (MDV1) but requires atleast 106 MW (MinMem).6、Q;Estimate disk for full transformation -677255533 words. Semi-Direct transformation. Bad length for file.E:MaxDisk has been set too low.7、Q:Error termination in NtrErr:NtrErr Called from FileIO.E:The calculation has exceeded the maximum limit of maxcyc.8、Q:Erroneous read. Read 0 instead of 6258688. fd = 4 g_readE:Disk quota or disk size exceeded. Could also be disk failure or NFS timeout.9、Q:Erroneous write. Write 8192 instead of 12288. fd = 4E:Disk quota or disk size exceeded. Could also be disk failure or NFS10、Q:orig len = 12288 left = 12288 g_writeE:timeout11、另有link错误:如:Error termination request processed by link 9999对于优化不收敛,即L9999错误,实际上是在规定的步数内没有完成优化,即还没有找到极小值点。
高斯留下的十大数学难题

高斯留下的十大数学难题
卡尔·弗里德里希·高斯被誉为数学史上最伟大的数学家之一,他在数学领域留
下了许多经典问题和难题。
其中,被认为是高斯留下的十大数学难题包括以下内容:
1. 平面上的三角形划分问题:给定一个平面上的任意三角形,能否将其划分为
尽可能多的小三角形,这些小三角形不相交且边长相同?
2. 整数三角形问题:给定一个正整数n,是否存在边长为整数的三角形,其三
个内角分别为n的倍数?
3. 质数的分布问题:证明素数分布的规律,即证明存在无穷多个素数。
4. 高斯圆数问题:研究高斯整数环中的素数分布规律。
5. 数学分析的基础问题:证明实数的完备性,即任何有界的实数集合必有上确界。
6. 几何的基础问题:证明欧几里德几何的五大公设的独立性。
7. 微分方程问题:研究微分方程的解的存在性和唯一性。
8. 算术的基础问题:证明算术的基本定理,即任何大于1的自然数都可以分解
为质数的乘积。
9. 算术和几何的关系问题:研究数学的两大基础分支的联系和互相补充。
10. 数学的未解问题:探索数学的边界,寻找新的数学难题和问题。
这些数学难题体现了高斯在数学领域的卓越贡献和深刻思考,也为后人提供了
许多研究的方向和启示。
数学的发展离不开数学家们的不懈努力和探索,高斯留下的数学难题也激励着数学家们不断挑战和突破自己的思维边界。
在数学的广阔领域里,高斯的数学难题仍然是许多数学家和研究者的探索目标和挑战,也为数学的发
展提供了无限的可能性和希望。
愿数学的光芒继续照耀着人类的智慧和未来,让数学的奥秘和魅力永不衰竭。
高斯错误修改总结讲解

A list of error messages and possible solutions -Gaussian calculations can fail with various error messages. Some error messages from .out and .log files - and possible solutions - have been compiled here to facilitate problem solving.-These are divided into:-Syntax and similar errors-语法类错误Memory and similar errors-内存类错误Convergence problems -不收敛错误Errors in solvent calculations -溶剂中的计算错误Errors in log files-错误文件-ERROR MESSAGES IN OUTPUT FILES-Syntax and similar errors:End of file in ZSymb.-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l101.exe Solution: The blank line after the coordinate section in the .inp file is missing. (输入文件空行丢失)Unrecognized layer "X".-(不识别层X)Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l101.exeSolution: Error due to syntax error(s) in coordinate section (check carefully). If error is "^M", it is caused by DOS end-of-line characters (e.g. if coordinates were written under Windows). Remove ^M from line ends using e.g. emacs. To process .inp files from command line, use sed -i 's/^M//' File.inp (Important: command does not work if ^M is written as characters - generate ^M on command line using ctrl-V ctrl-M).-QPERR --- A SYNTAX ERROR WAS DETECTED IN THE INPUT LINE.-Solution: Check .inp carefully for syntax errors in keywords -RdChkP: Unable to locate IRWF=0 Number= 522.-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l401.exe or-FileIO operation on non-existent file.-[...] Error termination in NtrErr:-NtrErr Called from FileIO.Solution: Operation on .chk file was specified (e.g.geom=check, opt=restart), but .chk was not found. Check that:-%chk= was specifed in .inp-.chk has the same name as .inp-.chk is in the same directory as .inp -run script transports .chk to temporary folder upon job start. Run scripts downloaded here should do this. -The combination of multiplicity N and M electrons is impossible.-(多重性)Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l301.exeSolution: Either the charge or the multiplicity of the molecule was not specified correctlyin .inp.-(电荷和多重性指定错误)Memory and similar errors: Out-of-memory error in routine RdGeom-1 (IEnd= 1200001 MxCore= 2500)-Use %mem=N MW to provide the minimum amount of memory required to complete this step-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l101.exe or-Not enough memory to run CalDSu, short by 1000000 words.-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l401.exe or-[...] allocation failure: -(表示配分失败)Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l1502.exe Solution: Specify more memory in .inp (%mem=Nmb). Possibly, also increase pvmem value in run script. Especially solvent calculations can exhibit allocation failures and explicit amounts of memory should be specified.-galloc: could not allocate memory.-(无法分配内存)Solution: The %mem value in .inp is higher than pvmem value in run script. Increase pvmem or decrease %mem. -Probably out of disk space(磁盘空间). Write error in NtrExt1 Solution: /scratch space is most likely full. Delete old files in temporary folder. -Convergence problems: Density matrix is not changing but DIIS error= 1.32D-06 CofLast= 1.18D-02.-(收敛问题)The SCF is confused. Error termination via Lnk1e in/global/apps/gaussian/g03.e01/g03/linda-exe/l502.exel Solution: Problem with DIIS. Turn it off completely, e.g. using SCF=qc, or partly by usingSCF=(maxconventionalcycles=N,xqc), where N is the number of steps DIIS should be used (see SCF keyword). -Convergence criterion not met. SCF Done: E(RHF) = NNNNNNN A.U. after 129 cycles -[...] Convergence failure -- run terminated. Error termination via Lnk1e in/global/apps/gaussian/g03.e01/g03/linda-exe/l502.exe Solution: One SCF cycle has a default of maximum 128 steps, and this was exceeded without convergence achieved. Possible solution: In the route section of input file, specify SCF=(MaxCycle=N), where N is the number of steps per SCF cycles. Alternatively, turn of DIIS (e.g. by SCF=qc) (see SCF keyword).--Problem with the distance matrix.-(距离矩阵)Error termination via Lnk1e in /pkg/gaussian/g03/l202.exe Solution: Try to restart optimization from a different input geometry. -(重新不同几何异构体的输入优化)New curvilinear step not converged(新曲线步骤不收敛). Error imposing constraints-Error termination via Lnk1e in /pkg/gaussian/g03/l103.exe-Solution: Problem with constrained coordinates (e.g. in OPT=modredun calculation). Try to restart optimization from a slightly different input geometry. -(一种稍微不同的输入几何)-Optimization stopped. -- Number of steps exceeded, NStep= N-[..] Error termination request processed by link 9999.-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l9999.exe Solution: Maximum number of optimization steps is twice the number of variables to be optimized. Try increasing the value by specifying OPT=(MaxCycle=N) in .inp file, where N is the number of optimization steps (see OPT keyword). Alternatively, try to start optimization from different geometry.--Errors in solvent calculations: AdVTs1: ISph= 2543 is engulfed by JSph= 2544 but Ae( 2543) is not yet zero!-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l301.exe Solution: Problem is related to building of the cavity in solvent calculations(溶剂效应优化计算错误). One possible solution is to change the cavity(腔) model (default in g03 is UAO, can be changed by adding RADII keyword in section below coordinates in the .inp file, e.g. RADII=UFF, see SCRF keyword).--Hydrogen X has 2 bounds. Keep it explicit at all point on the-potential energy surface to get meaningful results.Solution: In UAO cavity model, spheres are placed on groups of atoms, with hydrogens assigned to the heavy atom, they are bound to. If assignment fails (e.g. because heavy atom-H bond is elongated), cavity building fails. Possible solutions: a) use cavity model that also assigns spheres to hydrogens (e.g. RADII=UFF) or b) Assign a sphere explicity on problematic H atom (use SPHEREONH=N, see SCRF keyword)--ERROR MESSAGES IN LOGFILES =>> PBS: job killed: wall time N exceeded limit M-signal number 15 received. Solution: Job did not finish within specified wall time. Retrieve .out and .chk files from temporary folder /global/work/$USER/$JOB (or $PBS_JOBID) and restart calculation if possible (using e.g. opt=restart or scf=restart). -cp: cannot stat $JOB.inp: No such file or directory Solution: The .inp file is not in the directory from where the job was submitted (or its name was misspelled during submission. If error reads: cp: cannot stat $JOB .inp .inp, the .inp file was submitted with extension).-ntsnet: unable to schedule the minimum N workers Solution: The value of %N proc Linda=N in the .inp file is higher than the number of nodes asked for during submission. Make sure these values match.Connection refused [...] died without ever signing in-Sign in timed out after 0 worker connections. Did not reach minimum (N), shutting downSolution: Error appears if you run parallel calculations but did not add this file to your $HOME directory: .tsnet.config containing only the line: Tsnet.Node.lindarsharg: ssh (see also guidelines for submission). -Density matrix is not changing but DIIS error - Suggested solutions1/- SCF=qc will probably solve the problem, albeit at a cost- Change the SCF converger to either SD, Quadratic or Fermi2/- lower the symmetry of optimize with and optimizewith the "nosymm" keywordI solved the problem using a variation on the first suggestion. Normally the scf took less than 80 cycles to converge. So i used scf=(Maxconventionalcycles=100,xqc) which resulted in a good compromise between using scf=qc and optimisation speed.In the case of the DIIS error the scf always took more than 100 cycles before the error,so by adding scf=(Maxconventionalcycles=100,xqc) the scf switched to qc after 100 cycles in the standard DIIS mode.l9999错误是优化圈数不够,把out文件保存成gjf,修改后接着优化。
高斯消元法的适用条件

高斯消元法的适用条件
高斯消元法是求解线性方程组的一种常见方法,但是它并不是适用于所有线性方程组的。
具体而言,高斯消元法适用的条件如下:
1. 系数矩阵必须是一个方阵,即行数和列数相等。
2. 系数矩阵必须是可逆的,即行列式不为零。
3. 系数矩阵的各个元素必须都是已知数或常数。
4. 方程组的个数必须等于未知数的个数。
如果以上条件都满足,那么高斯消元法就可以被应用于求解该线性方程组。
在实际应用中,我们还需要注意到以下一些问题:
1. 如果系数矩阵的某个元素非常小,甚至接近于零,那么在进
行消元的过程中会出现除以零的情况,导致计算错误。
因此,我们需要对这种情况进行特别的处理,比如采用交换行或列的方式来避免出现除以零的情况。
2. 如果系数矩阵中某些元素具有相同的值,那么在进行消元的
过程中会出现多解或无解的情况。
这时,我们需要对系数矩阵进行一些变换,使其符合高斯消元法的要求。
总的来说,高斯消元法是一种非常实用的求解线性方程组的方法,但是在应用时需要注意以上所述的条件和问题。
- 1 -。
高斯使用中的问题汇总

⾼斯使⽤中的问题汇总如何从下⾯的Gaussian输出⽂件中找出轨道系数及轨道能!!(新⼿多谢),请帮忙标出来求助]如何从下⾯的Gaussian输出⽂件中找出轨道系数及轨道能!!(新⼿多谢),请帮忙标出来The electronic state is 1-A1.Alpha occ. eigenvalues -- -20.58265 -11.33946 -1.39265 -0.87259 -0.69715 Alpha occ. eigenvalues -- -0.63950 -0.52294 -0.44073Alpha virt. eigenvalues -- 0.13573 0.24842 0.33338 0.37329 0.73660 Alpha virt. eigenvalues -- 0.80783 0.84685 0.94689 1.10445 1.10700 Alpha virt. eigenvalues -- 1.13937 1.27145 1.33529 1.62050 1.78192 Alpha virt. eigenvalues -- 1.79416 1.99239 2.18347 2.23684 2.45514 Alpha virt. eigenvalues -- 2.64513 2.87165 2.97616 3.27576 4.09792 Alpha virt. eigenvalues -- 4.47637Molecular Orbital Coefficients1 2 3 4 5(A1)--O (A1)--O (A1)--O (A1)--O (B2)--O EIGENVALUES -- -20.58265 -11.33946 -1.39265 -0.87259 -0.697151 1 C 1S 0.00000 0.99566 -0.11060 -0.16262 0.000002 2S 0.00047 0.02675 0.20981 0.33995 0.000003 2PX 0.00000 0.00000 0.00000 0.00000 0.000004 2PY 0.00000 0.00000 0.00000 0.00000 0.420175 2PZ -0.00007 0.00066 0.17259 -0.18451 0.000006 3S -0.00024 -0.00743 0.08051 0.31309 0.000007 3PX 0.00000 0.00000 0.00000 0.00000 0.000008 3PY 0.00000 0.00000 0.00000 0.00000 0.157609 3PZ -0.00048 0.00135 -0.01160 -0.07970 0.0000010 4XX -0.00002 -0.00272 -0.01628 -0.01333 0.0000011 4YY -0.00006 -0.00202 -0.01365 0.03019 0.0000012 4ZZ -0.00074 -0.00123 0.03302 -0.00166 0.0000013 4XY 0.00000 0.00000 0.00000 0.00000 0.0000014 4XZ 0.00000 0.00000 0.00000 0.00000 0.0000015 4YZ 0.00000 0.00000 0.00000 0.00000 -0.0139416 2 O 1S 0.99472 -0.00038 -0.19672 0.08889 0.0000017 2S 0.02094 0.00025 0.44184 -0.20351 0.0000018 2PX 0.00000 0.00000 0.00000 0.00000 0.0000019 2PY 0.00000 0.00000 0.00000 0.00000 0.3212220 2PZ -0.00153 -0.00029 -0.13537 -0.14216 0.0000021 3S 0.00436 -0.00058 0.37895 -0.27048 0.0000022 3PX 0.00000 0.00000 0.00000 0.00000 0.0000023 3PY 0.00000 0.00000 0.00000 0.00000 0.1797624 3PZ 0.00006 0.00108 -0.04718 -0.06799 0.0000025 4XX -0.00418 0.00015 -0.00022 -0.00041 0.0000026 4YY -0.00383 -0.00011 -0.00073 -0.00413 0.0000027 4ZZ -0.00356 -0.00019 0.01969 0.00906 0.0000028 4XY 0.00000 0.00000 0.00000 0.00000 0.0000029 4XZ 0.00000 0.00000 0.00000 0.00000 0.0000030 4YZ 0.00000 0.00000 0.00000 0.00000 -0.0233931 3 H 1S -0.00002 -0.00020 0.03017 0.17902 0.1908232 2S -0.00013 0.00210 -0.00537 0.06479 0.1202633 4 H 1S -0.00002 -0.00020 0.03017 0.17902 -0.1908234 2S -0.00013 0.00210 -0.00537 0.06479 -0.120266 7 8 9 10(A1)--O (B1)--O (B2)--O (B1)--V (A1)--V EIGENVALUES -- -0.63950 -0.52294 -0.44073 0.13573 0.248421 1 C 1S 0.01942 0.00000 0.00000 0.00000 -0.122122 2S -0.06075 0.00000 0.00000 0.00000 0.148963 2PX 0.00000 0.32517 0.00000 0.40259 0.000004 2PY 0.00000 0.00000 -0.19811 0.00000 0.000005 2PZ -0.37597 0.00000 0.00000 0.00000 -0.210866 3S 0.03971 0.00000 0.00000 0.00000 1.980967 3PX 0.00000 0.21231 0.00000 0.71124 0.000008 3PY 0.00000 0.00000 -0.04477 0.00000 0.000009 3PZ -0.08856 0.00000 0.00000 0.00000 -0.7497710 4XX 0.00549 0.00000 0.00000 0.00000 -0.0027311 4YY 0.02734 0.00000 0.00000 0.00000 -0.0126512 4ZZ -0.01933 0.00000 0.00000 0.00000 -0.0045913 4XY 0.00000 0.00000 0.00000 0.00000 0.0000014 4XZ 0.00000 0.03558 0.00000 -0.03288 0.0000015 4YZ 0.00000 0.00000 0.06035 0.00000 0.00000Sample Text相关回复:作者: lixiaona158 发布⽇期: 2008-04-03EIGENVALUES 后⾯的数字就是这个轨道对应的能量,但是它的单位是HF,⼀般使的时候需要换成电⼦福特,⽤这个系数乘27.2116就可以了。
高斯安装报错-上传版本
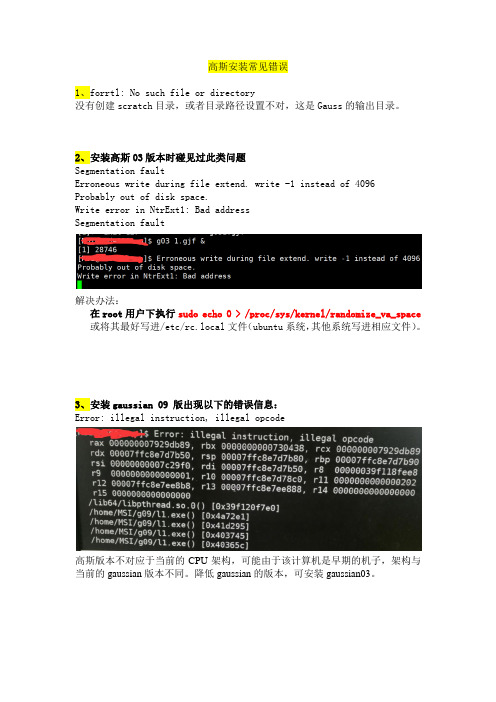
高斯安装常见错误
1、forrtl: No such file or directory
没有创建scratch目录,或者目录路径设置不对,这是Gauss的输出目录。
2、安装高斯03版本时碰见过此类问题
Segmentation fault
Erroneous write during file extend. write -1 instead of 4096
Probably out of disk space.
Write error in NtrExt1: Bad address
Segmentation fault
解决办法:
在root用户下执行sudo echo 0 > /proc/sys/kernel/randomize_va_space 或将其最好写进/etc/rc.local文件(ubuntu系统,其他系统写进相应文件)。
3、安装gaussian 09 版出现以下的错误信息:
Error: illegal instruction, illegal opcode
高斯版本不对应于当前的CPU架构,可能由于该计算机是早期的机子,架构与当前的gaussian版本不同。
降低gaussian的版本,可安装gaussian03。
4、错误
或者Last state="mem2"
TCursr= 260 LCursr= 10………………..
这是错误提示Last state="mem2"表示你的输入文件为dos格式而系统却是linux,故需用dos2unix转化后就可以了。
输入命令:dos2unix file (文件名)
2017.9.2。
高斯常见错误

近来一直在学习高斯,因为不精通常遇到各种错误。
结合自学的东西和查阅的资料总结出来一些错误,希望对和我一样的高斯初学者有所帮助。
1、Q:Error termination in NtrErr: ntran open failure returned to fopen. Segmentation faultE:Can't open a file.2、Q:Internal consistency error detected in FileIO for unit 1I= 4 J=0 I Fail= 1.E:Gaussian is limited to 16 GB of scratch space on the 32-bit nodes.3、Q:Out-of-memory error in routine UFChkP (IEnd= 12292175MxCore= 6291456)Use %Mem=12MW to provide the minimum amount of memory required to complete this step. Error termination via Lnk1e at Thu Feb 2 13:05:32 2006.E efault memory (6 MW, set in $GAUSS_MEMDEF) is too small for unfchk.4、Q:galloc: could not allocate memory.: Resource temporarily unavailableor Out-of-memory error in routine...or End of file in GetChg. Error termination via Lnk1e ...E:Not enough memory.5、Q:IMax=3 JMax=2 DiffMx= 0.00D+00Unable to allocate space to process matrices in G2DrvN:NAtomX= 58 NBasis= 762 NBas6D= 762 MDV1= 6291106 MinMem= 105955841.E:Gaussian has 6 MW free memory (MDV1) but requires at least 106 MW (MinMem).6、Q;Estimate disk for full transformation -677255533 words. Semi-Direct transformation. Bad length for file.E:MaxDisk has been set too low.7、Q:Error termination in NtrErr:NtrErr Called from FileIO.E:The calculation has exceeded the maximum limit of maxcyc.8、Q:Erroneous read. Read 0 instead of 6258688. fd = 4 g_readE:Disk quota or disk size exceeded. Could also be disk failure or NFS timeout.9、Q:Erroneous write. Write 8192 instead of 12288. fd = 4E:Disk quota or disk size exceeded. Could also be disk failure or NFS10、Q:orig len = 12288 left = 12288 g_writeE:timeout11、另有link错误:如:Error termination request processed by link 9999对于优化不收敛,即L9999错误,实际上是在规定的步数内没有完成优化,即还没有找到极小值点。
高斯计算常见错误及解决方案

GAUSSION计算常见错误及解决方案1. 自旋多重度错误2. 变量赋值为整数3. 变量没有赋值4. 键角小于等于0度,大于等于180度5. 分子描述后面没有空行6. 二面角判断错误,造成两个原子距离过近7. 分子描述一行内两次参考同一原子,或参考原子共线运行出错1. 自洽场不收敛 SCFa. 修改坐标,使之合理b. 改变初始猜 Guessc. 增加叠代次数SCFCYC=Nd. iop(5/13=1)2. 分子对称性改变a. 修改坐标,强制高对称性或放松对称性b. 给出精确的、对称性确定的角度和二面角c. 放松对称性判据 Symm=loosed. 不做对称性检查iop(2/16=1)3. 无法写大的Scratch文件RWFa. 劈裂RWF文件%rwf=loc1,size1,loc2,size2,……..,locN,-1b. 改变计算方法MP2=Direct可以少占硬盘空间c. 限制最大硬盘maxdisk=N GB4. FOPT出错原因是变量数与分子自由度数不相等。
可用POPT 或直接用OPT5. 优化过渡态只能做一个STEP 原因是负本征数目不对添加iop(1/11)=16. 组态相互作用计算中相关能叠代次数不够,增加叠代次数QCISD(Maxcyc=N)Default.Rou设置•在Scratch文件夹中的Default.Rou文件中设置G03程序运行的省缺参数:• -M- 200MW•-P- 4•-#- MaxDisk=10GB•-#- SCF=Conventional or Direct•-#- MP2=NoDirect or Direct•-#- OPTCYC=200•-#- SCFCYC=200•-#- IOPs 设置如iop(2/16=1)Default.Rou设置中的冲突•Default route: MaxDisk=2GB SCF=Direct MP2=Direct OPTCYC=200 SCFcyc=100 iop(2/16=1) iop(5/13=1)• ------------------• # ccsd/6-31G** opt• ------------------• L903/L905 and L906 can only do MP2.问题在于,MP2=Direct!去掉这个设置,CCSD的作业就能进行了。
高斯错误修改总结#精选.

A list of error messages and possible solutions -Gaussian calculations can fail with various error messages. Some error messages from .out and .log files - and possible solutions - have been compiled here to facilitate problem solving.-These are divided into:-Syntax and similar errors-语法类错误Memory and similar errors-内存类错误Convergence problems -不收敛错误Errors in solvent calculations -溶剂中的计算错误Errors in log files-错误文件-ERROR MESSAGES IN OUTPUT FILES-Syntax and similar errors:End of file in ZSymb.-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l101.exe Solution: The blank line after the coordinate section in the .inp file is missing. (输入文件空行丢失)Unrecognized layer "X".-(不识别层X)Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l101.exeSolution: Error due to syntax error(s) in coordinate section (check carefully). If error is "^M", it is caused by DOS end-of-line characters (e.g. if coordinates were written under Windows). Remove ^M from line ends using e.g. emacs. To process .inp files from command line, use sed -i 's/^M//' File.inp (Important: command does not work if ^M is written as characters - generate ^M on command line using ctrl-V ctrl-M).-QPERR --- A SYNTAX ERROR WAS DETECTED IN THE INPUT LINE.-Solution: Check .inp carefully for syntax errors in keywords -RdChkP: Unable to locate IRWF=0 Number= 522.-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l401.exe or-FileIO operation on non-existent file.-[...] Error termination in NtrErr:-NtrErr Called from FileIO.Solution: Operation on .chk file was specified (e.g.geom=check, opt=restart), but .chk was not found. Check that:-%chk= was specifed in .inp-.chk has the same name as .inp-.chk is in the same directory as .inp -run script transports .chk to temporary folder upon job start. Run scripts downloaded here should do this. -The combination of multiplicity N and M electrons is impossible.-(多重性)Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l301.exeSolution: Either the charge or the multiplicity of the molecule was not specified correctlyin .inp.-(电荷和多重性指定错误)Memory and similar errors: Out-of-memory error in routine RdGeom-1 (IEnd= 1200001 MxCore= 2500)-Use %mem=N MW to provide the minimum amount of memory required to complete this step-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l101.exe or-Not enough memory to run CalDSu, short by 1000000 words.-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l401.exe or-[...] allocation failure: -(表示配分失败)Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l1502.exe Solution: Specify more memory in .inp (%mem=Nmb). Possibly, also increase pvmem value in run script. Especially solvent calculations can exhibit allocation failures and explicit amounts of memory should be specified.-galloc: could not allocate memory.-(无法分配内存)Solution: The %mem value in .inp is higher than pvmem value in run script. Increase pvmem or decrease %mem. -Probably out of disk space(磁盘空间). Write error in NtrExt1 Solution: /scratch space is most likely full. Delete old files in temporary folder. -Convergence problems: Density matrix is not changing but DIIS error= 1.32D-06 CofLast= 1.18D-02.-(收敛问题)The SCF is confused. Error termination via Lnk1e in/global/apps/gaussian/g03.e01/g03/linda-exe/l502.exel Solution: Problem with DIIS. Turn it off completely, e.g. using SCF=qc, or partly by usingSCF=(maxconventionalcycles=N,xqc), where N is the number of steps DIIS should be used (see SCF keyword). -Convergence criterion not met. SCF Done: E(RHF) = NNNNNNN A.U. after 129 cycles -[...] Convergence failure -- run terminated. Error termination via Lnk1e in/global/apps/gaussian/g03.e01/g03/linda-exe/l502.exe Solution: One SCF cycle has a default of maximum 128 steps, and this was exceeded without convergence achieved. Possible solution: In the route section of input file, specify SCF=(MaxCycle=N), where N is the number of steps per SCF cycles. Alternatively, turn of DIIS (e.g. by SCF=qc) (see SCF keyword).--Problem with the distance matrix.-(距离矩阵)Error termination via Lnk1e in /pkg/gaussian/g03/l202.exe Solution: Try to restart optimization from a different input geometry. -(重新不同几何异构体的输入优化)New curvilinear step not converged(新曲线步骤不收敛). Error imposing constraints-Error termination via Lnk1e in /pkg/gaussian/g03/l103.exe-Solution: Problem with constrained coordinates (e.g. in OPT=modredun calculation). Try to restart optimization from a slightly different input geometry. -(一种稍微不同的输入几何)-Optimization stopped. -- Number of steps exceeded, NStep= N-[..] Error termination request processed by link 9999.-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l9999.exe Solution: Maximum number of optimization steps is twice the number of variables to be optimized. Try increasing the value by specifying OPT=(MaxCycle=N) in .inp file, where N is the number of optimization steps (see OPT keyword). Alternatively, try to start optimization from different geometry.--Errors in solvent calculations: AdVTs1: ISph= 2543 is engulfed by JSph= 2544 but Ae( 2543) is not yet zero!-Error termination via Lnk1e in /global/apps/gaussian/g03.e01/g03/l301.exe Solution: Problem is related to building of the cavity in solvent calculations(溶剂效应优化计算错误). One possible solution is to change the cavity(腔) model (default in g03is UAO, can be changed by adding RADII keyword in section below coordinates in the .inp file, e.g. RADII=UFF, see SCRF keyword).--Hydrogen X has 2 bounds. Keep it explicit at all point on the-potential energy surface to get meaningful results.Solution: In UAO cavity model, spheres are placed on groups of atoms, with hydrogens assigned to the heavy atom, they are bound to. If assignment fails (e.g. because heavy atom-H bond is elongated), cavity building fails. Possible solutions: a) use cavity model that also assigns spheres to hydrogens (e.g. RADII=UFF) or b) Assign a sphere explicity on problematic H atom (use SPHEREONH=N, see SCRF keyword)--ERROR MESSAGES IN LOGFILES =>> PBS: job killed: wall time N exceeded limit M-signal number 15 received. Solution: Job did not finish within specified wall time. Retrieve .out and .chk files from temporary folder /global/work/$USER/$JOB (or $PBS_JOBID) and restart calculation if possible (using e.g. opt=restart or scf=restart). -cp: cannot stat $JOB.inp: No such file or directory Solution: The .inp file is not in the directory from where the job was submitted (or its name was misspelled during submission. If error reads: cp: cannot stat $JOB .inp .inp, the .inp file was submitted with extension).-ntsnet: unable to schedule the minimum N workers Solution: The value of %N proc Linda=N in the .inp file is higher than the number of nodes asked for during submission. Make sure these values match.Connection refused [...] died without ever signing in-Sign in timed out after 0 worker connections. Did not reach minimum (N), shutting downSolution: Error appears if you run parallel calculations but did not add this file to your $HOME directory: .tsnet.config containing only the line: Tsnet.Node.lindarsharg: ssh (see also guidelines for submission). -Density matrix is not changing but DIIS error - Suggested solutions1/- SCF=qc will probably solve the problem, albeit at a cost- Change the SCF converger to either SD, Quadratic or Fermi2/- lower the symmetry of optimize with and optimizewith the "nosymm" keywordI solved the problem using a variation on the first suggestion. Normally the scf took less than 80 cycles to converge. So i used scf=(Maxconventionalcycles=100,xqc) which resulted in a good compromise between using scf=qc and optimisation speed.In the case of the DIIS error the scf always took more than 100 cycles before the error, so by adding scf=(Maxconventionalcycles=100,xqc) the scf switched to qc after 100 cycles in the standard DIIS mode.l9999错误是优化圈数不够,把out文件保存成gjf,修改后接着优化。
高斯消解 方程式不足

高斯消解方程式不足
高斯消元法是一种求解线性方程组的方法。
它的主要思想是通过逐次消元法将线性方程组化为等价的上三角形方程组,再求解这个上三角的方程组。
高斯消元法主要用于求解线性方程组的解,但由于算法相对复杂,因此常用于低阶稠密方程组。
高斯消元法的缺点是在消元过程中可能会出现误差较大甚至无法计算的现象。
这是因为在每次将主对角线上的元素化为1时,如果主对角线元素的绝对值很小时,就会产生很大的误差;如果主对角线元素为0时,则无法进行计算。
高斯中关于频率的几点问题

Gaussian 程序频率计算中的几个问题在Gaussian计算中,为了确定优化得到的几何结构是势能面上的局域极小点还是鞍点,或者要得到相关的热力学性质,经常需要对优化后的几何结构进行振动分析。
这里我们将讨论几个频率计算中常见的一些问题。
希望能对初学Gaussian的人有所帮助。
首先,原则上说,振动频率分析只对稳定结构有意义。
这里所说的稳定结构包括是势能面上的局域极小点和鞍点。
如下图1所示是一维自由度上的势能面,A和B处在势能面的局域极小点,而处在势能面的鞍点上。
他们在都处在平衡位置(原子核受力为零),不同的是,A和B来说离开平衡位置会受到指向平衡位置处的力,而C离开平衡位置会受到远离平衡位置的力。
因此A和B处在稳定平衡点,C处在不稳定平衡点。
实际上,一个分子可以有很多的自由度,如果在所有自由度上分子都处在稳定平衡,就是稳定的分子。
频率分析得结果是所有频率都是正的,表明这是一个局域的极小点。
如果分子只在一个自由度上处于不稳定平衡位置,其他自由度上都处在稳定平衡位置,说明该结构是一阶鞍点。
分子在稳定自由度方向上的振动才是真实的振动,在不稳定自由度方向上的实际上是不会有振动的。
不过我们可以对不稳定方向上的运动也按振动来做数学处理,会的到负的振动频率,我们称它为虚频。
虚频的出现表明该结构为鞍点。
图1 势能面上的局域极小点和鞍点第二,Gaussian计算中,频率的计算一定要在和分子结构优化相同的方法,基组下进行,否则计算的结果是没有意义的。
我们知道,任何理论水平下的计算,都是在一定的近似下进行的,不同的理论水平的近似程度是不同的。
在一种理论水平A下优化的稳定结构Geom_A会和另一种理论水平B下优化的稳定结构Geom_B有差别,也就是说Geom_A不会是理论水平B下的稳定结构。
根据前面我们所讨论的,在理论水平B下对一个不稳定的结构进行频率分析是没有意义的。
图2示意说明了不同理论水平下稳定点结构的不同。
图2 不同理论水平下优化的稳定结构是不同的第三,频率计算中可以考虑同位素效应(Freq=ReadIsotopes)。
高斯计算荧光能量误差

高斯计算荧光能量误差高斯计算荧光能量误差是指通过高斯方法计算得到的荧光能量与实际测量值之间的差异。
荧光能量是指物质受到光激发后发出的电磁辐射的能量。
高斯计算是一种理论计算方法,通过求解量子力学方程,可以预测物质的结构和性质。
在计算荧光能量时,高斯方法一般采用密度泛函理论(DFT)或者含时密度泛函理论(TDDFT)。
DFT方法利用波函数密度来描述物质的电子结构,TDDFT方法则考虑了电子的时间演化过程。
高斯计算也有其局限性,其中最大的问题之一就是计算误差。
高斯计算荧光能量误差主要有以下几个方面:荧光能量的计算需要精确的电子波函数描述。
在高斯计算中,电子波函数的精确描述受到基组的选择、基组尺寸和基组的形状等因素的影响。
如果基组选择不合适或者基组尺寸过小,那么得到的波函数可能与实际情况差异较大,从而导致荧光能量误差增大。
荧光能量计算中的近似方法可能引入误差。
高斯计算中常用的DFT 和TDDFT方法都是基于近似理论推导而来的,这些方法在处理电子间相互作用、电子关联效应等问题时都存在一定的近似。
这些近似方法可能无法精确描述体系的真实行为,从而导致荧光能量的计算结果存在误差。
还有,高斯计算中的数值精度也会对荧光能量的计算造成影响。
在计算中,常需要对电子波函数进行数值积分、矩阵对角化等操作,这些数值计算的精度直接影响到最终结果的精度。
如果数值精度不够,那么得到的荧光能量结果也会受到误差的影响。
除了以上几个方面,荧光能量计算误差还与实验条件的不确定性有关。
实际荧光实验中会存在仪器的误差、样品的不纯度、温度的变化等因素,这些因素都会对测量得到的荧光能量产生一定的影响。
如果高斯计算的结果与实验测量值有较大差异,那么很可能是实验条件的不确定性导致的。
为了减小荧光能量误差,可以采取以下几个方法:合理选择基组和基组尺寸。
基组的选择应该尽可能地包含物质体系中的电子行为,基组尺寸也应该足够大,以使计算结果更加准确。
可以尝试使用更精确的理论方法。
GeoIPAS使用中常见问题

GeoIPAS使用中常见问题1.数据检查数据检查的必要性,因为实验室的分析报告还是手工输入的,还是存在录入错误的,我们重点检查的是“>”,数据中间的空格等录入错误问题;另外还有畸变检查,数据的特大值,比如超过10倍变差,一般对这样的分析值实验室会很重视的,你也可以提出让他们再确认一下,做到心中有数。
另一类错误可能会是我们录入样号或者坐标时出现的错误,如:“56b”写成“56 b”,程序是以空格分开数据的,数据如果写成这样就会产生错误结果,有时在完成处理后才可能发现,这样一来我们前面的工作就作废了。
所以数据检查是非常必要的。
2.坐标投影变换在坐标投影变换和成图时经常出现的是将“源数据投影参数”的单位、比例尺弄错的情况,比如把数据直接转换成结果投影的单位等,这些是不需要做的,我们一般工作默认用的投影参数就是我们的地图参数,比如“投影平面直角坐标,北京54,高斯-克吕格投影坐标系”或者“投影平面直角坐标,西安80,高斯-克吕格投影坐标系”,实际工作的坐标单位一般用米,比如我们要成5万图,那参数设置就是:源数据投影参数,比例尺:1,坐标单位:米,13度带结果投影参数,比例尺:50000,坐标单位:毫米,13度带3.网格化一个工区的数据都要统一网格化,如计算模型、搜索范围和网格化参数,程序已经设置成批处理,可以一次全部做完。
这主要是为方了满足便组合异常,叠加处理时数据要求统一的要求。
问:在进行不规则网格化时,网格化参数、间距能采用默认值吗?为什么要取整?进行不规则网格化时,网格化参数中的默认值X、Y坐标为坐标数据的最小、最大值,初始行数设为50,初始的X间距为:(X最大值-X最小值)/50,Y间距等于X间距,然后(Y最大值-Y最小值)/Y间距,取整数作为列数,然后再如X 一样得到Y间距,这是程序先计算的一个初始值,而我们在实际工作中采样的密度是不同的,那我们在设定X、Y间距时就要按实际的密度来做调整,取整数这也是网格化时的一个要求,有时候按实际要求还有可能不是正方形的网格,而是矩形网格。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
优化1在优化时采用Scf(tight)的选项,增加收敛的标准。
再去计算频率。
如果还有虚频,参见下一步。
2.对称性的影响,很多情况下的虚频是由于分子本身的对称性造成的。
这样,在优化时,如果必要,要将对称性降低,还有,输入文件有时是用内坐标。
建议如果有虚频的话,将内坐标改成直角坐标优化。
3.如果上述方法还有虚频,看一下虚频,找到强度较大的,将在频率中产生的原子的振动坐标加到相应的输入文件中。
这样,重新计算。
直到虚频没有。
4实际上,如果分子柔性较大,很难找到最低点,这是电子结构计算的问题,这种情况下,需要动力学的东西,用构象搜寻的办法解决。
如:模拟退火,最陡下降法,淬火法等。
将得到的能量最低的构象做一般的电子结构计算,这样,应当没有问题。
不要讲你还没有得到最稳定的结构,那么,是你的分子有问题,要么计算错了,要么就是游离出现代计算的范畴1。
低频率振动模式时,力常数很小,必须使用opt=tight以确保适当的收敛和随后任务步骤中频率计算的可靠度。
比如我曾经算过一个体系,第一个频率振动的力常数只有10~20,很不放心,加入tight后,优出来的结构却与前面大相径庭,而且还出现了一个虚频。
2。
做IRC计算的时候,如果步长很小,必须要用verytight。
就曾有人步长设为1,而不加verytight,算出来的反应路径是v字形倒海鸥状的。
3。
使用int指定用于数值积分的积分网格来用于消虚频的时候,必须注意,在比较能量的时候,对所有的计算要使用相同的积分网格。
检查错误1.检查是否有初始文件错误在命令行中加入%kJob L301 or %kJob L302如果通过则一般初始文件ok。
常见初级错误:a.自旋多重度错误b.变量赋值为整数c.变量没有赋值或多重赋值d.键角小于等于0度,大于等于180度e.分子描述后面没有空行f.二面角判断错误,造成两个原子距离过近g.分子描述一行内两次参考同一原子,或参考原子共线2.SCF(自洽场)不收敛则一般是L502错误省却情况做64个cycle迭代(G03缺省128 cycles)a.修改坐标,使之合理b.改变初始猜Guess=Huckel或其他的,看Guess关键词。
c.增加叠代次数SCFCYC=N(对小分子作计算时最好不要增加,很可能结构不合理)d. iop(5/13=1)这样忽略不收敛,继续往下做。
3.分子对称性改变a.修改坐标,强制高对称性或放松对称性b.给出精确的、对称性确定的角度和二面角。
如CH4的角度给到109.47122c.放松对称性判据Symm=loosed.不做对称性检查iop(2/16=1)(最好加这个选项)iop(2/16=2)则保持新的对称性来计算4.Opt时收敛的问题a.修改坐标,使之合理b.增加叠代次数optcyc=N5.优化过渡态,若势能面太平缓,则不好找到。
iop(1/8=10)默认30(下一个结构和该结构的差别0.3Å),可改成10。
如果每一步都要用到小的步长,应该加opt(notrustupdate)6.在CI(组态)方法中如QCISD(T),CCSD(T),CID方法中,省却最大循环50,若出错(L913错误)解决方法:#P QCISD(maxcyc=N)注:N≤5127.优化过渡态opt=TS (给出过渡态)opt=qst2 (给出反应物和产物)opt=qst3 (给出反应物和产物和过渡态)a.用G03时的出错opt=ts必须加FC (force constant)写法:opt=(TS, calcFc)oropt=(TS,calchffc)计算HF力常数,对QCISD,CCSD等方法用;oropt=(TS,modRedundant)(最好写这个)b.如果计算采用QCISD计算(不好计算FC)则写为QCISD opt=(TS, calcHFFC) (用HF计算FC)8.无法写大的Scratch文件RWFa.劈裂RWF文件%rwf=loc1,size1,loc2,size2,……..,locN,-1b.改变计算方法MP2=Direct可以少占硬盘空间c.限制最大硬盘maxdisk=N GB,****MB,有些系统写2GB会出错,可以写2000MB9. FOPT出错原因是变量数与分子自由度数不相等。
可用POPT或直接用OPT10.优化过渡态只能做一个STEP原因是负本征数目不对添加iop(1/11)=1或者noeigentest过度态优化1.首先遇到的问题是,用哪种方法来寻找过渡态?GAUSSIAN提供的方法是QSTN和TSN方法。
两种方法各有优点和缺点。
QSTN方法特别QST3方法要求输入反应物,过渡态的猜测结构,产物这三者的结构。
特别麻烦。
但很管用,一般不会出现不收敛的情况。
对于TSN(对应关键词为OPT=TS)方法,只要求输入过渡态的初始结构,但这个初始结构非常的关键,如果结构不好,则很容易出现不收敛的情况。
所以我建议,如果是刚开始做过度态的话,用QSTN方法是好的选择,等有了“感觉”之后,再用TSN方法。
2.怎么解决经常出现的错误?在找过度态的时候,经常碰到的一些问题就是不收敛(1).,有一个错误的本征值(错误信息为:there isa wrong sign eigenvalue in hessian matrix.....)(2),和LINK9999错误导致退出。
(3)对于不收敛的情况,可以分为两类,比如提示信息里的CONVERGENCE FAILER提醒收敛到了10(-5),而此时你设定的SCF循环次数也仅仅是64步,那么完全有希望通过加大SCF循环次数来达到收敛的目的。
倘若只收敛到10(-3)或10(-2),此时加大循环次数可能就没用了。
结果还是CONVERGE FAILER。
此时可采用SCF=QC,来达到强制收敛的目的。
因为SCF=QC(LINK508)的计算量比默认的L502要大,所以不到万不得以就不用它了。
出现第二个错误可以直接用关键词OPT=NOEIGEN来实现。
LINK9999出错是因为已经走完了默认的步数,但还未完成。
系统会自动跳出。
出现这种情况大多数就是因为优化步数和SCF步数超过了默认值。
可用OPT(MAXCYCLE=100)和SCF(MAXCYCLE=300)来改错。
3.怎么样控制过渡态的优化,使得过渡态不至于收敛到其他的分子结构中去?我用GAUSSVIEW可以解决这个问题,当刚开始运行GAUSSIAN时,你用GVIEW去打开输出文件时,你可以看到你的过渡态的初始输入结构,当一个循环过后(从上一个LINK502到下一个LINK502),你再打开输出文件,你就可以清晰地看到优化一步后分子的构型,这样就可以随时监控过度态分子的结构,倘若已经有收敛到其他分子构型的趋势时,你就可以把它给KILL了,而不至于需要等全部工作结束后,打开输出文件才知道已经不是想要的过渡态了。
如果收敛到其它的构型上去,可以考虑缩小OPT的步长.iop(1/8=2或3)即可。
4.还需要加其它的关键词吗?建议在OPT中加入CALCFC。
这样可以加大找到过渡态的几率。
本人深有体会!先写这么多了,难免有错误和不恰当之处,还希望大家来指点和补充!虚频首先,什么是频率。
中学的时候我们学过简谐振动,对应的回复力是f=-kx,对应的能量曲线,是一个开口向上的二次函数E=kx^2/2.这样的振动,对应的x=0的点是能量极小值点(简单情况下也就是最小值点)。
这时的振动频率我们也会求:ω=2πsqrt(k/m)。
显然它是一个正的频率,也就是通常意义下的振动频率。
那么,一维情况下,如果能量曲线是一个开口向下的二次曲线呢?首先,从能量上看,这是个不稳定的点,中学的物理书上称为“不稳平衡”。
用现在的观点看,就是这一点导数是零(受力为0),且是能量极大值。
如果套用上面的公式,“回复力”f=-k'x(实际上已经不是回复,而是让x越来越远了),这里k'是个负数,ω=2πsqrt(k'/m)显然就是一个虚数了,即所谓的虚频。
Gaussian里面给出一个负的频率,就是对应这个虚频的。
实际情况下,分子的能量是一个高维的势能面,构型优化的时候,有时得到了极小值点,这样这个点的任意方向上,都可以近似为开口向上的二次函数,这样这里对应的振动频率就都是正的。
对于极大值点,在每个方向都是开口向下的二次函数,那么频率就会都是负的——当然一般优化很少会遇到这样的情况。
对于频率有正有负的情况,说明找到的点在某些方向上是极大值,有些方向上是极小值。
如果要得到稳定的能量最低构型,显然需要通过微调分子的构型,消去所有的虚频。
如何微调?要看虚频的振动方向。
想象着虚频对应的就是开口向下的二次函数,显然,把分子坐标按照振动的方向移动一点点,分子应该就可以顺着势能面找到新的稳定点,但是也不能太小。
而所谓的过渡态,则是连接反应物和产物之间的最低能量路径上的能量极大值。
好比山谷中的A,B两点,它们之间的一个小土丘,就是过渡态,从A到B的反应,需要越过的是这个小土丘,而不是两边的高山。
这样,过渡态就是在一个方向上是极大值,而在其它方向上都是极小值的点。
因此,过渡态只有一个虚频。
IRC在%section部分加上%chk=保存文件名其他的与一般输入的一样就可以啦。