第6讲 配合物的化学键理论-分子轨道理论
结构化学讲义教案6配位化合物的结构和性质

第六章配位化合物的结构和性质教学目的:通过学习,使学生对配位化合物的三大化学键理论(价键理论、晶体场理论、分子轨道理论)有所了解,并能够运用合适的理论对常见配合物的结构和性质进行理论分析和解释。
教学重点:1.晶体场理论;2.姜-泰勒效应;3.分子轨道理论。
引言:配位化合物简称配合物,又叫络合物,是一类含有中心金属原子(离子)(M)和若干配体(L) 的化合物(MLn)。
中心原子通常是过渡金属元素的原子或离子,具有空的价轨道;而配体则有一对或多对孤对电子。
在广泛的化学实践和量子化学巨大发展的基础上,提出了各种解释中心原子和配体之间化学键本质的理论,主要有价键理论、晶体场理论和分子轨道理论第一节价键理论1928年Pauling把杂化轨道理论应用到配合物中,提出了配合物的价键理论。
一、理论要点:配体的配位原子提供孤对电子进入中心原子(或离子)的空的杂化轨道形成配位键;配位键可分为电价配键和共价配键两种,相应的配合物叫做电价配合物和共价配合物。
二、杂化轨道与空间构型三、电价配键和共价配键1、电价配合物中心离子的电子层结构和自由离子的一样,它与配体是以静电作用力结合在一起,常采用spd外轨道杂化,形成高自旋配合物。
电价配合物特点:配体往往电负性大,不易给出孤电子对,中心离子的结构不发生变化。
配合物中配位键共价性较弱,离子性较强;键能小,不稳定,在水中易分解简单粒子;2、共价配合物中心离子腾出内层能量较低的空d轨道,进行dsp内轨道杂化,接受配体的孤对电子,形成低自旋共价配合物。
共价配合物特点:配体往往电负性较小,较易给出孤电子对,对中心离子的影响较大,使其结构发生变化。
配合物中配位键共价性较强,离子性较弱;由于(n-1)d轨道比nd轨道能量低,所以一般共价配合物比电价配合物稳定,在水溶液中不易解离为简单离子。
3.实验测定:通过测定络合物的磁化率,可判断中央离子与配体间化学键性质kTN x A 32μμ=, )()(反顺O M x x x +=μ磁矩cn ehn n e B B πμμμ4,)2(=+=(玻尔磁子) n 未成对电子数有摩尔磁化率X m 可计算络合物的磁矩μ,由μ可估算出n(未成对电子数),从而可判断此络合物是电价配键,或共价配键。
配合物的化学键理论

杂化
轨道 sp3d2 d2sp3
sp3
dsp2
配键 类型 外轨型 内轨型
外轨型
内轨型
Kf 1014
稳定性
<
1042
107. 96
1031. 3
<
磁性
Ni2+的d电子构型 杂化轨道 配键类型
未成对电子数 磁性
[Ni(NH3)4]2+ [Ni(CN)4]2 d8
sp3 外轨型
dsp2 内轨型
2 顺磁性
弱场配体
强场配体
——以上称为光谱化学序列
4. 电子成对能和配合物高、低自旋
电子在分裂后轨道上的分布遵循: 能量最低原理和洪特规则
如 Cr3+ d3
eg
E t2g
八面体场
d4d7构型的离子, d电子分布有高、低自旋两种方式。
如 Cr2+ d4
[Cr(H2O)6]2+
eg
△o t2g
[Cr(CN)6]4-
中心离子和配体之间以静电引力相互作用而形 成化学键。
中心离子的5个能量相同的d轨道受配体负电场 的排斥作用,发生能级分裂(有的轨道能量升 高,有的能量降低)。
2. 正八面体场中d轨道的能级分裂
无外电场作用下的d轨道 Edxy= Edxz= Edyz= Edx2-y2= Edz2
在带负电荷均匀球形场的作用下,d轨道能量 均升高相同值,能级不发生分裂。
请问: [Zn(NH3)4]2+、 [Ag(NH3)2]+呈现什么颜色?
中心离子d 轨道全空(d0)或全满(d10), 不能发生 d-d跃迁,其水合离子为无色。
解释配合物的稳定性
Eeg=+0.
分子结构和化学键的电子分子轨道理论

分子结构和化学键的电子分子轨道理论化学是研究物质变化和性质的科学,而分子结构和化学键的电子分子轨道理论是解释和预测分子性质的重要工具。
本文将探讨分子结构的形成以及化学键的形成机制,并介绍电子分子轨道理论在解释分子性质方面的应用。
一、分子结构的形成分子结构的形成是由原子之间的相互作用而产生的。
原子通过共用、转移或共享电子来形成分子。
其中最常见的是共价键和离子键。
共价键是由两个非金属原子共享电子而形成的。
在共价键中,原子通过共享电子对来达到稳定的电子构型。
例如,氢气分子(H2)中的两个氢原子通过共享一个电子对形成共价键。
共价键的强度取决于原子核吸引电子的能力和电子云的形状。
离子键是由金属和非金属原子之间的电荷吸引力而形成的。
在离子键中,金属原子失去电子,形成正离子,而非金属原子获得电子,形成负离子。
正负离子之间的电荷吸引力形成了离子键。
例如,氯化钠(NaCl)中的钠离子和氯离子通过离子键结合在一起。
二、化学键的形成机制化学键的形成机制可以通过量子力学的电子分子轨道理论来解释。
电子分子轨道理论认为,分子中的电子存在于分子轨道中,这些分子轨道是由原子轨道线性组合而成的。
共价键的形成可以通过氢原子的轨道重叠来解释。
当两个氢原子靠近时,它们的1s轨道发生重叠,形成一个σ(sigma)键。
σ键是最强的共价键,因为它是通过头对头重叠形成的。
离子键的形成可以通过金属和非金属原子之间的电子转移来解释。
金属原子失去一个或多个电子,形成正离子,而非金属原子接受这些电子,形成负离子。
正负离子之间的电荷吸引力形成了离子键。
三、电子分子轨道理论的应用电子分子轨道理论在解释分子性质方面有广泛的应用。
通过计算分子轨道的能量和形状,可以预测分子的稳定性和反应性。
例如,通过电子分子轨道理论可以解释分子的颜色。
分子吸收光的能力取决于分子轨道的能量差异。
当分子吸收特定波长的光子时,能量差异与光子能量相匹配,分子吸收光的颜色就会发生变化。
分子轨道理论

M
Cr
Mn
Fe
Co
Ni
价电子数 需要电子数
6 12
7 11
8 10
9 9
10 8
Ni(CO)4
形成的羰基配
位化合物
Cr(CO)6 Mn2(CO)10
Fe(CO)5 Co2(CO)8
谢
谢
!
e
g
配位 体群 轨道
反键MO
s
d
△ 非键MO σ
eg t1u
金属 a1g 络合物
成键 MO 配位体
分子轨道理论不像晶体场理论那样只考虑静电作用,也考虑 到了d轨道的能级分裂。
在晶体场理论中: 其差别在于: 分子轨道理论中:
E
0
eg
Et Et
2g
E
0
e g
2g
⑴ [FeF6]3-
中心金属和配位体之间σ配键和反馈∏键的形成是同时进 行的,而且σ配键的形成增加了中心原子的负电荷,对反馈 ∏键的形成更加有利,反馈∏键的形成则可减少中心原子的 负电荷,对σ配键的形成更加有利。两者互相促进,互相加 强,这就是协同效应。
大多数羰基配位化合物具有如下特点:
每个金属原子的价电子数和它周围配位体提供的价电子数加在
dx2-y2 dz2
eg Δ
这种π型轨道的形 成,使得体系的分裂能 Δ增大。 故,此类配合物常 是低自旋构型。 配体的π 空轨道
Δ=10 Dq
E0 3d
中央原子 轨道
t2g
dxy dxz dyz
t2g
受配位场微扰 d轨道分裂 分子轨道
例如,CN-、CO、NH3、NO2- 等就属于此类配体,其造
分子轨道理论

电子只填充在成键轨道中,能量比在原子轨道中低.这个能量差,就是分子轨道理论中化学键的本质.可用键级表示分子中键的个数:
键级= (成键电子数-反键电子数)/2
H2分子中,键级= (2 - 0)/2 = 1,单键
由于填充满了一对成键轨道和反键轨道,故分子的能量与原子单独存在时能量相等.故He2不存在,键级为零,He之间无化学键。
分子轨道理论
1.分子轨道理论在化学键理论中的位置
分子轨道理论是现代共价键理论的一个分支。其与现代共价键理论的重要区别在于,分子轨道理论认为原子轨道组合成分子轨道,电子在分子轨道中填充、运动。而现代共价键理论则讨论原子轨道,认为电子在原子轨道中运动。
2.理论要点
1)分子轨道由原子轨道线性组合而成
分子轨道的数目与参与组合的原子轨道数目相等。H2中的两个H有两个 1s,可组合成两个分子轨道。
4.异核双原子分子
非键轨道:无对应的(能量相近,对称性匹配)的原子轨道,直接形成的分子轨道.注意:非键轨道是分子轨道,不再属于提供的原子.
H的1s与F的1s, 2s能量差大,不能形成有效分子轨道.所以F的1s, 2s仍保持原子轨道的能量,对HF的形成不起作用,称非键轨道,分别为1σ和2σ.
当H和F沿x轴接近时,H的1s和F的2px对称性相同,能量相近( F的I1比H的I1大,故能量高),组成一对分子轨道3σ和4σ(反键),而2py和2pz由于对称性不符合,也形成非键轨道,即1π和2π.
4)分子轨道中的电子排布
分子中的所有电子属于整个的分子,在分子轨道中依能量由低到高的次序排布,同样遵循能量最低原理,保里原理和洪特规则。
3.同核双原子分子
1)分子轨道能级2, C2等分子.
化学键与分子轨道理论

分子轨道理论:分 子轨道理论的发展 趋势是向着更精确、 更全面的方向发展, 包括对分子电子结 构和性质更准确的 计算和预测。
计算方法的改进:随着计算机技术的进步,计算方法不断优化,使得分子轨道理论计 算更加准确和高效。
实验验证的加强:随着实验技术的不断发展,对分子轨道理论的实验验证越来越精 确,有助于理论的发展和完善。
化学键理论的优点:简单易懂,能 够解释许多化学现象。
分子轨道理论的优点:能够更准确 地描述电子云的分布和跃迁等化学 现象,预测分子的性质。
添加标题
添加标题
添加标题
添加标题
化学键理论的缺点:无法解释分子 轨道理论中的某些现象,如电子云 的分布和跃迁等。
分子轨道理论的缺点:较为复杂, 需要较高的数学基础和计算能力。
跨学科的融合:分子轨道理论正与其他学科领域如量子化学、生物学、环境科学等相 互融合,拓展了其应用范围。
人工智能的应用:人工智能技术正在逐渐应用于分子轨道理论的研究中,为理论的 发展提供了新的思路和方法。
化学键与分子轨道理论在药 物设计中的交叉发展
化学键与分子轨道理论在材 料科学中的应用
化学键与分子轨道理论在能 源领域的应用与交叉发展
添加标题
添加标题
添加标题
添加标题
原子轨道:构成分子轨道的基本单 元
分子轨道对称性:决定分子在空间 中的对称性和旋光性
分子轨道理论 的基本概念
电子填充规律
分子轨道的能 级顺序
分子轨道能级 的实际应用
分子轨道的对称元素:包括对称轴、对称面和对称中心等 对称性分类:根据分子轨道的对称性,可以将分子轨道分为对称与反对称两类
局限性:无法解释某些物质的性质,例如共价键和金属键的形成机制。
第六部分-配位键理论-1

试画出[BeF4]2–或[Be(H2O) 4]2+的结构。
排布没有变化,配位原子的孤对电子填在由外层轨道杂 化而得的杂化轨道上。这样一类络合物叫外轨型配合物 (Outer orbital complexes)。
同样是四配位,但对络合物[Ni(CN)4]2–就成了另一回事
[Ni(CN)4]2–的结构 3d
dsp2杂(CN)4]2-形成之前和之后, 中心原子的d电子排布发
因而有人说, 过渡元素化学就是d电子的配位化学
试按照价键理论分析下列两个化合物中心原子可能采取 的杂化类型 并画出其杂化轨道图示,判断其分属何种配合物 (外轨型和内轨型配合物)
[FeF6]3-
[Fe(CN)6]3–
配合物中的化学键一直是化学家们十分感兴趣的 一个领域,为了解释配位化合物中中心离子与配位体 间的成键本质,化学家们进行了长期的探索,先后提 出过多种理论。
有效原子序数规则
20世纪30年代初,鲍林将价键理论应用于配合物结构,能够解释一些 问题,但有些问题不能解释。 20世纪50年代,引入晶体场理论和分子轨道理论解释配合物中的化学 结合和化学结构,形成了配位场理论。
化学键概念经历了三个发展阶段:
经典理论阶段:仅仅指出了化学结合现象,并把这种结合现象称之 为化学键;
配位化合物的基本理论
➢价键理论(VBT) ➢分子轨道理论(MOT) ➢晶体场理论(CFT) ➢配位场理论(LFT)
配合物分子轨道理

Cr与CO的5σ轨道形成6个σ分子轨道,由6个 CO的12个5σ电子填充。同时Cr的一个电子被推
3d上去,使Cr有6个d电子。这6个d电子占用的
t2g轨道与配体CO的高能空π轨道形成的
t2gSALC组成成键π分子轨道。这样成键t1g分子
轨道由Cr的6个d电子占据。这相当于把中心Cr 原子的d电子送回到配体空轨道上去,使中心原 子上的过量负电荷减少,这样的π键叫反馈π键。
2 、环多烯配合物
许多环多烯具有离域π键的结构,离域π键 可以 作为一个整体和中心金属原子通过多中心π 键形成 环丙烯基 环丁二烯 环戊二烯基 苯 配位化合物。平面对称的环多烯有很多,下 C4H4 C6H6 图只列 C3H3 C5H5 出其中一些比较常见的环多烯的结构式和电 M M 子数。 M
M
3 、夹心化合物—二茂铁的结构
2、配体的π轨道是高能的空轨道。低能的
中心离子t2g轨道中的电子将进入成键π分 子轨道,使中心离子的t2g轨道能量下降, 增大Δ值,形成低自旋配合物。如P、As、 CN--、CO等都属于这种g *
π*
t2g
t2g M ML L
四、 σ– π键和羰基配合物的结构
分子轨道能级图
三、正八面体配合物中的π配键
1、配体的π轨道是低能的占有轨道,其 中的电子和高能的金属离子的t2g轨道中 的电子将在填满成键π分子轨道后,进 入反键的π*分子轨道,结果是降低了分 裂能Δ值。这类配合物都是高自旋 。如 卤素离子,H2O等配体.
eg *
eg *
Δo
Δo
t2g* t2g π t2g M ML L
CO的价电子组态: (1 σ )2(2 σ )2( 1π )4(3 σ )2(2 π )0 电子组态: kk(3 σ )2(4 σ )2( 1π )4(5 σ )2(2 π )0
配合物的化学键理论(讲义)

在不同场中的分裂情况:正方形>八面体>四面体
四面体场
八面体场
四方形场
分裂能与中心离子的关系电荷Z增大,增大;主量子数n 增大, o增大,3d < 4d < 5d。 例如:
[Cr(H2O)6]3+ o /cm-1 o /cm-1 o /cm-1 17600 [Fe(H2O)6]3+ 13700 [Cr(H2O)6]2+ 14000 [Fe(H2O)6]2+ 10400
[CrCl6]313600
[MoCl6]319200
分裂能与配位体的关系:光谱化学序列
[CoF6]3- [Co(H2O)6]3+ [Co(NH3)6]3+ o/cm-1 13000 18600 22900 [Co(CN)6]334000
各种配体对同一中心离子的晶体场分裂能的值由小到大的 顺序:
4.价键理论的局限性 配离子的杂化轨道类型,说明了配离子的空间构型和 配位数,以及配合物之间稳定性的差异。 (1)可以解释[Co(CN)6]4- 易被氧化[Co(CN)6]3- 但无法 解释[Cu(NH3)4]2+结构稳定的事实。 (2)对配合物产生高低自旋的解释过于牵强。 (3)无法解释配离子的稳定性与中心离子电子构型之 间的关系,未考虑配体对中心离子的影响。
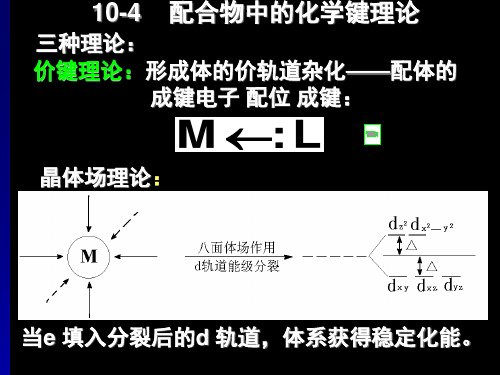
二、 晶体场理论
晶体场理论要点: 在配合物中,中心离子M处于带电的配位体L形 成的静电场中,二者完全靠静电作用结合在一起。 配位体对中心离子的影响 (a)中心离子M与配位体L成键时,配体的静电 场对中心离子的d 轨道电子的不同排 斥作用力, 使d 轨道能级发生了分裂。
(b)过渡金属的中心离子的5个d轨道在假想的球形 场(均匀电场) 中能量是简并的, 受到配位体负电 场作用时,会发生d 轨道的能级分裂。 晶体场理论的核心是配位体的静电场与中心离子 的作用引起的d轨道的分裂和d电子进入低能轨 道时所产生的稳定化能。 分裂类型与空间构型有关。
配合物中的化学键理论
④、成键过程:
17
[Ag(NH3)2]+的形成过程 解:Ag+的价电子构型为 4d10 5s0
5p 5s 4d
↑↓ ↑↓ ↑↓ ↑↓ ↑↓
SP杂化 5p
4d
↑↓ ↑↓ ↑↓ ↑↓ ↑↓
sp
:NH3 :NH3
↑↓ ↑↓ ↑↓
5p
2NH3
4d
↑↓ ↑↓ ↑↓ ↑↓
重叠
返回3
18
例:
[Ni(NH3)4]2+的形成 。
↑ ↑
3d
↑ ↑ ↑
4d
SP 3d2 杂化
3d
↑ ↑ ↑ ↑ ↑
sp3d2
23
6F重叠
4d
:F- :F- :F- :F- :F- ↑↓ ↑↓ ↑↓ ↑↓ ↑↓
↑ ↑
3d
↑ ↑ ↑
:F- ↑↓
sp3d2
返回5
24
例: [Fe(CN)6]3-的形成。 解:Fe3+ 的价电子构型为
4S 3d
↑ ↑ ↑ ↑ ↑
③规律:中心离子 SP3d2 与d2SP 3 杂化, 配离子的空间构型均为正八面体形。
7
3-
3-
3、 外轨型配合物和内轨型配合物 ①、外轨型配合物:
A、定义:指形成配合物时,中心离子全部采用 外层空轨道(ns, np, nd)进行杂化,并与配体结 合而形成的配合物。
B、特点:
a、中心离子仅采用外层空轨道(ns, np, nd) 进行杂化成键。 b、杂化类型为:sp3和sp3d2杂化。 c、配合物有较多的未成对电子。
4
B、以dsp2杂化轨道成键:
例:
成键结果分析比较: 2+ 2①Ni(NH3)4 ②Ni(CN)4 M 用以杂 4s 4p (4-1)d 4s 4s 化的轨道: ns np (n-1)d ns np 杂化特点:全部用外层轨道 使用内层轨道和 外层轨道 成键类型: 外轨配键 内轨配键 配合物的类型: 外轨型 内轨型 成单电子状态: 高自旋 低自旋 空间构型 正四面体 平面正方形
分子轨道理论介绍课件

未来研究方向
01
量子化学 计算方法 的发展
02
电子结构 理论的完 善
03
化学反应 机理的研 究
04
生物大分 子结构的 研究
分子轨道理论在无 机化学中的地位
● 原子结构:原子的电子排布和化学键的形成 ● 化学平衡:化学反应的平衡条件和平衡常数 ● 热力学:化学反应的热力学数据和热力学函数 ● 动力学:化学反应的速率和机理 ● 电化学:电化学反应的机理和电极电势 ● 结构化学:分子和晶体的结构和性质 ● 配位化学:配位化合物的形成和性质 ● 光谱学:原子和分子的光谱分析和结构鉴定 ● 量子化学:量子力学在化学中的应用和计算化学 ● 分子轨道理论:分子轨道理论的基本概念和原理,以及其在无机化学中的应用
04
预测分子稳定性和 反应活性
化学反应机理分析
分子轨道理论 可以解释化学 反应的机理
01
分子轨道理论 可以解释化学 反应的立体选 择性
03
02
通过分子轨道 理论可以预测 化学反应的产 物和速率
04
分子轨道理论 可以解释化学 反应的电子效 应
光谱学解释
分子轨道理论可以解释分 子的电子能级和光谱性质
分子轨道:原 子轨道的组合, 形成分子轨道
3
排布方式:根 据原子轨道的 能级和形状进
行排布
2
填充规则:遵 循能量最低原 理和保里不相
容原理
4
影响因素:分 子轨道的能级、 形状和重叠程
度
分子轨道理论的 应用
分子结构预测
01
利用分子轨道理论预 测分子的几何构型
02
预测分子轨道能级 和电子排布
03
预测分子光谱性质 和化学反应性质
04 指导无机材料设计和合成: 分子轨道理论可以指导无机 材料的设计和合成,为无机 化学研究和应用提供新的材 料和方法。
配合物-分子轨道理

t2g
M
eg*
Δo
t2g*
t2g ML
π L
2、配体的π轨道是高能的空轨道。低能的 中心离子t2g轨道中的电子将进入成键π分 子轨道,使中心离子的t2g轨道能量下降, 增大Δ值,形成低自旋配合物。如P、As、 CN--、CO等都属于这种配体。
eg* Δo
t2g
M
t2g*
eg* Δo
t2g ML
1 、不饱和烃络合物
此类化合物是以不饱和烃为配位体,通过
σ-π
配键与过渡金属形成的配位化合物。最早
制得
的此类配位化合物是 K[PtCl3(C2H4)]•H2O ,
称
Cl
为Zeise盐(1827年21,.
Zeise首先P 制得OC 2)15p,m C
其
Cl Pt
结构示于下图C。l
135pm
6、4 分子轨道理论
一、要点:用分子轨道理论和方法处理金属离子和 配体的成键作用:
cM M cLL
要满足对称性匹配,轨道最大重叠,能级相近 二、正八面体配合物中的σ-配键
对称性匹配线性组合:配位σ轨道能级比中心轨道 低,所以成键 σ轨道又要表现为配位σ轨道的性质,而 反键轨道则可以近似地认为2p金属轨道。故
π* L
四、 σ– π键和羰基配合物的结构
当中心金属是叁价或负价时,则晶体场作用对配 合物稳定性的贡献就不会太大,而主要贡献应归 结为共价键的形成。这样,当配体时电子给予体 时,中心金属就会带上较多负电荷而阻止合配体 的进一步结合,这与试验事实不符。所以,人们 认为在金属和配体之间还有其他类型的键,即 σ– π键。
Cr与CO的5σ轨道形成6个σ分子轨道,由6个 CO的12个5σ电子填充。同时Cr的一个电子被推 3d上去,使Cr有6个d电子。这6个d电子占用的 t2g轨道与配体CO的高能空π轨道形成的 t2gSALC组成成键π分子轨道。这样成键t1g分子 轨道由Cr的6个d电子占据。这相当于把中心Cr 原子的d电子送回到配体空轨道上去,使中心原 子上的过量负电荷减少,这样的π键叫反馈π键。
第六章 配合物结构

的热力学稳定性有如下规律:
d1 d 2 d 3 d 4 d 5 d 6 d 7 d 8 d 9 d10
如[M(H2O)6]2+ ,M2+如为第一过度金属离子
H kj / mol 的变化规律如下图所示
H
Ca2+ Sc2+
Ti2+
V2+
Cr2+
Mn2+ Fe2+ Co2+ Ni2+
中Fe2+的 d电子排布
CN p ,
Fe ( CN )6 4中Fe2+的 d电子排布
eg 0 t2 g 6
3、配合物的紫外可见光谱:
值在 10000cm-1~30000cm-1 之间
而 可见光区 10000cm-1~25000cm-1 之间 d电子在分裂的d轨道中跃迁, d— d跃迁的频率在紫外区和 可见光区。所以,一般过渡金属配合物有颜色。
配键和反馈
电子授受配键。
配键的形成是协同进行的,生成
具有 型空轨道的配体如CO,CN-,-NO2等。
这类配合物满足18电子规则:
如 Cr的价电子数(6)+配体CO提供的电子数(6×2=12)=18 又如: Mn2(CO)10 Mn: kk3d54s2 CO: kk 5
2
7×2=14
配体:两个戊环的离域 分子轨道也具有 对称性,
Fe和两个C5H5 能形成 配键,生成二茂铁 配合物。
对称性匹配的原则,组成配合物的离域分子轨
道。结合晶体场理论,讨论配合物的结构和性能。
配合物是物质存在的一种形式,具有广泛的应用价值。 如 [Fe(CN)6]3[Ag(CnH2n)]+ (CO)5Mn Mn(CO)5
有机化学中的分子轨道理论

有机化学中的分子轨道理论在有机化学中,分子轨道理论是一种重要的理论工具,用于解释有机分子的化学性质和反应机理。
分子轨道理论基于量子力学的原理,通过计算和描述分子中电子的运动状态,从而揭示了分子中化学键的形成和断裂、化学反应的进行等重要现象。
本文将介绍有机化学中的分子轨道理论的基本概念、应用以及研究进展。
一、分子轨道理论的基本概念分子轨道理论是基于原子轨道的概念,原子轨道是描述单个原子中电子运动状态的函数。
在一个分子中,原子之间通过共价键形成连接。
根据量子力学的原理,分子中的电子不再局限于单个原子,而是在整个分子中运动。
因此,分子的电子状态需要用一组轨道来描述,这组轨道被称为分子轨道。
分子轨道可以通过线性组合原子轨道(Linear Combination ofAtomic Orbitals,简称LCAO)的方法得到。
LCAO方法假设分子中的分子轨道是由原子轨道线性组合而成的,即每个原子轨道会形成分子轨道的一部分。
通过线性组合的过程,得到的分子轨道既保留了原子轨道的主要特征,又反映了分子中电子的运动状态。
分子轨道可以分为成键轨道和反键轨道。
成键轨道是由原子轨道线性组合形成的,对分子中的共价键的形成起着积极的作用;而反键轨道则是在原子轨道的基础上得到的,它们对共价键的形成没有帮助,反而会削弱共价键。
在分子中,成键轨道和反键轨道总是呈成对存在,它们之间通过分子中的原子核进行相互作用,形成了稳定的分子。
二、分子轨道理论的应用分子轨道理论在有机化学中有着广泛的应用。
它可以通过分析分子轨道的能级和电子分布,预测有机分子的性质和反应行为。
1. 能级结构分子轨道理论可以帮助确定分子中的能级结构。
不同的分子轨道具有不同的能级,电子会填充在低能级的轨道中。
通过计算和实验,可以确定分子中各个分子轨道的能级顺序,从而预测有机分子的稳定性、光谱性质等重要特性。
2. 共价键的形成和断裂分子轨道理论解释了共价键的形成和断裂过程。
键合理论与分子轨道理论

键合理论与分子轨道理论键合理论和分子轨道理论是描述化学键形成和分子结构的理论模型。
它们在解释化学反应、分子和材料性质方面起着重要作用。
本文将介绍这两个理论的基本概念、原理以及在化学领域的应用。
一、键合理论键合理论是描述分子中化学键形成和键的特性的理论模型。
它最早由保罗·迪里克(Paul Dirac)和罗伯特·桑德斯·穆兹利(Robert Sanderson Mulliken)等人在20世纪初提出,并得到了后来物理学家林纳斯·鲍林和化学家福克特等人的进一步发展。
键合理论的基本概念是通过原子间的电子转移和共享来解释化学键的形成。
它假设原子之间的化学键是通过Valence Bond(价键)形成的,即原子轨道(Atomic Orbital)中的电子在键形成过程中重叠和共享。
键合理论通过描述原子轨道的叠加和电子的共享来解释化学键的强度和性质。
键合理论主要包括以下几个核心概念:1. 来自原子的轨道(Atomic Orbitals):键合理论中,原子间的键通过来自原子的轨道上的电子形成。
原子轨道可以分为s、p、d、f等不同类型,它们具有不同的形状和能量。
2. 电子的自旋(Spin):键合理论中的电子带有自旋,自旋可以是向上或向下的。
电子自旋的方向影响了键的稳定性,特别是对于键的强度和反应性有着显著的影响。
3. 原子轨道叠加和电子共享:键的形成是通过原子轨道之间的叠加和电子的共享。
当两个原子间的原子轨道叠加时,会形成键和反键轨道。
键轨道中的电子寿命相对较长,而反键轨道中的电子寿命较短。
4. 化学键类型:键合理论可以解释不同类型的化学键,包括共价键、离子键和金属键。
通过电子的共享、转移和交换,原子之间形成不同强度和性质的化学键。
二、分子轨道理论分子轨道理论是描述分子中电子结构和分子性质的理论模型。
它是对键合理论的一个扩展和发展,最早由罗伯特·穆兹利等人在20世纪30年代提出。
分子轨道理

分子轨道理分子轨道理论是化学中的一个重要概念,用于描述分子中原子之间的电子运动。
它在有机化学、无机化学和物理化学等领域中广泛应用。
分子轨道理论指出,原子在分子中的电子不再是属于单个原子的轨道,而是分布在整个分子中的一组分子轨道中。
分子轨道理论主要包括以下几个方面:1. 原子轨道的组合:分子中各个原子的原子轨道将组合成一个新的轨道,用于描述整个分子中的电子运动。
原子轨道的组合方式可以是线性组合,也可以是简单的加法和减法。
2. 分子轨道的分类:分子轨道可分为成键轨道、反键轨道和非成键轨道。
成键轨道是分子中电子密度最高的轨道,由原子轨道的积极相互作用形成。
反键轨道是分子中电子密度最低的轨道,由原子轨道的消极相互作用形成。
非成键轨道则是分子中既不属于成键轨道也不与反键轨道有关联的轨道。
3. 轨道能级:分子中的分子轨道能级与原子轨道能级不同,原子轨道能级具有离散性,而分子轨道能级连续分布。
能级的顺序分别是成键轨道最低、反键轨道最高、非成键轨道在中间。
4. 轨道重叠:分子中的原子轨道之间会发生重叠,这会影响分子中的电子结构。
重叠程度越高,分子的稳定性就越高。
例如化学键就是由两个原子轨道之间的较强重叠形成的。
5. 分子轨道的描述:分子轨道可以用波函数来描述。
波函数可以用于计算分子中的能量、电子密度、电荷分布等物理性质。
在实际应用中,通常使用量子化学计算方法来获得分子轨道的波函数和参数。
总的来说,分子轨道理论为我们了解分子中的电子结构和化学反应提供了基础和框架。
在有机合成和物质设计中,分子轨道理论被广泛应用于分子的构建、反应性和化学性质的预测等方面。
配合物的分子轨道理论及配位场理论

个电子。
P(84kcal.mol −1) > ∆(40kcal.mol −1)
电子的排布为:
a12g
t16u
eg4
t
3 2g
e*g2
[Co(NH3)6]3+, P(60kcal.mol −1 ) < ∆(65kcal.mol −1 )
Co3+的价电子组态为d6,每个配位体L提供一对p电子,总 共 有18个电子
a12g
t16u
e
4 g
t
6 2gΒιβλιοθήκη e*g0未成对电子数n=0,是低自旋配合物。
配体可有三种类型的轨道: (a) 垂直于M—L所形成的 键轴的p轨道; (b) 与金属d轨道处于同一平面的配体的d轨道; (c) 与金属d轨道处于同一平面的配体的反键分子
轨道。
(a)
(b)
(c)
(1) 配位体的轨道是高能的空轨道。 增大了分裂能Δ值
如磷(P有3d轨道,但无3d电子)、砷(As有4d轨道, 但无4d电子);CO,CN-1等都属于这类配体,因而Δ值特别 大,是强场配位体,故常生成低自旋配合物。
±
1 23
(2σ 3
+
2σ 6
−σ1
−σ 2
−ο4
−σ5)
±
1 2
(σ 1
−σ4)
±
1 2
(ο 2
−σ5)
±
1 2
(ο 3
−σ6)
t2 g
3d xy ,3d xz ,3d yz
M
ML6
L
图6.2.2 配位化合物分子轨道能级图
例如,在[FeF6]3-中,Fe3+的价电子组态为d5,每个配位 体L提供一对p电子,共12个, 所以离域分子轨道中总共有17
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
除d9的Cu2+离子外,高自旋的d4型(如Cr2+ ,Mn3+), 及低自旋的d7型(Co2+ ,Ni3+)在形成六配位配合物时, 也有姜-泰勒效应,一般为拉长的八面体结构。
但也有例外,如[Cu(NO2)6]4 - [Mn(H2O)6] 3+都为 正八面体结构,没有姜-泰勒效应,原因还不清楚。
t2g
电子构型
洛阳师范学院
(2) o与配合物的颜色 溶液呈现不同的颜色,是由于溶液中有色粒子(分子 或离子)选择性的吸收某种颜色的光所引起的。
KMnO4溶液显紫色 K2CrO4溶液显黄色
洛阳师范学院
d1~d9构型过渡金属离子所生成的配合物一般具有颜 色,是由于eg或t2g轨道上有单电子,可以产生d-d跃迁。
o 改变引起颜色的改变: (颜色变深或变浅、变浓或变淡)
洛阳师范学院
[Ti(H2O)6]3+
在20400 cm-1附近有一最大吸收峰,这一吸收峰相当 于电子从t2geg,显示紫红色。 Ti3+(d1):在水溶液[Ti(H2O)6]3+配离子d电子先占据t2g 轨道,但吸收一定波长的可见光后,可以发生跃迁 t2geg轨道,这种跃迁所需能量为△。 ∴△0=10Dq = 20400 cm-1
②各种构型的CFSE均相等, 则此时三种构型都能稳 定存在。显然, 只有在d0、d10和弱场d5才有这种可能。 因此, 只有在d0、d5、d10及大体积配体时才生成Td 。
洛阳师范学院
从配合物的稳定化能大小可以看出,具有相同中 心原子和相同配位体的配合物,其稳定性应有如下顺 序:
平面正方形 > 正八面体 > 正四面体 但在配合物中,稳定化能与总键能(配位键的个 数)相比,只是很小的部分,只有当正八面体与平面 正方形两者的稳定化能差值很大时,才有可能形成平 面正方形配合物。 如:在弱场中d4 、 d9离子以及强场中的d8型离 子,两种构型的稳定化能差值最大,所以可形成平面 正方形构型的配合物, 如[Cu(H2O)4]2+ , [Cu(NH3)4]2+ , [Ni(CN)4]2- ,以及较重金属或带更高 电荷的金属离子Pt2+、 Au3+,可形成平面正方形结构。
洛阳师范学院
(6) 离子半径
120
r/ pm
100
M
M
80
Z*依次增大, r逐渐减小; Ca2+, Mn2+, Zn2+球形对称电子云; 其它d电子非球形分布。
HS
LS
HS d6 LS
60
V2+(d3),电子组态为t2g3eg0:由于t2g电子主要集中在远 离金属-配体键轴的区域,它提供了比球形分布的d电 子小得多的屏蔽作用, 故而半径进一步减小。 Cr2+(d4),电子组态为t2g3eg1:由于新增加的eg电子填入 位于金属-配体键轴区域,它的屏蔽作用增加,核对 配体的作用相应减小,故离子的半径有所增大。
洛阳师范学院
⑸ 配合物立体结构的变形现象 Jahn-Teller(姜-泰勒)效应
Oh场中电子在简并轨道中的不对称占据会导致分 子的几何构型发生畸变, 从而降低分子的对称性和轨 道的简并度, 使体系的能量进一步下降, 这种效应称为 Jahn-Teller效应。
通俗地讲:如果d 电子在eg和t2g轨道上呈不均匀分 布时,则配合物的构型要偏离理想构型,产生长、
短键之分。 Eg
d9: T2g
洛阳师范学院
例如:Cu2+(d9)八面体构型时:
有两种排布:
屏蔽作用
第一种 (dz2)2(dx2-y2)1
t2g6
能量有简并
第二种 (dz2)1(dx2-y2)2
t2g6
极限
极限
拉长八面体
平面正方形 压扁八面体
直线型
洛阳师范学院
● 结论: 较低能级(t2g)上d 电子不均匀时,发生小畸变。 较高能级(eg)上d 电子排布不均匀时,发生大畸变。
洛阳师范学院
第6讲 配合物的化学键理论
晶体场理论应用简介
洛阳师范学院
晶体场理论的核心
➢ d轨道分裂 ➢ o与P ➢ CFSE
洛阳师范学院
一、晶体场理论的应用
能解释配合物的磁性、稳定性、颜色、几何构型等。 (1) o与配合物的磁性
主要包括单电子的自旋运动和电子绕核的轨道运动。 在第一过渡系中,中心离子的d轨道受配体电场的影 响较大,而轨道运动对磁距的贡献被配体场所抑制, 因此,磁距主要是由电子的自旋运动所产生,可忽略 轨道运动对磁矩的贡献: m n(n 2)B n(n 2)e
把两者联系起来,可解释过渡金属配合物的稳定 次序:d0< d1 < d2 < d3 >or< d4 > d5 < d6 < d7 < d8 >or< d9 < d10。
洛阳师范学院
① 在八面体弱场中, d0、d5、d10处于全空、半 满、全满状态,晶体场稳定化能为0,因而生成的配 合物稳定性较差;
理想八面体
d 0 , d 3, t26ge*g2 , d10
d电子对
t23ge*g2 (HS),t26g (LS) Oh对称分布
变形八面体
大变形
t23ge*g1(HS ), t26ge*g3, t26ge*g1(LS ) t21g , t22g , t24ge*g2 (HS ), t25ge*g2 (HS ), t25g
பைடு நூலகம்
[Cu(H2O)4]2+吸收峰约在12600cm-1处(吸收橙红色光为 主),而显浅兰色;
[Cu(NH3)4]2+吸收峰约在15100cm-1处(吸收橙黄色光 为主),而显深兰色。
洛阳师范学院
解释:在光谱化学序列中,NH3位于水后面,NH3 是比H2O更强的配体,即△NH3 >△H2O。因此, [Cu(NH3)4]2+的吸收向短波(高波数)方向移动,发生 蓝移。
洛阳师范学院
八面体和四面体场只有在d0 、d10和弱场d5 ,两者 的稳定化能才相等,其余情况稳定化能都是八面体较 大。
∴在过渡金属的配合物中,八面体构型要比四面体 构型为多。
一般只有在下述情况时,才可能形成四面体构型:
①中心离子的电子构型为d0 、 d10 或d5 (弱场);
②配位体体积很大,在八面体场中过于拥挤;
CFSE
+4Dq +8Dq +12Dq +6Dq 0Dq +4Dq +8Dq +12Dq +6Dq 0Dq
排布
t2g1 t2g2 t2g3 t2g4 t2g5 t2g6 t2g6eg1 t2g6eg2 t2g6eg3 t2g6eg4
强场(LS)
单电子数 CFSE
1
+4Dq
2
+8Dq
3
+12Dq
2
+16Dq-P
② 在八面体弱场中,d3 、 d8离子的晶体场稳 定化能最大,其配合物就最稳定;
③ 在八面体强场中,d6 离子的晶体场稳定化能 最大,其配合物就最稳定;
④ d4 、 d9离子配合物的稳定性,有时也能大 于d3 、 d8的配合物,是由于姜-泰勒效应所引起的。
洛阳师范学院
⑷ 配合物的空间构型
M + mL → MLm
小变形
洛阳师范学院
发生Jahn-Teller效应可获得额外的稳定化能:
无论采用哪一种几何畸变, 都会引起能级的进一步 分裂, 消除简并, 从而获得额外的稳定化能。
洛阳师范学院
配位数为6的配合物,若中心离子的t2g、eg轨道 处于全空、半满或全满状态(t2g0eg0、t2g3eg0、 t2g 3eg2、t2g6eg0、t2g6eg2、 t2g6eg4六种情况 ),则d电子 的排布对八面体场是对称分布的。
d-d 跃迁:
E
0
h
h
c
0: 10000~30000 cm-1
可见光波数:14000~25000 cm-1。
所以 d-d 跃迁一般落在可见紫外光范 围,因此,过渡金属配合物绝大多数 是有颜色的。
洛阳师范学院
● 关于d-d 跃迁吸收强度
拉波特(Laporte)选择定则:
S=0
自旋定则
g↔u 但 g↔g u↔u 宇称定则
八面体配合物中 d 电子的排布和CFSE
组态
d1 d2 d3 d4 d5 d6 d7 d8 d9 d10
排布
t2g1 t2g2 t2g3 t2g3eg1 t2g3eg2 t2g4eg2 t2g5eg2 t2g6eg2 t2g6eg3 t2g6eg4
弱场(HS)
单电子数
1 2 3 4 5 4 3 2 1 0
在配合物磁性研究中,一般方法是:
m n 排布方式 成键性质
洛阳师范学院
对于正八面体型配合物, d1 、d2 、d3、d8 、d9 、d10 构型的中心离子只有一种排布方式。
但对于d4 ~ d7 构型的中心离子, 有高、低自旋两种排 布方式:
eg 弱场高自旋
(△小)
t2g
eg
强场低自旋
(△大)
Ti2+ Cr2+ Fe2+ Ni2+
Zn2+
Sc2+
V2+ Mn2+ Co2+ Cu2+
第一过渡系列金属离子(M2+)的水合热
洛阳师范学院
➢静电理论认为:随 d 电子数增多,Z*应增大,配 体(H2O) 与中心离子M2+的作用应单调增大,所以, 水合热也应是单调增大。