SQP质量验厂+GMP质量验厂审核所需文件及验厂详细要求解释以及验厂主要文件范本
GMP认证检查现场必备的文件、记录、档案、图纸等资料

一、文件管理档案⑴现行文件目录;⑵一套完整的存档受控文件并按文件编码分类排列;⑶文件发放记录、关键文件会审记录(如:文件管理程序、变更管理、偏差管理、物料管理、供应商管理、产品召回、退货等涉及部门多和规定范围广的文件);⑷文件变更台帐和变更记录;⑸过时、作废文件回收、销毁记录(过时、作废文件原件加盖过时或作废印章并与现行文件原件分开存放,以便专家追溯)。
二、质量档案:按《质量档案管理程序》执行,包括各品种生产批件三、印刷包材档案⑴印刷包材药监部门批件;⑵供应商印刷前彩稿(或墨稿)的QA签字审核件;⑶ 标准样张:① 包材实样经QA签字的盖章件和分发记录(旧版本留档一份并有回收和销毁记录);② 现行包材实样的分发:QA、QC、采购、库房、车间各一份,作为采购、验收、检验、使用的标准;四、机构职责⑴公司组织机构图、质量管理机构图、各部门/车间的组织机构图;⑵GMP认证领导小组、自检小组、供应商审计评估小组、药品质量风险管理小组、药品召回领导小组、变更控制小组、偏差处理领导小组、验证委员会组成人员名单及公司批准证明文件;⑶各级机构职能、岗位职责;员工花名册,可再细分管理人员列表、质量管理人员列表、⑷技术人员列表;内容包括编号、岗位、姓名、性别、出生年月、学历、毕业院校、专业、从药年限、职称、入司时间、备注;五、人事健康档案⑴ 人事档案:① 个人简历(包括毕业院校、专业和工作经验、从药年限)、毕业证、职称证;② 中层以上干部的任免文件,总经理授权副总主管产品放行人员的授权委托书,取样等指定人员的专项培训记录和授权书;③ 关键岗位外部资格证:化验员、锅炉工、电工、计量员、中药购销(验收)员;④ 主管质量的企业负责人、质量部门负责人、生产部门负责人的备案表等;⑵ 健康档案:①员工健康卡、历年县级以上单位健康体检证明(体检表、健康证等);②体检结果异常处理情况证明、复岗体检合格证明③注意:包括整个生产质量系统(生产、质量、库房、采购、动力、机修、行政后勤),容易漏掉清洁工、临时工、机修、厨师等;生产质量系统每年至少体检一次,其他建议两年一次;六、培训档案⑴公司培训档案:①公司培训规划、每年度培训计划、培训实施情况(培训记录)及培训效果评估表;②每次培训签到表和培训教材(若为书本或文件可不收集) ;③培训空白考卷(考试可为培训一次考一次,也可为培训一阶段考核一次,但文件培训必须在其生效日期前完成)⑵ 个人培训档案① 员工培训卡、考核试卷或其他方式的培训效果评价记录;② 外出培训总结、证明;③ 培训合格上岗证明、不合格调整工作岗位记录;④ 每人一档,企业分层次对全员培训。
GMP-审核需要的资料及程序文件

GMP-审核需要的资料及程序文件无菌医疗器械检查细则容GMP英文:Good Manufacturing Pratice,中文的意思:良好作业规(优良制造标准)。
医疗器械GMP(医疗器械生产质量管理规),它是一套适用于医疗器械行业的强制性标准,要求企业从原材料采购、人员、设施设备、生产过程、包装运输、设计开发等方面按国家有关法规形成一套可操作的作业规帮助企业改善企业生产环境,及时发现生产过程中存在的问题加以改善,确保最终产品的质量符合法规的要求。
一、总则为了加强医疗器械生产监督管理,规医疗器械生产质量管理体系,根据《医疗器械监督管理条例》和相关法规规定,制定本规。
医疗器械生产质量管理体系的基本准则,适用于医疗器械的设计开发、生产、销售和服务的全过程。
企业应根据产品的特点,按照本规的要求,简历质量管理体系,并保持有效运行。
作为质量管理体系的一个组成部分,生产企业应当在生产实现全过程中实施风险管理。
二、管理职责生产企业应建立相应的组织机构,规定各机构的职责、权限、明确质量管理职能。
生产管理部门和质量管理部门负责人不能互相兼任。
生产企业负责人应具有并履行的职责:(一)组织制定生产企业的质量方针和质量目标;(二)组织策划并确定产品实现过程,确保满足顾客要求;(三)确保质量管理体系有效运行所需的人力资源、基础设施和工作环境;(四)组织实施管理评审并保持记录;(五)制定专人和部门负责相关法律法规的手机,确保相应法律法规在生产企业部贯彻执行。
生产企业负责人应当确定一名管理者代表。
管理者代表负责建立、实施并保持质量管理体系,报告质量管理体系的运行情况和改进需求,提高员工满足法规和顾客要求的意识。
三、管理资源生产、技术和质量管理部门的负责人应当熟悉医疗器械相关法规,具有质量管理的实践经验,有能力对生产和质量管理中的实际问题作出正确的判断和处理。
从事影响产品质量工作的人员,应当经过相应的技术培训,具有相关理论知识和实际操作技能。
验厂所需资料

陳總經理:您好!關於驗廠,據我們現在拿到的資料所看,是非常嚴格,而且Detail,對我們目前而言,建立這樣的體系,工作量相當大,請趕快安排人建立,據我們的經驗而言,完全不能隻是依靠顧問公司幫我們搞定這件事。
以下是我們這邊根據GMP, TCPS,和WALMART驗廠資料與劉先生,副總開會總結出來的我們目前應該建立哪些方面的文件!以便達到目的!一、從這三個驗廠文件中我們不難看出他們的精髓在於:A. 做每一件事都要有記錄,並且要有方法確保其記錄的正確性;B. 要有每一個崗位的作業指導書,用以指導其崗位的操作,從文件中可以看到最重要的還不是總經理的作業指導書,而是底下員工,甚至工人的作業指導書;C. 工廠運作的每一個環節,都要有記錄,從進料檢驗開始,一直到出貨,這裡面涉及到很多方面,舉一個簡單的例子:很多工廠都在運行8S,來確保環境衛生的整潔,他會問到你怎麼確保你工廠天花板和掉燈的幹凈,誰負責清潔,多長時間進行一次,記錄在哪裡!二、質量記錄表單記錄時間:從現在開始往前推半年,即從2006年1月開始就要有相關資料的記錄;三、配備人員:目前劉先生指示你們那邊至少要有4名可以說漢語的文員一起來起草ISO文件及驗廠資料;四、以下是經我們討論,認為現在你們需要準備的崗位作業指導書應該具有那些要求(當然,你們需要根據你們的架構再做調整,但細節的問題不變)(一)採購部:A. 採購員(分深圳採購員和越南採購員)採購的項目分為:1、石蠟; 2.棕櫚蠟; 3.蠟芯; 4.蠟托; 5.香精; 6.色粉;7.AC6; 8. UV; 9.防火杯; 10.鋁杯; 11.紙盒; 12.紙卡; 13.PVC盒;14. glass holder; 15.metal holder;16. ceramic holder/plate/tart burner/pot,17. tin box; 18. carton (inner, master), 19增白劑, 20.各種decoration(包括RIBBON,BEADS等其它小配件); 21.bamboo container, 22 wooden container; 等要求:1.按照採購項目分類:(1)每種採購項目都需要建立編號(建議可以先按照大類分為不同代碼:如原料---RM、包裝物---PK,然後加流水號),而且編號要能夠進入將來的電腦採購系統,以便於倉管管理並按類別分發物料;(2)每種採購項目要求要有採購標準,每個採購項目都要有單頁的規定,這個標準最好設置成表格形式;採購記錄表單也按照這個採購標準來執行;(3)採購的項目要包括供應商的批號,採購單和CARTON箱上都要SHOW出來.(4)所有原材料採購的規格是否和供應商對材料的說明相一致?規定包括:-----物理、化學特性,例如:物理特性指採購物料的尺寸,重量,外觀等;化學特性指成份分析;-----強度及純凈度-----儲存條件,架子的壽命(每種採購回來的ITEM對存儲的要求)(5)成份的檢測,是否符合供應商也認同的要求?------檢測標準包括:-----圖片-----尺寸-----誤差-----物理特徵-----特別的運輸要求2〃有無供應商驗廠LIST ,需要建立供應商管理程序,要規定到對工廠的篩選、評選B. 倉管(倉庫管理員)倉管的項目分:1.配件;2.包裝;3.原料;4.半成品;5.成品要求:1〃有書面的庫存控制程式嗎?(要有入庫單)包括:(1)有無對入倉之產品進行記錄,對倉儲之產品加以區分及對入倉之產品進行跟蹤之程式?(如有確保最早被提供的產品或材料先被使用嗎?)(2)所有外來庫存及需處理的材料和部件都有進行防污染,混淆,和錯誤的保護措施嗎?(3)大貨有無按客人區分隔開,直到被驗貨通過?(4)原材料記錄保存包括:-----有無批號(次)-----有無被規定供應商的批號-----數量記錄-----檢驗記錄(5)庫存產品要有清楚的標識,如數量、合格狀態、供應商等(6)各類原材料存放標準?防潮、防蟲、防霉;與牆隔開的距離、溫度、濕度2. 半成品區要有亞克力板子制成的半成品區標識,上面要有中文、英文、越語,要有號碼,此號碼也是要能夠進入電腦系統;要設置不合格品隔離區(生產中的次品)3. 要求倉庫要有辦公室,要有電腦及放FILE的地方;C. 生管計劃:(生管計劃員)管理的項目:1.全廠的產能;2.每台機器的生產效率;3.每個工人的生產效率;要求:1.工廠的主要機械設備有多少台?列出設備清單,並對機器進行定期的維修保養,要有記錄;2.現有機器設備適合生產客人的產品嗎?主要生產的產品是什麼?3.為何使用外發廠?無法滿足生產量?商業的決定?無生產設備?列出外發商的名稱及相關聯系資料.及如何篩選。
验厂品质体系要求及文件

1 仓库空间,条件适合/通风、采光、摆放、清洁、安全性等
2 出入库记录,是否电脑化,是否包含名称、数量和不合格等相关记录
材 3 原材料出入库检验标准,检验系统,是否符合客人的要求 料 4 供应商考核,合格供应商名录 和 5 在生产过程中、仓存中,是否有合格品与不合格品的分类,产品分区
供 6 生产过程中的不合格品的返工和重检记录
管 4 品管部组织架构图
理 体 系
5 每天、周、月品质报表,主管签字确认/来料检验、半成品检验、成品检验、成品抽查等 6 每周品质例会记录表/生产经理或主管要参与
及 7 缺陷分类表
培 8 纠正措施表、客投诉资料分析处理
训 9 工厂QC的权限/发现不良品有权停止生产
10 独立的检验区域,条件适合/通风、采光、摆放、清洁等;
类别 序号 相应文件
1 ISO证书、CE证书
文 2 程序文件
件 3 质量手册
和 过
4 质量记录表格/测试报告、检测结果
程 5 内审记录
控 6 管理评审记录
制 7 外审记录 ,
8 公司组织架构图及公司简介
1 程序文件中包好的《质量检验控制程序》《生产过程记录表
内 6 机器设备台账 部 7 计量器具台账 测 8 计量器具检定计划 试 9 计量器具检定证书
10 内检人员资格证书
样 1 工厂是否有样品室,条件适合/通风、采光、摆放、清洁等 品 2 样品是否有经过检测、检验、分发记录表
开 3 样品设计明细表 发 4 是否以原版为参照,计算机模拟
1 生产车间是否条件适合,通风、采光、摆放、清洁等 2 备份电源发电机的管理程序,柴油管理程序(如适合或有此设备) 3 特殊岗位如割片、抛光、点焊、印字、车房等工序需佩戴耳塞、口罩、护目镜等防护工具 4 机器、设备维修计划、保养记录 5 每天或每周品质结果是否有在工厂张贴 6 生产排期/月排 7 检验环节/半成品、产品 日报表 8 产前会议记录、及分发/需包含生产经理或主管、品管;产品要求,试产样品的批准 9 试生产记录 10 交货期记录表 生 11 现场检验(半成品、成品)相关记录表,成品出货抽查记录表 产 12 AQL抽查标准,记录表中是否相符合(如适合) 车 13 机器状态标识 间 14 利器管理制度,利器收发记录 15 首件报告,首件办,纠正措施 16 水杯统一放置,统一服装 17 消防设施、应急灯的定期检查 18 电梯安全检验合格证 19 化学品的标识,二次防漏,清洗设备,MSDS 20 作业指导书 21 区域的标识 待产、合格、不合格等 22 温湿度记录表,清洁卫生自查表 23 急救药箱 24 金属检测设备及其使用指南
质量验厂全套程序文件1体系文件管理程序

质量验厂全套程序文件1体系文件管理程序质量验厂全套程序文件1体系文件管理程序一、背景介绍在当今竞争激烈的市场环境中,企业为了保持竞争优势,提高管理水平以及确保产品质量成为了至关重要的一环。
为了有效应对各种挑战,企业需要建立起一套完整的质量验厂程序。
这不仅有助于提高产品质量,还将有助于企业防范潜在风险,确保持续稳定的发展。
本文将详细介绍质量验厂全套程序文件中的体系文件管理程序。
二、体系文件概述体系文件是质量验厂全套程序文件中的重要组成部分,它涵盖了企业质量管理的各个方面,包括质量手册、程序文件、作业指导书和记录表格等。
体系文件管理程序明确了这些文件在编写、审批、发布、修订、保管和销毁等环节的管理要求,以确保文件内容的完整性、准确性和统一性。
三、程序文件管理流程1、文件的创建与修订:由各个部门负责创建和修订各自领域的体系文件,根据实际工作需求进行更新和完善。
2、文件审批:经过审批流程,确保文件内容与相关标准、规定以及企业实际情况相符。
审批流程应包括申请、审核、批准等环节。
3、文件发布:经过审批后的文件应正式发布,以供全体员工学习和执行。
4、文件修订:根据实际需要对文件进行修订,并按照审批流程对文件进行审核与批准。
5、文件保管与销毁:按照规定的保管期限对文件进行保存,对于过期的文件应进行销毁处理。
四、程序文件审批流程1、申请:创建或修订体系文件的部门向质量管理部门提出申请。
2、审批:质量管理部门对申请的体系文件进行审核,确保文件内容符合相关标准和规定。
3、编号与发布:经过审批后的体系文件应给予编号,并正式发布。
五、程序文件分发流程1、领取:各部门负责人向质量管理部门领取已发布的体系文件。
2、安装:各部门负责将体系文件放置于指定位置,确保员工能够方便地查阅。
3、培训:企业应组织相关培训,确保员工了解并掌握体系文件的内容和要求。
4、执行:员工应按照体系文件的规定执行各项操作,确保产品质量和生产过程的规范性。
工厂验厂需准备的资料

各部门验厂需准备的资料1、人事档案:内容有员工简历、身份证复印件(是本人,必须要有)和相关资格证书。
注解:员工入厂时的年龄要在18周岁以上,否则被视为招有未成年人或童工:未成年人是指年满16周岁未满18周岁的人,必须要到当地的劳动部门进行备案及体检,体检的费用全部有厂方支付直到年满18周岁以上,入厂进行体检一次,每隔半年要复查一次。
童工是指未年满16周岁的人,是严禁招聘的。
档案表中的年龄、地址、身份证号码要同身份证复印件一致,员工档案人数要同当时验厂的人数即考勤表、工资表、劳动合同、保险名单一致。
请假、休假、辞职等要有书面的记录(请假条、休假单、辞工书等),必须要同考勤表、工资表一致的记录。
2、员工手册:包含工厂简介、厂规厂纪、奖惩制度、安全管理制度、自愿加班指引、招聘管理制度、培训制度、考勤管理制度、薪酬管理制度、工资计算方案、有薪假期制度、晋升与降职制度、员工建议与投诉程序、请假制度、离职管理制度。
注解:奖惩制度里面不能有涉及到罚款及侵犯员工权益的内容,只能警告、记过。
3、工厂制度:不虐待与不骚扰政策、不歧视政策、不使用强迫劳工指引、行为规范管理制度、员工代表选举程序、童工拯救政策、未成年人保护政策、女职工劳动保护政策、宿舍管理规定、员工饭堂管理制度、综合环境健康安全程序、保安职责、化学品仓管理若干规定、急救的护理常识、紧急医疗救护计划、新员工培训计划、车间制度、消防演习方案。
4、保险:分为养老、医疗、工伤、失业、住房五种。
注解:要根据客户验厂的要求购买养老、医疗、工伤保险。
比如:要求70%的员工购买就可以,当地的社保部门会有一个有关购买保险的年审证明很重要,购买保险发票也要出示。
5、合同:员工入厂时就必须要跟厂方签定劳动合同一式二份注解:此劳动合同从当地劳动部门购买,签定后厂方要盖章,员工签字,工厂和员工各保留一份。
注意签定的劳动合同数量要同人事档案人数一样,及签字日期和进厂日期一致。
6、考勤:员工每天上下班,必须要打卡,要保留最近至少一年的考勤记录。
SQP验厂专用文件-质量管理体系管理评审报告

7.9.3在物理性风险方面,板房在产品开发时要特别留意产品的外形与内部构造,确保产品不会有尖点、利边和小物件等产品风险;在生产过程中,品质部要定期对产品进行检测和试验,发现有安全问题时,立即召集相关部门开会讨论解决方案;各部门严格依照“利器管理程序”要求对利器进行管控;车缝部/手工部严格依照“利器管理程序”要求对针类进行管控;所有车缝产品要100%过针机方可进入下一生产工序,对有怀疑或断针未打齐的产品严格按照《利器管理程序》执行;严格执行公司玻璃碎品管理规定;
①
建议:如何加强公司的企业文化建设,减少中层管理干部的流失问题。
跟进:现在全国各地都在进行工业园的开发,工厂的管理干部回流内地非常大,这是东莞企业普遍存在的问题。
②
建议:生产安全是企业的生命,公司需要以“生产安全标准化”为契机,加强员工的安全操作培训;适当增加投入来减少工厂的安全隐患。
跟进:2017年依据东莞市“企业生产安全标准化”要求建立了安全生产标准化体系,但是安全生产是个不断优化的过程,工厂在持续改进中。
2、2017年3月,ITS对本公司进行了ISO9001-2015管理体系认证,发现3个不符合项,工厂已经做了改善,详细资料可以参考《ITS审核报告》。
7.3
顾客满意和投诉信息(负责人:管理者代表)
7.3.1顾客满意度
7.3.1.1在2017年9月我们对顾客满意度进行了调查,满意率得分(平均)是90.3分(详见2017年《客户满意度调查汇总表》),达到了工厂的【客户满意度85分以上】的目标。
验厂所需文件清单

验厂所需文件清单第一篇:验厂所需文件清单验厂所需文件清单1、工卡或考勤记录(过去十二个月)。
如果是使用电子考勤,审核员可能需要抽样。
2、工资表(过去十二个月)3、请假条(近三个月的)4、辞职申请表(近三个月的)5、辞职人员工资表(近三个月的)6、员工花名册及员工个人档案(含身份证复印件)7、劳动合同8、社会保险收据、参保人员花名,当地参保要求文件,或合格证明文件等。
9、工商营业执照。
10、建筑工程消防验收意见书/消防检查报告。
11、消防演习记录、紧急疏散计划及工伤记录等。
12、特种设备注册登记及检验文件,如电梯、起重机械、机动叉车、锅炉及压力容器(含气瓶,压力表及安全阀)等13、特种作业人员操作证,如电工、焊接工等;特种设备作业人员操作证,如电梯司机、叉车司机、起重机械操作工、司炉工等。
14、环保文件(如建设项目环评、环评批复、建设项目竣工环境验收报告,排放污染物申报登记表等)15、危险废物处置商的经营许可证,危险废物转移联单等。
16、厂规或员工手册17、政府有关当地最低工资规定文件。
18、当地劳动局关于综合计算工时工作制批文或延长加班批文。
19、未成年工体检及劳动局登记记录。
20、厂房平面图。
21、化学品清单。
22、机械设备维修计划第二篇:2009-11-22一般技术验厂所需的文件清单一般技术验厂所需要的文件清单1,公司简介2,公司组织框架图3,公司平面图4,供应商评审记录5,营业执照6,员工手册7,厂纪厂规8,宿舍管理制度9,招工指引或者程序10,人事记录,员工入厂登记(全体员工)11,劳动合同(全体员工)12,体检记录13,未成年工的登记以及工作安排14,综合计时15,社会保险交付记录16,请假单17,离职记录18,警告信19,考勤卡20,记件(台产(记录21,当地政府最底工资发放标准22,一年的员工工资发放表23,有工人签字的工资条24,工资扣除或者罚款记录25,厂房建筑结构安全合格证26,厂房消防合格证27,公司健康安全政策28,(消防、安全)安全主任资格证明29,消防(急救)人员资格证书30,消防、紧急疏散、急救的培训计划以及记录(照片)31,电梯、起重机等设备登记准用、验收、年检合格证32,特种作业人员资格证书33,设备维修工技术资格证书34,锅炉、压力容器使用登记证、年检合格证35,化学危险品仓库(张贴MSDS;危险品使用须知)36,化学危险品仓库管理员上岗培训证书37,车间有毒有害作业环保测试报告38,发电机噪音测试报告39,排污许可报告、环保检测报告40,食堂卫生许可证、工作人员健康证41,医护人员资格证书42,工伤事故处理记录以及防范措施43,定期杀虫记录44,保安守则、条例、上岗证45,工会组织会议记录、工会代表选举程序以及职责B安全生产(现场评估)1,所有部门消防通道、箭头方向正确(主消防通道:2米,次0。
质量验厂全套程序文件体系文件管理程序

质量验厂全套程序文件体系文件管理程序随着全球贸易的不断发展,质量验厂成为各行各业中至关重要的环节。
质量验厂是一个系统性的过程,需要准确、高效地管理各种程序文件。
这些文件包括质量控制计划、检测方法、验收标准、质量记录等。
为了确保质量验厂的顺利进行,建立一套完善的程序文件体系文件管理程序是非常必要的。
一、程序文件体系概述程序文件体系是质量验厂的重要组成部分,它用于规范和指导各项质量控制活动,确保产品质量符合相关标准和要求。
程序文件体系包括标准程序文件、操作程序文件和记录文件三个层级。
标准程序文件是由相关质量标准和要求制定的文件,包括质量控制手册、质量程序文件和质量作业指导书等。
它们规定了各项质量控制活动的基本要求和操作步骤。
操作程序文件是根据标准程序文件编制的具体操作规程文件,包括工艺操作程序、设备操作程序、检测操作程序等。
它们详细描述了各项质量活动的具体操作方法和要求。
记录文件用于记录各项质量活动的实施情况和结果,包括检验记录、检验报告、溯源记录等。
记录文件是质量活动过程中的重要依据,它们用于记录和证明质量活动的有效性和合规性。
二、程序文件体系文件管理要求程序文件体系的有效管理对质量验厂的顺利进行至关重要。
以下是程序文件体系文件管理的要求:(一)文件分类和编码根据程序文件体系的要求,将各类文件进行分类和编码。
分类可以按照文件类型(标准程序文件、操作程序文件和记录文件),也可以按照质量活动内容进行分类。
编码的目的是便于文件的查找和管理,可以采用数字编码或字母编码。
(二)文件编号和版本控制对于每个文件,应当给予独立的编号,并对文件的版本进行控制。
文件编号应当服从一定的规则,以确保文件的唯一性和辨识性。
版本控制是为了确保文件在修订过程中的一致性和可追溯性。
(三)文件审批和发布在文件编制完成后,应当进行审批和发布。
审批是为了验证文件的正确性和合规性,发布是为了使文件生效并为相关人员所知晓。
审批和发布的流程可以根据实际情况进行设计,确保文件的有效性和权威性。
验厂审核前文件准备

1. 公司组织架构图
2.5 工艺流程图、CCP监控点
3.1 质量手册,程序文件,作业文件
3.4 内审文件,外部审核文件,SC(审核不符合报告,日常监督检查报告)
3.5 供应商名单,供应商资质,原材料检测报告
3.7纠正预防
3.8 不合格品处理流程
3.9 可追溯性(整套追溯记录从原料到产成品)
3.10 客户投诉处理
3.11 产品召回程序
4.3 厂区平面图,人流物流图
4.5水检测报告
4.6 设备清单
4.7设备保养计划和记录(如有冷库,请提供冷库管理制度)
4.8 卫生清洁、设施、设备、建筑定期进行检查记录
4.14虫害控制图、虫害控制计划、记录
4.15食品添加剂管理
5.3 产品中过敏源控制
5.6实验设备清单、外部检定、有毒有害物品清单、管理程序、使用记录
5.7产品出厂报告、型式检验报告、产品标准
6.1 生产过程控制程序、操作规程、生产线记录6.4监控设备校准
7人员健康证、培训计划、培训记录。
SQP验厂所需文件总结分析全面版

DG(SQP全面版)验厂所需文件1.营业执照,厂区平面图,2.品保科架构图/职责3.质量手册/质量目标及方针/产品安全目标及方针4.设备清单,设备维护保养计划,设备维修及保养记录(可不用做点检)5.程序文件\管理制度\岗位职责(从总经理到清洁员也要)\ 文件发放记录.(要最新的,包括更改,做废)---不能出现二套不一样的资料6.质量方面的证书(ISO9000)7.内部审核(计划表/检查表/报告/及改善措施追踪/改善后的效果确认) ——最好出个通告8.管理评审(计划表/报告/及改善措施追踪/改善后的效果确认)——最好出个通告9.与行业产品安全相关的法律法规的收集(按出口国家来分别收集)——像重金属测试等10.内部/测试程序(步骤)/或外部的测试报告(从原材料,半成品,最终品.)——缩率测试(特别是针织品)、拉力测试、尖边尖角的测试、甲醛含量、色劳度等.11.风险评估程序(规定多长时间一次)/评估表(风险等级及改进措施)/评估报告(按程序规定的时间做)——要做到从采购开始,到原材料,生产车间的整个环节,到成品出货,到运输,及客户,到最终消费者的风险都要做预先评估.12.合格供应商名单(最新的)/供应商管理程序(选定及评估)/初次评估记录(要有数据)/供应商的年度绩效考核(也就是年度评估,要有仓库记录数据)/采购程序/采购合同(采购合同开始就要注明必须要给此批货的原辅料的测试报告,如没注明那就得要立保证书)13.来料检验记录/生产过程的检验记录/不合格品的控制程序(检验台也不能出现不合格品)/不良率分析表14.客户投诉记录/程序(怎么沟通与处理)/召回程序,15.应急计划——生产过程突发事件(停水,停电,自然灾害等因素)可能会影响无法如期交货,在这种情况下怎么确保实时出货.16.利器收发记录/入库记录/领料记录/更换记录(车间要当天发放并收回.有几人要用就发几把)17.断针记录/入库及领用(专人去仓库更换断针,记录要放仓库保管)/台帐(车间收发员也要),18.产前会议记录/品质周会/大货的试产记录19.原料的虫害控制(要有记录)/原辅料检验程序与AQL标准(标准里明确可接受范围%比例)20.卫生管理制度/检查记录(包括车间,厂区所有范围及周围)----日检21.测量仪器设备校验证书/校验计划/内校员证书/计量仪器清单(检测面料的磅精确0.01g)22.培训制度/记录(新进员工培训/品质部培训)/年度培训计划表/考核制度.23.化学品管理制度/清单(包括灭四害的药及品牌与生产商)24.包装后入库检验程序/抽检AQL标准.25.车间的工艺单(要有复核人签名或弄个章盖)/设计图纸板样(要有复核人签名或弄个章盖)——设计部跟实验室(设计时可以做上去)26.个人用品管理制度27.产品质量事故管理规定(事故定界)首次会议记录:首先审核员在会议首选描述了这个SQP它是有两个版(基础版/全面版)基础板审的跟FCCA差不多,也会比FCCA审的东西要多,全面版的是从采购合同开始到原材料到半成品到成品制作中的每个环节,到入库,到出库到运输到客户到送到消费者为止中间预先评估联想出其中所可能会发生的所有质量安全风险.所以说有很多不同于FCCA,审核也就是照这样来推理出来的,这就是他们的审核思路.不完全是在乎产品的批次号,到产品有问题到召回到追溯.这不是最重点.这个要9000要求文件还要多,还要细,还要全.给我们的具体思路如下:1.基础设施-----基础设施的建立进入车间要有可洗手的洗手台提供,进车间要洗手,要戴帽子(包装部必须要)前面的还可以控制不一定要.2.建立制度----从建立一个工厂首先要考虑建立一个品质目标及方针与产品安全目标与方针,全面的规章制度,质量手册,程序文件,各岗位职责(从总经理直至清洁工的也要有岗位职责)3.人员招聘及培训----那就要招人了,招了人首先要做入职培训,关键岗位的人员要有进来要依据考核制度进行技术能力考核,(涉及到培训计划的制定,培训了肯定要能力考核,再就要记录在案)4.收集法律法规----以上的都建立起来了,再就会有编制作业指导书,管理层要收集产品所涉及的所有的法律法规有什么检测要求,要打印出来成册保存,及时收集更新.去意大利只要做个扣子及拉链的拉力测试,去美国的还要有缩率测试,(童装里用的钮扣,拉链等小金属件必需要做尖边尖角的测试,重金属含量,甲醛含量.色劳度等)最好是做个培训5.原料测试----再说执行要从采购开始签合同最好注明要有各种测试报告提供,没注明就得立下保证书也行,(厂家的就是重金属含量,甲醛含量.色劳度等这些供应商自己有设备测试就不会多麻烦工厂自己做就麻烦多了.6.利器控制----到车间制作过程利器控制(戒刀是禁止车间使用的)折的小片片是无法控制的,7.质量控制----到车间要有的检验过程这中间就要关注不良品区分控制,8.内部测试----到内部的测试(原料进来前,车间制成中要有测试)或外部测试9.检验记录----再到成品的入库检,接着就出货时出货抽检或客户验货报告(单做质量可不用做日报表)10.风险评估----做了段时间的工厂要做产品从原材料到消费者之间的每个环节过程中多多少少肯定会有质量事故出现,得做风险评估,11.内部审核----以后过程中肯定自己要对之前的执行情况做内审(这内审比9000内容要细,要多,要细分到每个车间,做一个审查表做记录,) 对上次内审的问题作结论,要包括对风险评估做的报告问题, 然后就要对这次审查的问题做分析调查,整改措施,跟踪实施的结果做确认)这是对车间员工用的, 要有年度内审计划表,首次跟末次会议记录及会议签到,出个通告.12.管理评审----最后就是针对管理体系的一个整个大方位的执行情况做管理评审(包括号今年的内审报告,加上内审的内容)管理评审只是要有一个计划然后加个评审报告,再就审查的问题做分析,改进措施,结果要确认.(评审是没有调查表的)-要有年度内审计划表,首次跟末次会议记录及会议签到,出个通告。
SQP验厂要求及判定标准

SQP验厂的具体要求是什么?标准是怎么样的?如何准备SQP验厂?SQP是ITS整出来的合格供应商评估计划85分以上通过,可以取得证书,有效期一年,目前DG客人对此有要求。
审核分8个部分,管理承诺与持续改进、风险评估、质量管理体系、现场与设施管理、产品控制、产品测试、过程控制与员工培训,除第二点以外都是ISO9001的要求。
风险控制是QS9000里的要求,要利用FMEA,RPN等一些基本工具。
SQP验厂的中文清单SQP 文件清单1. 组织架构图2. 责任和/ 或职责描述3. 质量体系程序(包括:质量政策、目标、质量管理体系手册和程序,以及其它流程)4. 管理层审查记录5. 内部审核文件(审核计划、报告等)6. 供应商监管文件(供应商核准程序/ 标准、已核准的供应商清单、供应商评估记录、持续表现监督等)7. 文件监管程序和记录(包括记录保管)8. 产品规格/ 要求9. 检验要求说明、可接受的标准、检验和测试报告(包括IQC 的阶段、过程中和最终检验)10. 工作要求说明/ 每项生产工序的工艺技术标准11. 生产日程安排/ 记录12. “事故”的界定和报告程序13. 产品召回程序14. 客户投诉记录15. 整改行动报告(关于事故、内部审核、投诉等)16. 追溯系统中的测试报告17. 设备维护文件(计划、程序、记录等)18. 监督和测试设备的校准(计划、程序、记录等)19. 清理日程安排和程序20. 已核准的化学品清单,附带相应的品牌/ 生产商21. 有害物管控文件(受过培训的管控人员的名单、外部有害物管控机构的联系方式、有害物管控检查记录、投饵记录,等)——》虫害控制程序22. 整个生产流程的“风险评估”记录/ 计划23. 最终产品的风险评估记录24. 产品测试步骤/ 程序25. 实验室测试报告(包括涂料、涂层和非涂料部件中的铅和重金属、硬件、标签、最终产品,等)26. 夹杂物监控记录(如:金属探测记录、金属探测器的日常敏感物检查记录,等)27. 断针处理程序(如适用的话)28. 生产前会议记录29. 程序控制计划30. 培训(程序、培训需求和记录。
GMP认证检查工作指南

GMP认证检查工作指南GMP(Good Manufacturing Practice)是一种制药行业质量管理标准,用于确保药品的安全性、有效性和质量。
GMP认证是指通过对企业的生产过程和质量管理体系进行评估,判断其是否符合GMP标准,从而获得认证。
1.核查文件:确认企业是否有完备的质量管理体系文件,包括GMP规程、SOP(标准操作程序)文件、记录表等。
这些文件应当涵盖企业的各个环节,从原材料采购到生产过程再到产品包装等。
2.生产现场:检查企业的生产现场,包括贮存区域、生产线和设备等。
确认设备是否符合GMP要求,如设有有效的清洁程序和维护计划,以及是否具备适宜的温湿度控制系统和通风设施。
3.原材料管理:核实企业的原材料管理流程,确保采购的原材料符合GMP标准,并且有适当的存储和追踪措施。
检查样品的采样和检测程序,以确保原材料的质量和纯度。
4.生产记录:审查企业的生产记录,包括生产工艺参数、生产过程的记录、生产人员的签名等。
这些记录应当清晰、规范,并能够证明产品的质量和合规性。
5.检验和检测:检查企业的检验和检测设备,包括仪器设备和实验室人员。
确保检验方法的准确性和可靠性,并核实设备的校准和维护记录。
6.不合格品管理:检查企业的不合格品处理程序,包括不合格品的隔离、调查和处理等。
确保企业有合适的纠正和预防措施,以确保不合格品不会影响生产和产品的质量。
7.培训和教育:检查企业的员工培训和教育计划,包括GMP相关知识的培训和员工技能的认证。
确保员工了解和遵守GMP要求,并具备适当的技能和知识。
GMP认证检查是一个具有挑战性的过程,需要企业倾力以赴,并严格按照GMP要求进行检查和改进。
只有通过合格的GMP认证,企业才能证明其生产过程和质量管理体系达到了国际标准,并且可以提供安全和有效的药品产品给消费者。
SQP(GMP)记录和部分程序文件责任表
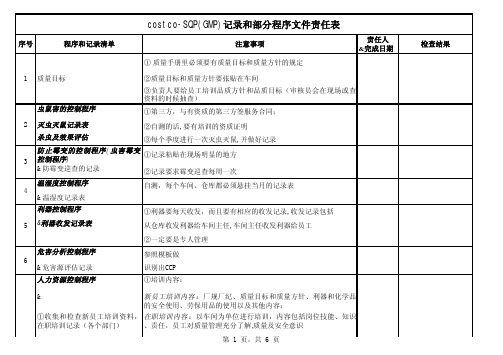
责任人&完成日期①质量手册里必须要有质量目标和质量方针的规定②质量目标和质量方针要张贴在车间③负责人要给员工培训品质方针和品质目标(审核员会在现场或查资料的时候抽查)虫鼠害的控制程序①第三方,与有资质的第三方签服务合同;灭虫灭鼠记录表②自测的话,要有培训的资质证明杀虫及效果评估③每个季度进行一次灭虫灭鼠,并做好记录防止霉变的控制程序(虫害霉变控制程序)①记录粘贴在现场明显的地方&防霉变巡查的记录②记录要求霉变巡查每周一次温湿度控制程序自测,每个车间、仓库都必须悬挂当月的记录表&温湿度记录表利器控制程序①利器要每天收发,而且要有相应的收发记录,收发记录包括&利器收发记录表从仓库收发利器给车间主任,车间主任收发利器给员工②一定要是专人管理危害分析控制程序参照模板做&危害源评估记录识别出CCP人力资源控制程序①培训内容:&新员工培训内容:厂规厂纪、质量目标和质量方针、利器和化学品的安全使用、劳保用品的使用以及其他内容;①收集和检查新员工培训资料,在职培训记录(各个部门)在职培训内容:以车间为单位进行培训,内容包括岗位技能、知识、责任,员工对质量管理充分了解,质量及安全意识注意事项检查结果costco-SQP(GMP)记录和部分程序文件责任表序号1 2 3程序和记录清单质量目标4 5 6②电工、机械工程师、机械操作工、质检人员、测试人员的技能培训&定期培训资料② 每种培训都必须要有考核成绩,也就是成绩汇总表③年度培训计划表③ 每种培训一定要有相应的试卷,做试卷要用黑色签字笔或原珠笔④验厂当天提供的花名册④ 要有培训人员的亲自签名⑤ 考试的方式有笔试、口试和实操⑥ 特殊工种不能内训的可以通过外训,外训人员必须有相关证书⑦花名册里出现的人员必须要有入职培训资料,在职培训根据实际确认⑧年度培训计划表要与所有的培训资料对应得上⑨新员工入职,要有培训试卷出货控制程序书&出货准时性记录采购合同/采购订单① 要有对方的回签 ③相关采购标准件摆放在仓库里采购规格说明(参考样品)②资料的源头,给审核员看的订单必须要有供应商控制程序①《供应商名册》要包括合格跟不合格供应商&②至少每三个月评估一次①供应商名册③供应商控制程序中应有对新供应商的选择、评估的规定②合格供应商名册③供应商评估记录&跟踪评估记录④外发供应商评估记录910811准时性记录及准时性比率的统计及跟踪评估记录⑤供应商配合协议书顾客投诉控制程序&①客户投诉记录表②客户投诉的纠正预防措施表有关设备控制的程序① 每台机器设备都必须有设备点检表&①设备清单② 每台机器设备要有一个维修卡,记录机器维修的时间和内容②设备维修保养年度计划③设备都必须要有编号,每台设备需要张贴标示卡,标示③设备清洁及维护保养记录卡要含有设备的名称、编号、维护日期④设备维修申请单和维修单⑤机器配件清单⑥模具清单⑦电工的工具清单⑧生产过程中使用到的工具清单① 仪器清单① 仪器必须送外校正② 仪器年度校正计划表② 送外校正的正书必须是管方的① 测试仪器定期校准报告③ 仪器清单只能出现已经校准并且合格的仪器名称,验厂当天不能出现的仪器必须收藏起来。
2023年质量验厂审核新要求的程序文件-产品检验控制程序

2023年质量验厂审核新要求的程序文件-产品检验控制程序引言本程序文件旨在规定2023年质量验厂审核中产品检验的控制程序要求。
本程序的目的是确保产品的质量符合最新验厂审核要求,并且能够满足客户和市场的需求。
所有参与产品检验的人员应遵守本程序,以确保一致的检验标准和结果。
1. 检验计划1.1 为了确保产品质量的合规性,公司应制定详细的检验计划,包括检验的频率、标准和方法。
1.2 检验计划应基于相关法律法规、客户要求和市场标准,确保产品满足所有适用的质量要求。
2. 检验设备和工具2.1 公司应确保所有使用的检验设备和工具符合相关的标准和规范,并保持其准确性和可追溯性。
2.2 所有检验设备和工具应定期校准和维护,并记录相应的校准证书和维护记录。
2.3 检验设备和工具的使用人员应经过培训,并具备相应的技能和知识,以确保准确和可靠的检验结果。
3. 检验过程3.1 检验过程应严格按照检验计划和相关标准进行,并记录检验结果。
3.2 检验过程中应注意防止混淆、交叉污染和错误判定,确保产品检验结果的准确性和可信度。
3.3 对于不合格的产品,应采取适当的措施进行处理,包括但不限于返工、报废或修正。
4. 检验记录和报告4.1 检验记录应准确、完整地记录所有的检验过程和结果,包括检验日期、检验人员、检验方法和检验结果等信息。
4.2 所有检验记录应保存并归档,以备将来的审核和追溯要求。
4.3 根据客户要求和市场标准,公司应编制相应的检验报告,并及时提供给相关方。
5. 持续改进5.1 公司应定期评估和改进产品检验程序,以确保其符合最新的质量验厂审核要求。
5.2 通过内部和外部审核、持续培训和技能提升,公司应不断改进产品检验的效率和准确度。
结论本文档规定了2023年质量验厂审核中产品检验的控制程序要求。
公司应遵守本程序文件的要求,并持续改进产品检验过程,以确保产品的质量符合最新的要求,满足客户和市场的需求。
质量验厂程序文件目录

质量验厂程序文件目录一、前言质量验厂是对供应商生产能力、质量控制体系等方面进行评估的重要环节。
为了确保验厂工作的规范化、标准化和有效性,特制定本质量验厂程序文件目录。
二、目的本程序文件的目的在于明确质量验厂的流程、要求和标准,为验厂人员提供清晰的指导,确保验厂结果的客观、公正和准确,以促进供应商不断提升产品质量和管理水平。
三、适用范围本程序文件适用于对所有潜在供应商和现有供应商的质量验厂。
四、职责分工(一)采购部门负责发起质量验厂需求,与供应商沟通协调验厂时间和相关事宜。
(二)质量部门主导质量验厂工作,制定验厂计划,组织验厂团队,实施验厂评估,撰写验厂报告。
(三)技术部门提供技术支持,评估供应商的生产工艺和技术能力。
(四)生产部门协助质量部门对供应商的生产现场进行评估。
五、质量验厂程序文件清单(一)《质量验厂策划程序》1、明确验厂的目标、范围和时间安排。
2、确定验厂团队的成员和职责。
3、制定验厂的方法和流程。
(二)《供应商信息收集程序》1、收集供应商的基本信息,包括企业概况、生产能力、质量管理体系等。
2、评估供应商提供的信息的真实性和完整性。
(三)《验厂前准备程序》1、研究供应商的相关资料,了解其产品特点和生产流程。
2、准备验厂所需的文件和工具,如检查表、测量仪器等。
(四)《现场验厂程序》1、对供应商的生产现场进行实地考察,包括生产设备、工艺流程、工作环境等。
2、检查原材料、半成品和成品的质量控制情况。
3、审核供应商的质量管理文件和记录。
(五)《员工访谈程序》1、随机抽取供应商的员工进行访谈,了解其对质量控制的认识和执行情况。
2、关注员工的工作条件和福利待遇。
(六)《数据分析与评估程序》1、对验厂收集的数据和信息进行整理和分析。
2、按照预定的标准和指标对供应商进行评估。
(七)《验厂报告编写程序》1、总结验厂的结果和发现的问题。
2、提出改进建议和后续跟进措施。
(八)《验厂结果沟通与反馈程序》1、将验厂结果及时反馈给供应商。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
20.风险评估程序(规定多长时间一次)/评估表(风险等级及改进措施)/评估报告(按程序规定的时间做)——要做到从采购开始,到原材料,生产车间的整个环节,到成品出货,到运输,及客户,到最终消费者的风险都要做预先评估;特殊要求:每个产品或每类产品都要有风险评估、既包括产品的风险评估也包括产品的过程风险评估、产品的风险评估要包括各类潜在的风险如适合使用的人群、安全性能、物理性能、环保等等、过程风险评估要包括监控检查情况
8.设备清单
9.设备维护保养计划(年度季度或月度的保养计划及按计划的保养记录)
10.设备维修记录,要有设备关键零部件备件清单
11.设备保养记录(每天日常的保养记录)
12.程序文件及管理制度
13.岗位职责(公司每个岗位包括从总经理到清洁员也要)
14.文件发放记录.(要最新的,包括更改,做废)---不能出现二套不一样的资料
22.来料检验记录/生产过程的检验记录/成品检验记录/不合格品评审记录/纠正及持续改进记录、不合格品的控制程序(检验台也不能出现不合格品)/不良率分析表/柏拉图/SPC/QC月报/追溯演练记录/控制计划注意:追溯演练记录要完整、要包括各个环节的记录单据记录表格全部都附上。
23.客户投诉记录/程序(怎么沟通与处理)/召回程序,召回演练记录
SQP质量验厂+GMP质量验厂审核所需文件及验厂详细要求解释以及验厂主要文件范本
1.营业执照
2.厂区平面图
3.品保部架构图
4.品保部职责
5.质量手册
6.质量目标及方针(包括目标的监视测量(即每月的统计)情况)
7.产品安全目标及方针(这个跟质量目标及方针是不一样的,关注的是产品安全,包括目标的监视测量(即每月的统计)情况)
8.1产品风险评估汇总表
8.2产品风险评估效果验证表
资料范本:
1.产品追溯演练报告
为验证公司追溯系统的有效性,江门市xx有限公司于2016年01月12日组织一次产品追溯的模拟演练。
模拟演练人员:周xx、杨xx、何xx
一、追溯演练过程:
1、追溯人员随意追溯在库的一批产品。生产单号为:KS-K2015135
4、追查BOM表,本批产品的最新BOM明细表更新日期为2015.11.24
5、前往采购部查找采购合同,其中采购合同编号为KS-150301203的采购单,所采购的塑胶件正是本批产品所有的配件。
34.个人防护品管理(所有设计危害岗位必须佩戴劳动防护用品如:手套口罩耳塞眼镜围裙等)
35.产品质量事故管理规定(事故定界)
附件资料版本:
1.追溯演练报告
2.生产恢复规定(灾难重建计划)
3.紧急采购控制程序
4.产品安全方针目标
5.管理评审要求输入的内容
6.消杀化学品清单及农药MSDS
7.控制计划
8.产品风险评估记录
31.化学品管理制度/清单(所有化学品必须有16项完整信息的MSDS),现场按化学品要求如化学品有害标识、有效期标识、存量记录、使用记录、第二容器;使用现场不得超过10公斤)
32.包装后入库检验程序/客户验货的程序(未检验、QA已检、客户已检状态表标识要明确)
33.车间的工艺单(要有复核人签名或弄个章盖)/设计图纸板样(要有复核人签名或弄个章盖)——设计部跟实验室(设计时可以做上去);作业指导书要求必须跟实际一致;检查岗位必须有检查记录;检验员要有明确标识;要有关键岗位的标识(人员及作业指导书都要明确标识);要有关键岗位替岗名单;
2、查实本批产品的生产单,单号KS-K2015135,客户单号为:32650,下单日期为20150831,更新日期为2016.01.06,出货日期为2016.01.15;生产数量为2616pcs,客户是JCS,型号为KS-1008A电热水壶。
3、追查生产计划排期(2015年01月04-15日生产周计划表),本批产品计划开拉时间2016.01.12,完成时间2016.01.05
24.应急计划——生产过程突发事件(设备故障、劳动力不足、质量异常、供应不足、停水,停电,自然灾害等因素)要全面,可能会影响无法如期交货,在这种情况下怎么确保实时出货.也要有灾难重建计划(要有外部联络人或联络小组)
25.利器收发记录/入库记录/领料记录/更换记录(车间要当班发放并收回.有几人要用就发几把,当事人签名零用及归还),利器丢失的处理及记录,利器要求统一编号并固定位置使用。玻璃制品的清单(包括玻璃产品及玻璃基建设施及玻璃损坏的记录)
26.断针记录/入库及领用(专人去仓库更换断针,记录要放仓库保管)/台帐(车间收发员也要),断针丢失的处理及记录
26.产前会议记录/品质周会/大货的试产记录
27.虫害控制(控制措施如:灭蝇灭蚊灯、防鼠笼、纱窗及防止一切虫害进入生产车间的设施,特别注意出口也要有防虫设施;另外要有杀虫记录、杀虫的农药按有包含16项完整信息的MSDS)
15.质量方面的证书(ISO9001-2015)
16.内部审核(计划表/检查表/报告/及改善措施追踪/改善后的效果确认)
17.管理评审(计划表/报告/及改善措施追踪/改善后的效果确认)—管理评审输入的内容比ISO体系内容要多,例如:产品的安全目标及方针;产品安全的风险评估;以后客户审核及验厂的情况
18.与行业产品安全相关的法律法规的收集(按出口国家来分别收集)——像重金属测试等
21.合格供应商名单(最新的)/供应商管理程序(选定及评估)/初次评估记录(要有数据)/供应商的年度绩效考核(也就是年度评估及月度考核,要有质量、交期、服务及价格等记录数据)/采购程序/采购合同(采购合同开始就要注明必须要给此批货的原辅料的测试报告,如没注明那就得要立保证书),要规定紧急采购的控制,要提供样板给供方并要有记录
28.卫生管理制度/检查记录(包括车间,厂区所有范围及周围)----日检
29.测量仪器设备校验证书/校验计划/内校员证书/30.培训制度/记录(新进员工培训/品质部培训/在职员工/专业技术人员)/年度培训计划表/考核制度.特别要包括产品风险评估的培训,要有培训需求记录