13线粒体疾病mitochondrialdiseases.ppt
合集下载
线粒体疾病MitochondrialDiseasePPT

细胞凋亡
线粒体功能障碍可以触发细胞凋亡,导致细胞死亡,从而引发各种 疾病症状。
流行病学
发病率
线粒体疾病的发病Hale Waihona Puke 较高,但具体发病率因地区和人 群而异。
遗传因素
线粒体疾病大多为母系遗传,即由母亲遗传给后代。
性别差异
线粒体疾病在女性中的发病率略高于男性,可能与性 染色体有关。
02
线粒体疾病的临床表现
神经系统症状
提高公众意识
提高公众对线粒体疾病的认知和理解,有助于早期发现和干预线粒体疾病。同时,加强相关研究,深 入了解线粒体疾病的发病机制和治疗方法,为预防和治疗线粒体疾病提供更多科学依据和技术手段。
05
线粒体疾病的研究进展
新药研发进展
靶向药物
针对线粒体疾病的特定分子靶点,开发出具有针对性的药物,以改 善线粒体功能或减轻疾病症状。
影像学检查
MRI检查
MRI可显示线粒体疾病患者的脑部病变,如脑萎 缩、脑白质病变等。
心电图和心脏超声
线粒体疾病患者可能出现心肌病、心律失常等心 脏病变,需进行相应检查。
X线和CT检查
可发现线粒体疾病患者的骨骼异常、肺部病变等。
鉴别诊断
其他遗传性疾病
01
线粒体疾病需与其他遗传性疾病进行鉴别,如神经退行性疾病、
运动康复
运动康复
适当的运动康复可以帮助线粒体疾病患者提高身体素质和心肺功能。建议在专业指导下进行低强度、有氧运动, 如散步、游泳等。
运动强度与频率
运动强度和频率应根据患者的具体情况而定,避免过度疲劳和不适。同时,运动康复应与药物治疗和饮食调整相 结合,以达到最佳的治疗效果。
基因治疗
基因治疗
基因治疗是一种新兴的治疗线粒体疾病的方法,通过修改或替换病变基因来纠正 线粒体功能异常。目前,基因治疗仍处于研究阶段,但未来有望为线粒体疾病患 者提供更有效的治疗方法。
线粒体功能障碍可以触发细胞凋亡,导致细胞死亡,从而引发各种 疾病症状。
流行病学
发病率
线粒体疾病的发病Hale Waihona Puke 较高,但具体发病率因地区和人 群而异。
遗传因素
线粒体疾病大多为母系遗传,即由母亲遗传给后代。
性别差异
线粒体疾病在女性中的发病率略高于男性,可能与性 染色体有关。
02
线粒体疾病的临床表现
神经系统症状
提高公众意识
提高公众对线粒体疾病的认知和理解,有助于早期发现和干预线粒体疾病。同时,加强相关研究,深 入了解线粒体疾病的发病机制和治疗方法,为预防和治疗线粒体疾病提供更多科学依据和技术手段。
05
线粒体疾病的研究进展
新药研发进展
靶向药物
针对线粒体疾病的特定分子靶点,开发出具有针对性的药物,以改 善线粒体功能或减轻疾病症状。
影像学检查
MRI检查
MRI可显示线粒体疾病患者的脑部病变,如脑萎 缩、脑白质病变等。
心电图和心脏超声
线粒体疾病患者可能出现心肌病、心律失常等心 脏病变,需进行相应检查。
X线和CT检查
可发现线粒体疾病患者的骨骼异常、肺部病变等。
鉴别诊断
其他遗传性疾病
01
线粒体疾病需与其他遗传性疾病进行鉴别,如神经退行性疾病、
运动康复
运动康复
适当的运动康复可以帮助线粒体疾病患者提高身体素质和心肺功能。建议在专业指导下进行低强度、有氧运动, 如散步、游泳等。
运动强度与频率
运动强度和频率应根据患者的具体情况而定,避免过度疲劳和不适。同时,运动康复应与药物治疗和饮食调整相 结合,以达到最佳的治疗效果。
基因治疗
基因治疗
基因治疗是一种新兴的治疗线粒体疾病的方法,通过修改或替换病变基因来纠正 线粒体功能异常。目前,基因治疗仍处于研究阶段,但未来有望为线粒体疾病患 者提供更有效的治疗方法。
第四章线粒体遗传病课件
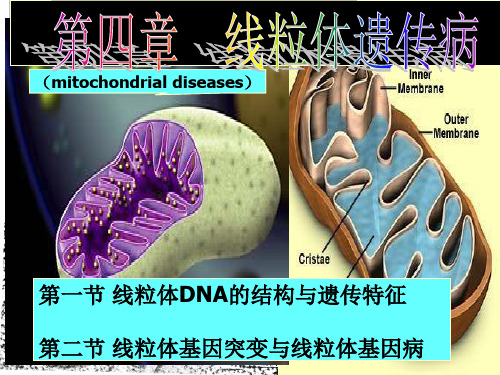
•线粒体的代谢障碍,则不能产生足够的能量而导 致细胞功能衰退,出现一系列临床症候。人群患病 率约为1/8,500
线粒体基因突变 表现的临床特征:
线粒体突变导致的疾病主要 累及中枢和外周神经系统, 肌病和脑病症状。与贫血和
糖尿病等疾病也相关。
问题:线粒体疾病主要受累的器官是哪些
•(一)碱基突变
•1、错义突变:称氨基酸替换突 变。 Leber遗传性视神经萎缩.
• 线粒体病有累加效应因此线粒体病 有随着年龄的增加病情会越来越严重 的特征。
• 问题:什么叫阈值效应?
(六)mtDNA的突变率极高
mtDNA的突变率比核DNA高10~20倍。 但因为都是中性和中度有害的mtDNA的 突变,有害的突变会通过选择(例如遗传 瓶颈) 而消除,故线粒体遗传病并不常 见。
相关,如线粒体肌病和脑肌病、线粒体眼病, 老年性痴呆、帕金森病、Ⅱ型糖尿病、心肌 病及衰老等,有人统称为线粒体疾病。
•
复习思考题:
• 1、线粒体DNA的结构特征如何?
• 2、简述线粒体遗传的特点。
• 3、线粒体疾病主要受累的器官是哪些?
• 4、常见的线粒体遗传病有哪几种?它们有什么共同特征?
• 5、何谓多基因遗传方式?
•2、蛋白质生物合成基因突变: 为tRNA基因突变,
(二)缺失、插入突变: 例如,肌阵挛性癫痫
(MERRF综合征)等
•例如,眼肌病,如KSS综 合征(无家族史、散发)
mtDNA拷贝数目低 于正常。例如致死 (三)mtDNA拷贝数目突变:性婴儿呼吸障碍等
突变 mt-3243 mt-3256 mt-3271 mt-3303 mt-3260 nmt-4269 mt-5730 nmt-8344 mt-8356 mt-15990 mt-8993 mt-11778 mt-4160 mt-3460 mt-7444 mt-14484 mt-15257
线粒体疾病PPT.

ACTCCTGAGGAG
1350 1150
200
分析mtDNA
取自5个地理区域(Africa、Asia、Australia、 New Guinea、Europe)的147人; 使用12种不同的限制性内切酶消化; 通过RFLP分析mtDNA的限制性酶谱; 共获得133种限制性酶谱型; 通过mtDNA的演化,分析人类的起源。
核DNA大10-20倍。
The Eve Hypothesis
Possible phylogenetic tree of human mitochondria based on analysis of presence or absence of restriction enzymes in mitochondrial DNA from 147 persons.
The common ancestor of all human mitochondrial DNA who lived in Africa between 14,000-28,000 years ago.
线粒体疾Байду номын сангаас分类
(1)线粒体遗传病 (2)核基因突变引起的线粒体疾病
Leber遗传性视神经病( Leber hereditary optic neuropathy, LHON)
GAG CTC
GAA CTT
人群中出 现三种基 因型
GAG, GAG GAG, GAA GAA, GAA
RFLP分析DNA
N代表任一核苷 酸
限制性内切酶Mst II 的识别序列 CCTNAGG
ACTCCTGAGGAG(可酶切)
ACTCCTGTGGAG(不能切)
AA AT TT
1350 bp 1150 bp
1350 1150
200
分析mtDNA
取自5个地理区域(Africa、Asia、Australia、 New Guinea、Europe)的147人; 使用12种不同的限制性内切酶消化; 通过RFLP分析mtDNA的限制性酶谱; 共获得133种限制性酶谱型; 通过mtDNA的演化,分析人类的起源。
核DNA大10-20倍。
The Eve Hypothesis
Possible phylogenetic tree of human mitochondria based on analysis of presence or absence of restriction enzymes in mitochondrial DNA from 147 persons.
The common ancestor of all human mitochondrial DNA who lived in Africa between 14,000-28,000 years ago.
线粒体疾Байду номын сангаас分类
(1)线粒体遗传病 (2)核基因突变引起的线粒体疾病
Leber遗传性视神经病( Leber hereditary optic neuropathy, LHON)
GAG CTC
GAA CTT
人群中出 现三种基 因型
GAG, GAG GAG, GAA GAA, GAA
RFLP分析DNA
N代表任一核苷 酸
限制性内切酶Mst II 的识别序列 CCTNAGG
ACTCCTGAGGAG(可酶切)
ACTCCTGTGGAG(不能切)
AA AT TT
1350 bp 1150 bp
线粒体脑肌病PPT课件

7
四、临床表现
线粒体病的临床分型 最常见的三种临床分
PEO)Kearns-Sayre综合征(KSS) 线粒体脑肌病、乳酸中毒以及卒中样发作(mitochondrial encephalomyopathy, lactic acidosis and strokelike episodes, MELAS) 肌阵挛性癫痫发作伴破碎红纤维(myoclonus epilepsy with ragged red fibers,MERRF) Leber遗传性视神经病 Leigh综合征 常染色体显性/隐性进行性眼外肌瘫痪(PEO) 线粒体神经胃肠性脑肌病 阿尔佩斯病
影像学特点
头CT扫描
病变呈现低密度改变,形态与脑 血管支配分布不一致,称为“皮 层样坏死”,系本病的特征性影 像学改变。 这种影像学改变数月后会完全消 失,而在复发时又会出现在另一 部位的皮质。 部分患者可有双侧基底节对称性 钙化。
10
线粒体脑肌病伴乳酸中毒和卒中样发作综合症(MELAS)
21
五、辅助检查
2、肌肉活检
冰冻切片组织化学染色, 显示RRF及糖、脂肪异常堆 积, 如RRF超过4%则有重要的诊断价值。电镜下观察, 除有大量形态异常的线粒体外,并在这些线粒体内有 结晶状、板层状及同心圆样的包涵体, 同时尚发现大 量脂滴或糖原颗粒堆积。
22
五、辅助检查
3、影像学检查
CT或MRI检查:线粒体脑肌病患者可见白质脑病、 基底节钙化、脑软化、脑萎缩和脑室扩大等。
32
二 、药物治疗 肌酸、L-肉毒碱和coQ10补充疗法通常混合为“鸡尾酒”来使 用以治疗线粒体病,是人体内三种参与ATP合成的自然物质 。 有报道用硫辛酸(300 mg/d)+辅酶Q10(240 mg/d)+肌苷 (3 g/d)治疗可降低线粒体脑肌病患者血乳酸水平、氧化应 激反应以及增加踝关节背屈力量。
四、临床表现
线粒体病的临床分型 最常见的三种临床分
PEO)Kearns-Sayre综合征(KSS) 线粒体脑肌病、乳酸中毒以及卒中样发作(mitochondrial encephalomyopathy, lactic acidosis and strokelike episodes, MELAS) 肌阵挛性癫痫发作伴破碎红纤维(myoclonus epilepsy with ragged red fibers,MERRF) Leber遗传性视神经病 Leigh综合征 常染色体显性/隐性进行性眼外肌瘫痪(PEO) 线粒体神经胃肠性脑肌病 阿尔佩斯病
影像学特点
头CT扫描
病变呈现低密度改变,形态与脑 血管支配分布不一致,称为“皮 层样坏死”,系本病的特征性影 像学改变。 这种影像学改变数月后会完全消 失,而在复发时又会出现在另一 部位的皮质。 部分患者可有双侧基底节对称性 钙化。
10
线粒体脑肌病伴乳酸中毒和卒中样发作综合症(MELAS)
21
五、辅助检查
2、肌肉活检
冰冻切片组织化学染色, 显示RRF及糖、脂肪异常堆 积, 如RRF超过4%则有重要的诊断价值。电镜下观察, 除有大量形态异常的线粒体外,并在这些线粒体内有 结晶状、板层状及同心圆样的包涵体, 同时尚发现大 量脂滴或糖原颗粒堆积。
22
五、辅助检查
3、影像学检查
CT或MRI检查:线粒体脑肌病患者可见白质脑病、 基底节钙化、脑软化、脑萎缩和脑室扩大等。
32
二 、药物治疗 肌酸、L-肉毒碱和coQ10补充疗法通常混合为“鸡尾酒”来使 用以治疗线粒体病,是人体内三种参与ATP合成的自然物质 。 有报道用硫辛酸(300 mg/d)+辅酶Q10(240 mg/d)+肌苷 (3 g/d)治疗可降低线粒体脑肌病患者血乳酸水平、氧化应 激反应以及增加踝关节背屈力量。
线粒体遗传病PPT课件

线粒体肌病脑病伴乳酸中毒及脑卒中样 发作综合征临床表现:为突发呕吐、乳酸 中毒、肌肉组织病变、有碎红纤维。少 数患者伴痴呆、周围性偏头痛、眼外肌 无力或麻痹等。
80% MELAS病例mtDNA编码的tRNA基因 3243位点有A→G。
一些少见的突变还可能出现在该基因的 3291、3271、3256和3252等位点。
(三)MELAS综合征
(四)MERRF综合征
又称为肌阵挛性癫痫伴碎红纤维病,是一种线粒体肌 病,它除了具有破碎的肌红纤维和形态异常的线粒体外 ,还伴有失调的阵挛性癫痫(周期性抽搐)。本病有明 显的母系遗传特点,患者的母系亲属常表现一定症状如 脑电图异常、扩张性心肌病等。
80% ~ 90% 的 患 者 的 mtDNA 的 tRNALys 基因 8344 bp A→G突变;小部分病人 是 该 基 因 的 8356 位 存 在 T→C突变。
二、线粒体基因组的遗传特征
1、母系遗传 2、同质性与异质性
3、阈值效应
4、突变率高 5、mtDNA可以稳定地整合到核基因组中
在特定的条件下,核DNA序列和mtDNA序列可 以在细胞内游走,从而造成mtDNA对核基因组的插 入。
二、线粒体基因组的遗传特征
1、母系遗传 2、同质性与异质性 3、阈值效应 4、突变率高 5、mtDNA可以稳定地整合到核基因组中 6、mtDNA在有丝分裂和减数分裂期间复制分离 的瓶颈现象
① 在异质性的细胞中,突变型和野生型的比 例决定了细胞是否能量短缺。 ②不同组织阈值差异很大,决定于特定细 胞或组织对能量的依赖程度: 脑>骨骼肌>心>肾>肝
③不同的 mtDNA 突变阈值的大小不同, 如 tRNA 点 突 变 的 阈 值 为 90% , 而 mtDNA大片段缺失的阈值为60%。
线粒体肌病理课件

治疗---采用酶调解药物治疗
? 辅酶Q10可使乳酸和丙酮酸水平降低 ? 核黄素对累及脂质代谢的病人有效 ? 激素可减轻乳酸中毒的症状 ? 可应用VitB族和VitK3药物
病例 二
病例特点:
? 1.患者少年男性,缓慢起病,逐渐加重。
? 2.半年前出现左颞顶部阵发性跳痛,伴一 过性视物模糊,一周前头痛加重伴恶心 呕吐。
301病例Leabharlann 论病例 一一.病例特点:
? 1.患儿女性,13岁,急性起病反复病程。 ? 2. 3年来反复出现头痛,视物不清,抗
炎及激素治疗好转。入院前再次出现头 痛,言语不清伴双耳听力下降,抽搐发 作。 ? 3.既往史:此次起病前有流脑疫苗接种史, 无其它病史。
? 4.查体:BP110/60mmHg,自动体位查体不 合作。神志清楚,表情淡漠,言语不清。 脑膜刺激征(-),双瞳孔直径3mm,等圆等 大,光反应存在,眼动充分,无眼震, 双侧额面纹对称,张口伸舌不偏,无吞 咽困难。双眼底视乳头边界清,无出血 及渗出。双侧听力检查欠合作。
MELAS型病理表现及基因定位
? 1、脑组织病理: CNS的改变以出现灶状坏死 性病变为特征,病变主要累及双侧半球后部皮 层:多位于脑回顶部,其它部位病变轻微,病 变周围星型胶质细胞增生,并且小血管异常增 多,增生血管管腔大小不等,厚薄不均,颅内 大血管未见异常。 MELAS临床表现的卒中样 发作,可能就是这些远端的异常血管网的局部 渗出出血或循环障碍所致,称线粒体性血管病, 与动脉源性梗塞范围不同。 另一个常见的病理 学改变是铁质沉积,以基底节,尤其是苍白球, 其次丘脑,齿状核和间脑,也可以出现脑组织 海绵状改变,并累及大脑皮层及脊髓后索和侧 索。
? 血抗中性粒细胞抗体:IgG增高。
13线粒体疾病mitochondrialdiseases37页PPT

Medical Genetics
Mutations (General)
– 3 Mutations account for 96% of cases
• All in Complex I genes • Mutations: G11778A (69%), G3460A (13%),
T14484C (14%)
Medical Genetics
13 线粒体疾病
mitochondrial diseases
Medical Genetics
Mutations (changes) in the mitochondrial chromosome are responsible for a number of disorders.
In mammals, 99.99% of mitochondrial DNA (mtDNA) is inherited from the mother. This is because the sperm carries its mitochondria around a portion of its tail and has only about 100 mitochondria compared to 100,000 in the oocyte.
• Disease signs especially manifest in
– Tissues with a high energy expenditure: Dependent on oxidative metabolism
– Specific tissues: Brain, Heart & Muscle
• 40% of patients with commonest mutation (G11778A) have negative family history
相关主题
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
• Muscle pathology
– No ragged red fibers – EOM mitochondria: Diffuse increase in
number and size; Disorganized cristae – Preservation of myofibrils
• MRI: Optic nerve may enhance on T2 weighted images
2020/9/30
The incidence about 1:3000-4000 individuals in the US.
2020/9/30
Characteristics
Unlike nuclear genes, which are inherited from both parents, mitochondrial genes are inherited only from the mother.
2020/9/30
4. MERRF
MERRF=Myoclonic Epilepsy; Ragged Red Fibers
2020/9/30
mtDNA point mutations: • tRNA Lys : A8344G (Frequent); T8356C;
G8363A; G8361A
– Syndromes: MERRF or MERRF/MELAS overlap
2020/9/30
Genetic counseling: A3423G mutation
• % of affected offspring: Increased with higher mutant load in maternal blood
• Mutant load 1% to 19%: 20% chance of affected offspring
2020/9/30
There are many forms of mitochondrial disease. Mitochondrial disease presents very differently from individual to individual.
2020/9/30
Mitochondrial derent ways.
• Hearing loss (40%) • Myopathy • Short stature (10%) • Lipomata (10%)
2020/9/30
2020/9/30
Laboratory • Lactic acidosis: Variable • Pathology of muscle
2020/9/30
Laboratory
• Lactic acidosis: Blood & CSF • EMG: Mormal or Myopathic • Serum CK: Normal to 2x high (32%) • MRI: Strokes • Biochemistry
– Respiratory chain dysfunction – Reduced activity of Complexes I & IV
– Ocular pathology
Other features: Some families
Cardiac conduction defects; Spastic dystonia; Spastic paraparesis; Dystonia
2020/9/30
2020/9/30
Laboratory
• Painless • Visual loss pattern • Severity: May deteriorate to 20/200 or less • Progression: Mean 4 months; • Interval between eyes affected: ~ 2 months • Tendency to recover depends on mutation • Pupillary reactions: May be relatively spared for degree of visual loss
There may be one individual in a family or many individuals affected over a number of generations.
2020/9/30
If there is a mutation in a mitochondrial gene, it is passed from a mother to all of her children; sons will not pass it on, but daughters will pass it on to all of their children, and so on.
Clinical features (General)
Male predominance:No relation to any X-linked genes Onset :Midlife: Mean 30 years; Range 1 to 70 Visual loss
– Clinical features
of onset; 3291
2020/9/30
Clinical Syndrome • Onset
– Mean = 10 years; Range = 2 to 40
• Encephalopathy: Often episodic • Systemic features • Myopathy • Polyneuropathy • More common in patients with myopathy • Mean life span with full clinical syndrome ~ 2 to
13 线粒体疾病
mitochondrial diseases
2020/9/30
Mutations (changes) in the mitochondrial chromosome are responsible for a number of disorders.
2020/9/30
Mitochondrial disease is a chronic, genetic disorder that occurs when the mitochondria of the cell fails to produce enough energy for cell or organ function.
• 40% of patients with commonest mutation (G11778A) have negative family history
• Large families with maternal inheritance: G11778 & T14484C mutations
2020/9/30
In mammals, 99.99% of mitochondrial DNA (mtDNA) is inherited from the mother.
This is because the sperm carries its mitochondria around a portion of its tail and has only about 100 mitochondria compared to 100,000 in the oocyte.
4 decades
2020/9/30
Scattered abnormal, vacuolated fibers with clear rim: H & E
2020/9/30
Scattered "ragged red" muscle fibers: Gomori trichrome
Ragged red muscle fibers: Gomori trichrome
• tRNA Ser
– Syndromes: MERRF/MELAS overlap; Epilepsia Partialis Continua;
• tRNA Leu
2020/9/30
Onset Late adolescence - Early adult
2020/9/30
Clinical syndrome:
• Mutant load > 20%: 50% chance of affected offspring • Full expression of phenotype in multiple family members:
Rare • Partial expression in multiple family members: Common
2020/9/30
Threshold effect
• % of mutant mtDNAs must be above a threshold to produce clinical manifestations
• % of mutant mtDNAs needed to cause cell dysfunction varies according to tissue oxidative requirements
2020/9/30
Mutations (General)
– 3 Mutations account for 96% of cases
• All in Complex I genes • Mutations: G11778A (69%), G3460A (13%),
T14484C (14%)
2020/9/30
• CNS
– Myoclonus (60%) – Epilepsy (45%) – Cerebellar dysfunction: Ataxia – Dementia – Optic atrophy (20%)
– No ragged red fibers – EOM mitochondria: Diffuse increase in
number and size; Disorganized cristae – Preservation of myofibrils
• MRI: Optic nerve may enhance on T2 weighted images
2020/9/30
The incidence about 1:3000-4000 individuals in the US.
2020/9/30
Characteristics
Unlike nuclear genes, which are inherited from both parents, mitochondrial genes are inherited only from the mother.
2020/9/30
4. MERRF
MERRF=Myoclonic Epilepsy; Ragged Red Fibers
2020/9/30
mtDNA point mutations: • tRNA Lys : A8344G (Frequent); T8356C;
G8363A; G8361A
– Syndromes: MERRF or MERRF/MELAS overlap
2020/9/30
Genetic counseling: A3423G mutation
• % of affected offspring: Increased with higher mutant load in maternal blood
• Mutant load 1% to 19%: 20% chance of affected offspring
2020/9/30
There are many forms of mitochondrial disease. Mitochondrial disease presents very differently from individual to individual.
2020/9/30
Mitochondrial derent ways.
• Hearing loss (40%) • Myopathy • Short stature (10%) • Lipomata (10%)
2020/9/30
2020/9/30
Laboratory • Lactic acidosis: Variable • Pathology of muscle
2020/9/30
Laboratory
• Lactic acidosis: Blood & CSF • EMG: Mormal or Myopathic • Serum CK: Normal to 2x high (32%) • MRI: Strokes • Biochemistry
– Respiratory chain dysfunction – Reduced activity of Complexes I & IV
– Ocular pathology
Other features: Some families
Cardiac conduction defects; Spastic dystonia; Spastic paraparesis; Dystonia
2020/9/30
2020/9/30
Laboratory
• Painless • Visual loss pattern • Severity: May deteriorate to 20/200 or less • Progression: Mean 4 months; • Interval between eyes affected: ~ 2 months • Tendency to recover depends on mutation • Pupillary reactions: May be relatively spared for degree of visual loss
There may be one individual in a family or many individuals affected over a number of generations.
2020/9/30
If there is a mutation in a mitochondrial gene, it is passed from a mother to all of her children; sons will not pass it on, but daughters will pass it on to all of their children, and so on.
Clinical features (General)
Male predominance:No relation to any X-linked genes Onset :Midlife: Mean 30 years; Range 1 to 70 Visual loss
– Clinical features
of onset; 3291
2020/9/30
Clinical Syndrome • Onset
– Mean = 10 years; Range = 2 to 40
• Encephalopathy: Often episodic • Systemic features • Myopathy • Polyneuropathy • More common in patients with myopathy • Mean life span with full clinical syndrome ~ 2 to
13 线粒体疾病
mitochondrial diseases
2020/9/30
Mutations (changes) in the mitochondrial chromosome are responsible for a number of disorders.
2020/9/30
Mitochondrial disease is a chronic, genetic disorder that occurs when the mitochondria of the cell fails to produce enough energy for cell or organ function.
• 40% of patients with commonest mutation (G11778A) have negative family history
• Large families with maternal inheritance: G11778 & T14484C mutations
2020/9/30
In mammals, 99.99% of mitochondrial DNA (mtDNA) is inherited from the mother.
This is because the sperm carries its mitochondria around a portion of its tail and has only about 100 mitochondria compared to 100,000 in the oocyte.
4 decades
2020/9/30
Scattered abnormal, vacuolated fibers with clear rim: H & E
2020/9/30
Scattered "ragged red" muscle fibers: Gomori trichrome
Ragged red muscle fibers: Gomori trichrome
• tRNA Ser
– Syndromes: MERRF/MELAS overlap; Epilepsia Partialis Continua;
• tRNA Leu
2020/9/30
Onset Late adolescence - Early adult
2020/9/30
Clinical syndrome:
• Mutant load > 20%: 50% chance of affected offspring • Full expression of phenotype in multiple family members:
Rare • Partial expression in multiple family members: Common
2020/9/30
Threshold effect
• % of mutant mtDNAs must be above a threshold to produce clinical manifestations
• % of mutant mtDNAs needed to cause cell dysfunction varies according to tissue oxidative requirements
2020/9/30
Mutations (General)
– 3 Mutations account for 96% of cases
• All in Complex I genes • Mutations: G11778A (69%), G3460A (13%),
T14484C (14%)
2020/9/30
• CNS
– Myoclonus (60%) – Epilepsy (45%) – Cerebellar dysfunction: Ataxia – Dementia – Optic atrophy (20%)