第9章 开环歧化聚合
第9章 开环歧化聚

卡宾型或亚甲基型催化剂: 这些催化剂引发机 理清晰, 催化活性高, 产物的立体选择性强
9.2 开环岐化聚合原理
目前最重要的催化剂有三大类:
1. 传统催化剂 20世纪50年代,Anderson等人开发的,后经
Laverty等人发展的。这类催化剂除了对普通的环 烯烃有较好的催化活性外,对极性单体,如共轭二 羰基化合物也有很好的催化活性。通过所得到的聚 合物的立构规整性受催化剂的立体构型控制, 可在 一定范围内调节,因此具有较好的分子结构可设计 性。
开环歧化聚合
9.1 概述
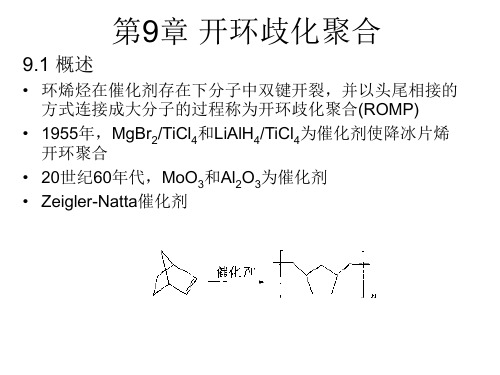
开环歧化聚合:环烯烃在催化剂存在下分子中双 键开裂,并以头尾相接的方式连接成大分子的过 程称为开环歧化聚合(Ring-Opening Metathesis Polymerization,ROMP)
催化剂
n
降冰片烯
9.1 概述
环烯烃的开环聚合研究开始于20世纪50年代。 1955年,Anderson和Merckling等人分别以 MgBr/TiCl4和LiAlH4/TiCl4为催化剂降冰片烯开 环聚合。
20世纪60年代,Eleuterio等以氧化钼和氧化铝, 也使降冰片烯开环聚合。
Truett等人证明用Zeiger-Natta催化剂也可使将冰 片烯聚合为完全相同结构的聚合物,并提出了σ建 (环双键旁)断裂的开环的机理。
9.1 概述
将冰片烯的开环聚合本质上是双键不断易位,分 子链逐渐扩大的过程,因此也称为开环易位(移位) 聚合。
利用开环岐化聚合可开发出许多其他方法无法 实现的聚合物新品种, 如上面介绍过的严格交 替三元共聚物的制备。
开环岐化聚合的出现,为高分子材料,尤其是 功能高分子的结构设计又提供了一种有用的手 段。 开环岐化聚合已成为进行新型高分子材料 结构设计、裁制和分子组装的强有力的工具。
有机化学 开环聚合

传统酚醛树脂
苯并噁嗪树脂固化过程 中进行开环聚合,不释 放出低分子物,改变了 酚醛树脂传统的工艺路 线,成型工艺简单,原 料广泛。
但 是
3 新型材料的制备
2.1苯并噁嗪的合成和聚合
合成:
酚醛式结构
聚合:
芳香醚式结构
3 新型材料的应用
1986年Gilliom等人发表了以Ti
杂烷丁环为催化剂的降冰片烯 开环易位聚合,所得产物的分 子量分布窄。并跟踪聚合反应 发现开环易位聚合是活性聚合。
烯烃的均相催化歧化,他将这一
反应命名为“烯烃易位反应”。
1
3
2
1976年开环易位聚降冰片
4
至今,利用开环易位聚合除了可以获 得特殊结构的均聚物外,还可以得到 严格交替的共聚物,而且反应速度很
分子结构中实际含有卡 宾部分,活性大,能使低 环张力的单体发生聚合。 同,Grubbs催化剂对极 性官能团、痕量氧和湿 气都不敏感。
烯烃复分解反应式 反应产物保留了 C=C
3 新型材料的制备
环烯烃的开环易位聚合是环烯烃通过链聚合转化成聚合物材料 的过程。聚合的机理主要是依据烯烃的易位反应,金属中间体 和 C=C 之间交换,生成一种含有不饱和 C=C 的聚合物。该过 程区别于典型的烯烃加成反应,如乙烯加成得到聚乙烯。
1967年,Calderon用WCl6Et2OH催化体系由2-戊烯得到了 2-丁烯和3-己烯,成功地实现了
1
开环聚合的分类和机理
——能否开环及聚合能力的大小
环状化合物很多,开环聚合的倾向各异: 三、四元环容易开环聚合; 五、六元环能否开环聚合与环中杂原子有关,由于杂原子提 供了引发剂亲核或亲电进攻的位置,所以在动力学上它们比 环烷烃更有利于开环聚合。 七元环以上的聚合可能性又加大,七、八元环也能开环聚合, 但环与线性聚合物往往构成平衡。
开环歧化聚合反应的原理

开环歧化聚合反应的原理
开环歧化聚合反应是一种通过引入外部骨架或功能单体,将环状分子转变为线性或支化高分子的反应。
开环歧化聚合反应的原理可分为两个步骤:
1. 开环反应:环状分子的环被打开,生成开链的中间产物。
通常是通过引入外部的剂来破坏分子之间的环状结构。
开环剂可以是氧化剂、还原剂、酸、碱或其他特定的引发剂。
开环反应的结果是生成具有反应位点的链状或链状中间产物。
2. 歧化反应:开链中间产物上的反应位点进行聚合反应,形成线性或支化的高分子。
歧化反应可以是自由基聚合、阴离子聚合、阳离子聚合等不同机制的反应。
在聚合反应中,开链中间产物上的反应位点与引发剂或催化剂发生反应,引发聚合反应,将分子进行连接或交联,形成高分子链。
总的来说,开环歧化聚合反应通过打破环状分子的结构,引入外部的剂来生成开链的中间产物,然后通过桥接或连接反应将这些中间产物进行聚合,形成线性或支化的高分子链。
第9章聚合反应工程

8 化学反应工程
2.1 缩聚反应
第9章 聚合反应工程
• 由具有两个或两个以上反应性官能团的低分子化 合物(单体)相互作用生成大分子的过程。
• 单体有两个反应性官能团时得到线形高分子化合 物。
• 单体有两个以上反应性官能团时得到非线形或网 状聚合物。
21 化学反应工程
反应的四个阶段
第9章 聚合反应工程
• 诱导期
当聚合体系中含阻聚剂或其他杂质是,生成自 由基先被消耗掉。
• 等速阶段 • 加速阶段 • 减速阶段
转化率在10%~20%以下,黏度较低,体系 中自由基数目不变,稳态阶段。
体系黏度加大,单体可自由扩散到长链自由基 处反应,但链自由基不易自由扩散使链终止难 发生,聚合速度上升,平均聚合度增大,为自 动加速效应,放热严重,易引起爆炸。
黏度更大,扩散困难,单体浓度下降,聚合速度 减慢。
22 化学反应工程
第9章 聚合反应工程
聚合速度:单体消失速度
Rp K p M fkd I Kt 1/ 2 Rp K p M Ri 2Kt 1/2
Rp是链增长速度,[M]单体浓度,[I]引发剂浓度,Kt为链终止 速度常数,f为引发剂效率,一般0.5~1之间,Ri为引发速度。 影响Rp的除了单体浓度就是引发速度Ri,若单体浓度相同, 比较聚合速度就是比较引发速度。 另外从能量消耗上看,主要消耗在引发阶段即引发剂分解,单 体聚合活化能很低在21kJ/mol。低温可聚合,但是关键在于低 温是否可以引发。
6 化学反应工程
第9章 聚合反应工程
4. 聚合反应工程与其他反应工程的区别: a. 总是包含多步的复杂反应,除原料单体转化率外还要
开环歧化聚合

高分子现代合成方法与技术
• 卡宾型或亚烷基型催化剂
高分子现代合成方法与技术
在卡宾型或亚烷基型催化剂中,最重要的是Schrock 型和 Grubbs Ru型两大类。
Schrock型催化剂 Schrock型催化剂的主要优点是其引发机理 清晰。它的催化活性、产物的立体选择性以 及与杂原子的相容性取决于ห้องสมุดไป่ตู้氧取代基的性 质。其配体的种类繁多。与同类型的钨系卡 宾催 化剂相比,副反应较少。 Grubbs Ru型催化剂 Ru盐作为开环歧化聚合催化剂已经有很长 的时间。80年代后期,Grubbs发现含水的 Ru盐化合物用作开环歧化聚合催化剂不仅 可耐痕迹量的水,而且用于水性体系的开环 歧化聚 合有很高的催化活性。
高分子现代合成方法与技术
The Modern Methods and Technology of Polymer Synthesis
高分子现代合成方法与技术
教材: 王国建 同济大学
高分子现代合成方法与技术
第8章 开环歧化聚合(4学时)
Ring Opening Metathesis Polymerization
高分子现代合成方法与技术
8.1 概述
• 环烯烃是烯烃类化合物中一个重要的组成部分,包括单环 和多环烯烃化合物。前者以环戊烯为代表,后者则以降冰 片烯为代表。
• 降冰片烯的开环聚合本质上双键不断易位,分子链逐渐扩 大的过 程。因此也称为开环易位聚合。聚合后,单体中 的双键、单键和环结构在所得的聚合物的重复单元中依然 保持不变,只是连接方式发生了变化,因此是一种完全不 同于传统聚合的新的聚合 方法,所得的聚合物具有十分 特殊的性能。
(2)增长的大分子卡宾络合物与终止剂反应生成无引发活性的金属卡 宾络合物。
化工新材料课程教学大纲(共享).doc

化工新材料课程教学大纲第一部分大纲说明一、课程性质和任务《化工新材料》是江苏广播电视大学开放教育应用化工技术专业(专科)的一门选修课。
本课程共1学分,18学时,开设一学期。
木课程的主要内容包括:化工新材料的教学内容包括功能聚合物载体、离了交换树脂、高效高质高了交换膜材料、聚合物负载试剂、聚合物负载有机催化剂、聚合物负载金属催化剂、吸附分离功能树脂、基于模板印迹的分了识别材料、有机薄膜光伏材料与器件、新能源材料、先进功能陶瓷和智能型医用高分了材料等教学内容。
通过该课程的学习,使学生能够建立起对化工新材料的全面认识。
本课程介绍多种功能材料的新知识、新理论、新技术和新工艺,通过本课程的学习,使得学生对近代化学材料研究领域的一些新理论、新方法以及新材料有较深入的了解,基本掌握主要功能材料的种类、性能特点及应用。
通过对典型的功能材料的介绍,使学生了解如何从化学角度去开发材料的功能特性,并具备运用所学的知识来开发与设计新的功能材料的基本素质。
通过木课程的学习培养学生良好的学习习惯、严谨的治学态度、实事求是的科学作风和分析解决问题的能力,使其逐步成为具备较强的实际工作能力的化工类高技能、可持续发展的应用性人才。
二、与其它课程的联系《化工新材料》是一门综合性课程,涉及到《无机化学》、《有机化学》、《分析化学》《物理化学》、《化工原理》等基础课程,通过学习,学生们能对现代新材料的结构、性能和应用有所了解,对将来学习和从事工作有所裨益。
三、课程教学基本要求本课程要求学生掌握材料在人类生活和社会发展中的重要地位和作用,了解材料的基本类型,尤其是材料的分类、组成与结构、性能与应用、制备与合成的知识,了解高新技术材料的最新发展趋势,具备认识和比较各种材料的能力,为今后从事材料的研究与开发、选择和使用打下坚实的基础。
四、教学方法及教学形式建议在讲授课程内容应木着“必需”、“够用”的原则。
在讲解重点、难点内容时应充分利用共享课程的资源,合理地利用文字教材、录像教材和网上教学等手段,使之更好地为教学服务。
开环聚合课件

1、自由基开环活性的机理
以DTBP作引发剂,TEMPO作增长链稳定剂,2-亚 甲基-1,3-二氧环庚烷可进行活性开环聚合,具 体反应过程如下:
以α,α’-二溴代二甲苯为引发剂, 溴化亚铜 /2,2’-联吡啶为催化剂, 首次实现了5, 6-苯并-2亚甲基-1, 3-二氧环庚烷的活性自由基开环聚合反 应[40]。
第七节
开环歧化聚合
在催化剂存在下,环烯烃分子中双键开 裂,并以头尾相接的方式连接成大分子的 过程称为开环歧化聚合。环烯烃按结构可 分为单环和多环烯烃。前者以环戊烯为代 表,后者则以降冰片烯为代表。
降冰片烯的开环聚合如下:
对环烯烃的开环歧化聚合的机理,目前普遍接受的 观点是金属卡宾配位化合物引发、增长机理。
CH2CH2O
n
O Na + ROH
R
CH2CH2O
n
+ RO-Na+ OH
交换反应生成的醇盐可继续引发聚合反应。从形 式上看,交换反应与链转移反应相似,但与链转移 反应不同,交换反应生成的端羟基聚合物并不是 “死”的聚合物,而只是休眠种,可和增长链之间 发生类似的交换反应再引发聚合反应:
R CH2CH2O n OH + R CH2CH2O m O-Na+ + R R CH2CH2O CH2CH2O m OH n O-Na+
2、聚合方法比较
方法 因素 推动力 结构单元 特殊基团 开环聚合 连锁聚合 逐步聚合 官能团性质的改 变 聚合过程中有小 分子放出 单体分子间官能 团的相互作用 形成的 苛刻 单体的环张力 化学键键型的 改变 与单体的化学 与单体的化学 组成相同 组成相同 单体分子固有 的 温和 一般 无
聚合条件
引发反应首先从环烯烃中的双键与金属卡宾M=CHR配 位后生成金属杂环丁烷过渡态开始。该过渡态以易 位方式裂解,形成新的增长金属卡宾配位化合物。 新形成的的金属卡宾络合物可继续与环烯烃单体上 的双键形成金属杂环丁烷过渡态,断裂后又形成新 的增长金属卡宾配位化合物,如此反复进行,即得 到高分子聚合物。
第9章 开环歧化聚合
• 1986年,Gilliom发现以Ti杂环丁烷为催化剂的降冰片烯开 环歧化聚合为活性聚合 • Nguyen 以三苯基膦卡宾型Ru络合物为催化剂进行降冰片 烯的开环歧化聚合
9.3.2 降冰片烯衍生物的开环歧化聚合
• 含氟降冰片烯的开环歧化聚合
• 含硼降冰片烯衍生物的开环歧化聚合
• 乙酰氧基降冰片烯的开环歧化聚合
• ③大分子卡宾配位化合物与体系中外加无环烯烃发生交叉 歧化反应
可能发生的链终止反应 • ①增长的大分子卡宾型配位化合物活性链端发生α氢转移, 生成带末端双键的大分子,并还原出金属原子
• ②增长的大分子卡宾型配位化合物与终止剂反应生成无引 发活性的金属卡宾配位化合物: 三甲基乙烯基硅烷、1,1二苯乙烯、乙烯基醚等。
• ③单体与催化剂的活性中心金属原子配位后发生双键上氢 转移反应,直接形成金属卡宾配位化合物。
引发反应:链增长反应:来自由此可见,开环歧化聚合的活性中心实际上是由金属卡宾 配位化合物与环烯烃单体上的双键形成的金属杂环丁烷过 渡态。
可能发生的链转移反应 • ①自身反咬转移 • ②大分子金属卡宾配位化合物与体系中的其他大分子发生 交叉歧化反应
• 带有不饱和侧基的聚合物与环烯烃进行歧化反应得到接枝 共聚物
9.4 开环歧化聚合在聚合物分子设计方面的应用
9.4.1 恒比共聚物的合成
9.4.2 理想交替共聚物的合成
9.4.3 全顺式聚合物和全反式聚合物的合成
9.4.4 全同立构全顺式共聚物和间同立构全反式聚合物的合 成
9.4.5 嵌段共聚物和接枝共聚物的合成
• 利用开环歧化聚合除了可以得到特殊结构的均聚物外,还 可以得到严格交替的 共聚物:
9.2 开环歧化聚合原理
9.2.1 开环歧化聚合催化剂 • 1 传统催化剂:MXn(其中,M=W、Mo、Re、Os 等过渡金属,X=Cl、Br等卤素原子) • 2 水溶性催化剂:K2RuCl3 H2O • 3 卡宾型或亚烷基型催化剂
开环易位(歧化)聚合-ROMP

高分子化学进展
三、开环易位(歧化)聚合
开环聚合:
环状单体开环相互连接形成线型聚合物的过程,称为开环 聚合。开环聚合为链式聚合反应,包括链引发、链增长和链终 止等基元反应。但开环聚合反应与乙烯基单体的链式聚合反应 有所区别,其链增长反应速率常数与许多逐步聚合反应的速率 常数相似,而比通常乙烯基单体的链式聚合反应低数个数量级。
他亲自走出讲台,邀请身边的 皇家科学院教授和两位女工作 人员一起在会场中央为大家表 演烯烃复分解反应的含义。最 初两位男士是一对舞伴,两位 女士是一对舞伴,在“加催化 剂”的喊声中,他们交叉换位 ,转换为两对男女舞伴,在场 记者随即发出了笑声。
烯烃复分解反应最初应用在石油工业中,以SHOP法的产物α-烯烃为原料, 高温高压下生产高级烯烃。传统的反应催化剂如WCl6-EtOH-EtAlCl2,由 金属卤化物与烷化剂反应制取。 烯烃复分解反应是个循环反应,过程为:首先金属卡宾配合物与烯烃反应, 生成含金属杂环丁烷环系的中间体。该中间体分解,得到一个新的烯烃和 新的卡宾配合物。接着后者继续发生反应,又得到原卡宾配合物。
开环易位聚合的单体是环烯烃,如果是开环烯烃, 生成什么产物?
瑞典皇家科学院2005年10月5日宣布,将2005年诺贝尔化 学奖授予法国化学家伊夫·肖万(Yves Chauvin)、美国化学家 罗伯特·格拉布(Robert H. Grubbs)和理查德·施罗克(Richard R. Schrock),以表彰他们在烯烃复分解反应研究领域作出的贡 献。在宣布仪式上,诺贝尔化学奖评委会主席佩尔·阿尔伯 格将烯烃复分解反应描述为“交换舞伴的舞蹈”。
金属卡宾
高活性,聚合反应控制能力强,可进行活性聚合
(3)可能存在的链转移:
开环聚合——精选推荐

开环聚合第⼋章开环聚合8.1 概述⾼分⼦化学中,以环状单体通过开环聚合来合成聚合物,同样具有重要的地位。
在这种聚合过程中,增长链通过不断地打开环状结构,形成⾼聚物:以环醚为例,环氧⼄烷经开环聚合反应,得到⼀种聚醚,即聚氧化⼄烯。
这在⼯业上已得到应⽤。
能够进⾏开环聚合的单体很多,如环状烯烃,以及内酯、内酰胺、环醚、环硅氧烷等环内含有⼀个或多个杂原⼦的杂环化合物。
开环聚合既具有某些加成聚合的特征,也具有缩合聚合的特征。
由开环聚合得到的聚合物,重复单元与环状单体开裂时的结构相同,这与加成聚合相似;⽽聚合物主链中往往含有醚键、酯键、酰胺键等,与缩聚反应得到的聚合物常具有相同的结构,只是⽆⼩分⼦放出。
开环聚合与缩聚反应相⽐,还具有聚合条件温和、能够⾃动保持官能团等物质的量等特点,因此开环聚合所得聚合物的平均分⼦质量,通常要⽐缩聚物⾼得多。
有些单体如乳酸,采⽤缩聚反应⽆法得到⾼分⼦质量的聚合物;⽽采⽤乳交酯的开环聚合,就能够获得⾼分⼦质量的聚乳酸。
但是,与缩聚反应相⽐,开环聚合可供选择的单体较少,例如⼆元酸与⼆元醇能够通过缩聚获得聚酯;⽽开环聚合,只有相当于α,ω-羟基酸的环内酯可供选择。
聚酰胺的情况也是如此。
另外,有些环状单体合成困难,因此由开环聚合所得到的聚合物品种受到限制。
开环聚合就机理⽽⾔,有些属于逐步聚合,有些属于连锁聚合。
8.1.1 聚合范围及单体可聚性如前所述,环醚、环酯、环酰胺、环硅氧烷等能够进⾏开环聚合。
此外,环胺、环硫化物、环烯烃、以及N-羧基-α-氨基酸酐等同样也能进⾏开环聚合。
环状单体能否转变为聚合物,取决于聚合过程中⾃由能的变化情况,与环状单体和线形聚合物的相对稳定性有关。
Dainton 以环烷烃作为环状单体的母体,研究了环⼤⼩与聚合能⼒的关系。
表6-1列出了环烷烃在假想开环聚合时的⾃由能变化ΔG lc 0、焓变ΔH lc 0、及熵变ΔS lc 0。
R X [ R X ]n n [ CH 2 CH 2 O ]n n H 2C CH 2O聚合过程中,液态的环烷烃(l )转变为⽆定型的聚合物(c )。
化学反应工程第九章

C6H5C O O C O O
CH=CH2
C6H5
Heat
2C6H5● + 2CO2
C6H5—CH—CH2●
b.链的引发
2C6H5● +
襄樊学院
7
《化学反应工程》
C6H5—CH—CH2●
CH=CH2
C6H5—CH—CH2— CH—CH2●
c.链的生长
+
C6H5—CH—CH2●
...
n
d[ M ] R总 Ri RP RP dt
襄樊学院
28
《化学反应工程》
4.普适方程 链引发速率: Ri 2 fkd [ I ] d[ M ] 链增长速率: RP dt k P [ M ][ RM ] d [ RM ] 2 R k [ RM ] 链终止速率: t t dt 1 fkd 1 2 ) [ M ][ I ]2 普适方程: R总 RP k P ( kt
襄樊学院
《化学反应工程》
§9.1.3 聚合物的分子量、聚合度和分子量分布 (一) 数均平均值
——把用端基滴定法、冰点下降法或渗透压法测得 的值,称为数均平均值
Mn
M jN j
j 1
Nj
j 1
Pn
j[ Pj ]
j 1
[ Pj ]
j 1
襄樊学院
17
《化学反应工程》
——此式可作为自由基聚合反应的判据,亦称平方根定则
襄樊学院
29
《化学反应工程》
由上可见,只要知道反应机理和各基元反应的速率 常数与单体及引发剂的浓度就可以算出分批操作时的反 应速率及转化率,进而求出聚合物的平均分子量与分子 量分布。 此外,按表9-4至表9-7可知,尽管kt较kp大3~5数量 级,但 [RM.] 极低 ( 约 10-7 ~ 10-8 mol/L) ,远较 [M]( 约 10 mol/L)要小,所以 rP比 rt仍要大3~4数量级,因此能够 生成聚合度为103~104的高聚物。
9开环聚合热力学

第9 章9.2.1 相变条件相变过程的推动力根据热力学理论,相变过程的推动力是相变前后体系自由能的差值,0T P G ∆≤当ΔG=0时,相变达到平衡当ΔG <0时,相变过程自发进行相变过程的温度条件由热力学可知,在等温等压下有: ST H G ∆-∆=∆则0H S T ∆∆=平衡条件下,△G =0,则有:00H T S ∆-∆=式中 T 0——相变的平衡温度 △H ——相变热相变过程的温度条件若在任意一温度T 的不平衡条件下,则有: G H T S ∆=∆-∆≠0若△H 与△S 不随温度而变化,将△S 表达式代入上式得:000T T H T T T H T H T H G ∆∆=-∆=∆-∆=∆相变过程要自发进行,必须有△G <0,则: 0T T H 0<∆∆讨论①若相变过程放热(如凝聚过程、结晶过程等)△H<0,要使△G<0,必须有△T>0,即△T=T-T>0,即T0>T,这表明系统必须过冷,即系统实际相变温度比理论相变温度要低,才能使相变过程自发进行。
讨论②若相变过程吸热(如蒸发、熔融等)△H>0,要满足△G<0,则必须△T<0,即T<T,这表明系统要自发发生相变必须过热。
讨论相变驱动力可以表示为过冷度(过热度)的函数,相平衡理论温度与系统实际温度之差即为相变过程的推动力。
相变过程的压力和浓度条件根据热力学理论,在恒温可逆不作有用功,即非体积功为零时: dG VdP=1212ln P P P RT G VdP dP RT P P ∆===⎰⎰对理想气体而言: 当过饱和蒸汽压力为P 的气相凝聚成液相或固相时(其平衡蒸汽压力为P 0)时,有: ln P G RT P∆=0讨论,要使相变能自发进行,必须有△G<0,即P>P也即要使凝聚相变自发进行,系统的饱和蒸汽压应。
大于平衡蒸汽压P这种过饱和蒸汽压差为凝聚相变过程的推动力。
对溶液而言,可以用浓度C 代替压力P ,上式可写成:CC RT G 0ln =∆若是电解质溶液还要考虑电离度α,即1mol 能电离出αmol 离子,则: ln ln()C C C G RT RT RT C C C∆∆∆=α=α+≈α⋅01式中C 0——饱和溶液浓度 C ——过饱和溶液浓度,若相变过程自发进行,则△G<0,故△C<0,即C>C)即即液相要有过饱和浓度,它们之间的差值(C-C为这一相变过程的推动力。
高分子问答11-15

11.简述基团转移聚合单体和引发剂的结构特点
12什么是开环歧化(易位)聚合?
开环歧化聚合是指:环烯烃的双键和金属卡宾配位,生成金属杂环丁烷过渡态,键被活化后断裂生成增长型金属卡宾配合物,该配合物继续和环烯烃的双键形成环状过渡态,再断裂,一直保持链的增长,得到高聚物。
由于形成环前后,键断裂的位置发生变化,因而叫易位或移位。
13 开环歧化聚合主要有哪些催化体系
主要由以上三种催化体系
14 以金属卡宾催化体系为例,简述开环歧化聚合机理
非环二稀的聚合机理
15 什么是酶催化聚合?简述酶催化聚合特点与应用。
开环聚合

这是一个平衡反应,必须真空除去副产物BH, 使平衡向右移动。然后,内酰胺阴离子与单体 反应而开环,生成活泼的胺阴离子(II)。
(2)内酰胺阴离子活性种(I)与另一己内酰胺单 体分子反应,形成活泼的胺阴离子活性种(II):
O C (H2C)5 (I) N M HN O C (CH2)5
慢
O C (H2C)5 N C O (CH2)5 (II) (反应2) H N M
8.4环醚的阳离子开环聚合 (3、4、5元环)
8.4 环醚的阳离子开环聚合机理
有些环醚阳离子开环聚合具有活性聚合的特性,如活 性种寿命长,分子量分布窄,引发比增长速率快,所 谓快引发慢增长。但往往伴有链转移和解聚反应,使 分子量分布变宽;也有终止反应。结合四、五元环醚 阳离子开环聚合,介绍各基元反应的特征。 (1)链引发与活化 有许多种阳离子引发剂可使四、 五元环醚开环聚合。 ①质子酸和Lewis酸。如浓硫酸、三氟乙酸、氟磺酸、 三氟甲基磺酸等强质子酸(H+A-),以及BF3、PF5、 SnCl4、SbCl5等Lewis酸,都可用来引发环醚开环聚合。 • Lewis酸与微量共引发剂(如水、醇等)形成络合物, 而后转变成离子对(B+A-),提供质子或阳离子。有些 Lewis酸自身也能形成离子对。
M O N C (CH2)5 NH
(反应4)
(反应4)
增长反应首先是活性较高的N—酰化内酰胺与内 酰胺阴离子反应,使N—酰化内酰胺开环。
O C (CH2)3 N M O C (CH2)3 N O C (CH2)5 + O C (CH2)5 N O C (CH2)5 NH2
( III )
M N O C (CH2)5
胺阴离子(II)无共轭作用,较活泼,很快夺取 另一单体己内酰胺分子上的一个质子,生成二聚 体( III ),同时再生内酰胺阴离子(I)。
化学反应工程 第九章 聚合反应过程

化学反应工程
9.1.2 聚合方法与设备
②管式
化学反应工程
9.1.2 聚合方法与设备
③塔式
化学反应工程
9.1.2 聚合方法与设备
④特殊形式的聚合反应器
化学反应工程
9.1.3 聚合物的分子结构、分子量和 分子量分布
用端基滴定法,冰点下降法,沸点上升法或渗透压法测得
的是数量平均分子量
简称数均分子量
化学反应工程
9.6 聚合过程的拟定和调节
1
混合过程的拟定 聚合过程的调节
2
化学反应工程
9.6.1 混合过程的拟定
实施一项聚合过程,其目的是要以最有效的方式获得所
需要的产品,针对一定的聚合体系,需要 ①选定合适的聚合方法; ②选定合适的操作方法; ③规定合适的工艺条件;
④确定合适的反应器型式;
⑤确定反应器的结构尺寸; ⑥确定反应系统的调节和控制方案。
化学反应工程
9.6.1 混合过程的拟定
化学反应工程
9.6.1 混合过程的拟定
化学反应工程
9.6.2 聚合过程的调节
聚合过程中的主要可调因素是:
①物料的浓度;
②反应的温度; ③反应的时间(即相当于转化率)。 调节物料浓度的方案可以举出下列一些: ①改变初始物料的浓度,包括单体及引发剂; ②共聚合时改变两种单体的比例; ③在分批聚合时,分多次陆续添加物料或者连续添加;
或黏均聚合度
至于分子量的分布,亦可按数量或重量作基准而定义
如下:
数量基准的分布 重量基准的分布
化学反应工程
9.1.3 聚合物的分子结构、分子量和 分子量分布
对于不同的聚合方法所得聚合物的分散指数大致如下
:
瞬间数均聚合度
第八章(更新) 开环岐化聚合

8。离子交换树脂的制备
9。特种材料的制备
减震、隔音、阻尼、密封材料
9.特种材料的制备
减震、隔音、阻尼、密封材料
CH2
CH2
RuCl3 /HCl BuOH n
n
催化剂 n
结语
思考题
1. 环烯烃开环聚合所用的催化剂是什么? 2.环烯烃开环聚合过渡态是什么? 3. 写出降冰片烯开环聚合的产物。 4. 烯烃开环聚合的合成上有些什么用途?
用RuCl3 为催化剂时,产物为反式
用RuCl3做催化剂时,产物为反式
RuCl3
n
2。降冰片烯衍生物的开环岐化聚合
内式
外式
2.降冰片烯衍生物的开环歧化聚合
例如以RuCl3.3H2O为催化剂进行endo-DCPD的开环歧化 聚合,得到的是全顺式双键结构聚合物,而对endoDCPD应用开环歧化聚合,得到的则是以反式双键结构为 主的聚合物。下列反应式为两种双环戊二烯异构体的开环 歧化聚合:
CH3
CH2 R WF6 -RCH3 Cl4W R CH2 R
引发反应首先从环烯烃中的双键与金属卡宾配 位后生成金属杂环丁烷过渡态开始。配位后, 环烯烃中的双键与金属卡宾键均被活化,进而 断裂生成新的增长金属卡宾配位化合物:
R R CH M HC R CH M CH R CH M CH HC R M CH
P
CH2 P CH
CH
M Me3Si CH2 Me3Si
CH CH2
CH2 CH M
③增长的大分子卡宾型配位化合物与其他含双键分子发生
环丙烷化反应而终止:
P CH2 -M
CH
M
R1 CH
CH R1
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
• ③单体与催化剂的活性中心金属原子配位后发生双键上氢 转移反应,直接形成金属卡宾配位பைடு நூலகம்合物。
引发反应:
链增长反应:
由此可见,开环歧化聚合的活性中心实际上是由金属卡宾 配位化合物与环烯烃单体上的双键形成的金属杂环丁烷过 渡态。
可能发生的链转移反应 • ①自身反咬转移 • ②大分子金属卡宾配位化合物与体系中的其他大分子发生 交叉歧化反应
• 降冰片烯的开环聚合本质上是双键的不断易位、分子链逐 渐扩大的过程,因此也称为开环易位聚合。聚合后,单体 中的双键、单键和环结构在所得的聚合物的重复单元中保 持不变,只是连接方式发生了改变。 • 1967年,Calderon用WCl6-EtOH/Et2AlCl催化体系实现了 烯烃的均相催化歧化,将这个反应命名为“烯烃易位反 应”。 • 用同样的催化剂进行了环烯烃的开环聚合,提出了聚合反 应是通过环烯烃分子中双键断裂进行的 • 1974年,Dolgoplosk提出了链式卡宾机理 • 1986年,Gilliom等人以Ti杂烷丁环为催化剂进行了降冰片 烯的开环歧化聚合
• 活性开环歧化聚合与其他活性聚合反应联用
开环歧化活性聚合物与含有特定官能团的其他聚合物进行偶 合反应
活性开环歧化聚合与原子转移自由基聚合联用
• 活性开环聚合制备接枝共聚物
9.4.6 梳状共聚物和星状共聚物的合成 梳状聚合物的合成
“梳齿”通过C-C键连接在聚合物主链上的梳状聚合物的合成
合成带功能基团的星状聚合物
• 1986年,Gilliom发现以Ti杂环丁烷为催化剂的降冰片烯开 环歧化聚合为活性聚合 • Nguyen 以三苯基膦卡宾型Ru络合物为催化剂进行降冰片 烯的开环歧化聚合
9.3.2 降冰片烯衍生物的开环歧化聚合
• 含氟降冰片烯的开环歧化聚合
• 含硼降冰片烯衍生物的开环歧化聚合
• 乙酰氧基降冰片烯的开环歧化聚合
其中,最重要的是(c)类和(e) 类,共催化剂主要有 CpTiCl2/RMgX(其中Cp为环戊 二烯,R=CH3,X=Cl、Br、I等)、 二茂钛化合物/MeLi等。
• Schrock型:它的催化活性、产物的立构选择性以及与杂 原子的相容性取决于烷氧取代基的性质。
• Grubbs Ru型
9.2.2 开环歧化聚合的活性中心及其引发机理 • 环烯烃的开环歧化聚合机理:是金属卡宾配位化合物引发、 增长机理。 • 金属卡宾配位化合物获得途径: • ①事先制备:Schrock型配位化合物 • ②由主催化剂(WF6、[Mo(CO)5Cl]等)与助催化剂 (Et2AlCl、Me4Sn)的原位反应制得:
• ③大分子卡宾配位化合物与体系中外加无环烯烃发生交叉 歧化反应
可能发生的链终止反应 • ①增长的大分子卡宾型配位化合物活性链端发生α氢转移, 生成带末端双键的大分子,并还原出金属原子
• ②增长的大分子卡宾型配位化合物与终止剂反应生成无引 发活性的金属卡宾配位化合物: 三甲基乙烯基硅烷、1,1二苯乙烯、乙烯基醚等。
• 利用开环歧化聚合除了可以得到特殊结构的均聚物外,还 可以得到严格交替的 共聚物:
9.2 开环歧化聚合原理
9.2.1 开环歧化聚合催化剂 • 1 传统催化剂:MXn(其中,M=W、Mo、Re、Os 等过渡金属,X=Cl、Br等卤素原子) • 2 水溶性催化剂:K2RuCl3 H2O • 3 卡宾型或亚烷基型催化剂
• ③增长的大分子卡宾型配位化合物与其他含双键分子发生 环丙烷化反应而终止
9.3 降冰片烯及其衍生物的开环歧化聚合
9.3.1降冰片烯的开环歧化聚合 • 1955年,Anderson等人以MgBr2/TiCl4和LiAlH4/TiCl4为催 化剂实现了降冰片烯的开环聚合 • 法国科学家Larroche发现:W系催化剂对降冰片烯的聚合 具有很好的立体选择性;而直链烯烃聚合时,立体选择性 很差。分析原因,有两种观点: • ①开环歧化过程中的立体选择性可能与环烯烃双键向金属 配位时具有一定的方向性以及配体使中心金属产生一定的 位阻有关; • ②立体选择性取决于配位烯烃与金属环丁烷两种结构的能 量大小,如果后者大于前者,体系将失去立体选择性。
4.4.7 遥爪聚合物的合成 以(ArO)2WC4-SnMe4为催化剂,以3-己烯-1,6-二羧酸 甲酯为链转移剂
9.4.8 导电高分子的合成
9.4.9 离子交换树脂的制备
9.4.10 特种性能聚合物的合成 • 反式聚降冰片烯
• 反式聚环辛烯
• 聚环戊烯
• 聚双环戊二烯
9.3.3 降冰片烯及其衍生物的共聚物的合成 1 降冰片烯及其衍生物的直接开环歧化共聚
2 通过降冰片烯及其衍生物的活性开环歧化制备嵌段共聚物 ABA型三嵌段共聚物的合成
星状聚合物的合成
交联聚双环戊二烯的合成
3 活性开环歧化聚合转为其他聚合方式制备嵌段共聚物 • 降冰片烯聚乙烯嵌段性共聚物 • 降冰片烯(乙烯-丙烯)嵌段性共聚物 • 接枝性共聚物
第9章 开环歧化聚合
9.1 概述
• 环烯烃在催化剂存在下分子中双键开裂,并以头尾相接的 方式连接成大分子的过程称为开环歧化聚合(ROMP) • 1955年,MgBr2/TiCl4和LiAlH4/TiCl4为催化剂使降冰片烯 开环聚合 • 20世纪60年代,MoO3和Al2O3为催化剂 • Zeigler-Natta催化剂
• 带有不饱和侧基的聚合物与环烯烃进行歧化反应得到接枝 共聚物
9.4 开环歧化聚合在聚合物分子设计方面的应用
9.4.1 恒比共聚物的合成
9.4.2 理想交替共聚物的合成
9.4.3 全顺式聚合物和全反式聚合物的合成
9.4.4 全同立构全顺式共聚物和间同立构全反式聚合物的合 成
9.4.5 嵌段共聚物和接枝共聚物的合成