脊髓小脑性共济失调
脊髓小脑性共济失调康复训练

脊髓小脑性共济失调康复训练脊髓小脑性共济失调(SCA)是一种遗传性疾病,主要影响脊髓和小脑,导致患者在运动协调和平衡方面出现困难。
康复训练是帮助SCA患者恢复功能和提高生活质量的重要手段。
本文将探讨脊髓小脑性共济失调康复训练的方法和效果。
一、认识脊髓小脑性共济失调(SCA)脊髓小脑性共济失调是一组由基因突变引起的疾病,主要影响小脑的神经元和脊髓。
SCA患者常常出现运动协调异常、步态不稳和姿势不良等症状。
这些问题会影响患者的生活质量和日常活动能力。
二、康复训练的目标康复训练的目标是帮助SCA患者提高肌肉控制能力、恢复平衡能力和提高运动协调性。
通过系统的康复训练,患者可以改善步态、减少姿势异常、增强肌肉力量和提升日常活动能力。
三、康复训练的方法1. 平衡训练:平衡是SCA患者康复训练的重点,因为平衡问题是其主要症状之一。
平衡训练包括静态平衡和动态平衡训练。
静态平衡训练可以通过单脚站立、平板支撑和闭目站立等方式进行。
动态平衡训练可以采用行走训练、平衡板训练和瑜伽等方式进行。
2. 运动协调训练:运动协调是SCA患者恢复日常活动能力的关键。
运动协调训练包括近远景训练、传球训练和目标跟随训练等。
这些训练可以通过改善患者的感知能力和运动规划能力来提高运动协调性。
3. 力量训练:肌肉力量的提升可以增加身体的稳定性和平衡能力。
力量训练可以包括站立推重、坐位推重和体力活动等。
逐渐增加训练强度和持续时间,有助于增强SCA患者的肌肉力量。
4. 柔韧性训练:柔韧性训练可以改善肌肉张力和关节活动范围,有助于减少姿势异常。
常见的柔韧性训练包括伸展运动、瑜伽和普拉提等。
这些训练可以通过拉伸身体各部位来达到改善柔韧性的效果。
四、康复训练的效果脊髓小脑性共济失调的康复训练是一个长期过程,需要患者和康复师的共同努力。
通过持续的康复训练,SCA患者可以明显提高运动协调性和平衡能力,减少步态异常和姿势异常,提高肌肉控制能力和生活质量。
然而,康复训练的效果可能因人而异。
脊髓小脑性共济失调Ⅲ型的临床特点及分子遗传学特征

[ 要 ] 目的 摘 探 讨 脊 髓 小 脑 性 共 济 失 调 Ⅲ型 ( C ) 临 床 特 点 及 分 子 遗 传 学 特 征 。方 法 S A3 的 采 用 聚 合
酶链 反应 一 聚丙 烯 酰胺 凝 胶 电泳 ( C — A ) 术 , 一 S A3家 系 的 8 中 的 3例 患 者 行 S A3 C ) P R P GE 技 对 C 例 C ( AG n的
S CA3 f miy o te t s q e e a no ma ne, n na y i n c i c lf a ur sby f m i a l f3 pa i n s, e u nc b r lge a d a l ss o lnia e t e a l y i e tg ton o nv s i a i fSCA3 pa i n s te t .Re uls Fort hr e pa i n s t e e tnu b r CAG r s t he t e te t he r p a m e ae 7 6 7 i e a dt r 6, 8, 4 tm s, n heno malpe pl r — 4 i e .Co c u in SCA3 i he mo tc m — o ea e1 0 tm s n l so st s o
S As 亚 型 的 临 床 表 现 相互 重叠 , 确 诊 有 赖 于 基 因 诊 断 。 C 各 其 [ 键 词 ] 脊 髓 小 脑 共 济 失 调 Ⅲ型 ; 因突 变 关 基 [ 中图 分 类 号 ] R 4 . 747 [ 献标 志码 ]B 文 [ 章 编 号 ] 1 0—4 7 2 1 ) 30 4 —2 文 0 93 2 (0 0 0 —130
型( E 即 A1和 E ) 是 遗 传 性 共 济 失 调 的 主 要 类 A2 ,
脊髓小脑性共济失调护理PPT课件

合作内容:疾病知识普及、康复训练指导、营养支持等
合作方式:定期会议、联合评估、共同制定护理计划
患者教育
03
心理支持:关注患者的心理需求,提供心理支持和疏导
02
指导康复训练:教授患者进行康复训练,帮助改善症状和功能
01
介绍疾病知识:让患者了解脊髓小脑性共济失调的病因、症状、治疗方法等
04
健康教育:指导患者进行自我管理,提高生活质量和健康水平
病因和症状
病因:遗传因素、环境因素、病毒感染等
症状:运动协调障碍、平衡失调、言语不清、眼球震颤等
诊断和治疗方法
诊断方法:神经科检查、影像学检查、基因检测等
治疗方法:药物治疗、康复治疗、心理治疗等
02
药物治疗:左旋多巴、金刚烷胺等
康复治疗:物理治疗、言语治疗、作业治疗等
心理治疗:认知行为疗法、心理教育等
05
定期监测生命体征,预防病情恶化
06
提高生活质量
关注个人卫生,如保持个人清洁、定期更换衣物等
04
保持良好的家庭和社会支持,如与家人、朋友保持联系,参加社会活动等
05
保持良好的生活习惯,如规律作息、饮食均衡等
01
定期进行康复训练,如进行肢体功能锻炼、语言训练等
03
保持良好的心理状态,如保持乐观、避免焦虑等
脊髓小脑性共济失调护理PPT课件
刀客特万
01
脊髓小脑性共济失调概述
02
护理要点
03
护理措施
04
护理团队协作
目录
脊髓小脑性共济失调概述
1疾病定义Fra bibliotek01脊髓小脑性共济失调是一种神经系统疾病
03
导致运动协调和平衡功能障碍
脊髓小脑性共济失调--御方生髓汤

脊髓小脑性共济失调临床表现:1.初期走路时步履不稳,肢体摇晃。
动作反应迟缓及准确性变差。
2.中期说话时发音含糊不清,无法控制音调。
眼球转动不平顺,影像容易产生“重叠”。
肌肉不协调感加重,无法写字,有时感到吞咽困难,进食时容易呛咳。
3.晚期说话极不清楚,甚至无法语言。
肢体乏力,不能站立,需靠轮椅代步。
理解能力逐步下降,最后失去意识,昏睡不醒。
【方药】御方生髓汤加黄芪、白术、甘草补脾,丹参、白芍、龙眼肉补气血,升麻、柴胡降浊,陈皮调气、当归、补血,姜枣调和营胃卫,山药健脾健胃,使脾胃健康,虚弱自除。
(注:以上中药为御方生髓汤的一部分。
具体的处方应该以医生的处方为准。
未经允许,请勿服药。
)【饮食注意】忌辛辣、油腻、生冷、不易消化食物,忌绿豆、白萝卜、柿子、浓茶,忌鱼虾蟹等海鲜类发物。
脾主筋骨,胃为水谷之海,气血生化之源。
如果脾虚为原发体或虚证所引起的长期疾病,则可视为阳痿证。
例如,“素文、太阴、阳明论”说:“脾脏病了,何不用四肢呢?四肢都是生而有气的胃,但不是经络,因为脾脏,它们是生而有气的。
如今,脾病不能像胃液一样治,四肢不能有水和谷气,气一天比一天衰,脉脉不畅,肌肉不活。
”如果气虚不能在脸上美化,脸就会无色;如果舌轻,苔藓薄白,脉细,则是脾胃虚弱的表现。
御方生髓汤疗法:历代医家对痿证的认识为“内脏不足”,“阴血不足”,“使之太过,肾精枯竭”,“元气败伤,精血虚不能灌溉”等,气血津液所伤而致,其中肝肾之虚为本病主要病机,气血津液不足是形成痿证的主要因素。
御方生髓汤本着“治痿独取阳明”的原则,选用党参、黄芪.白术、甘草补脾气;参以《丹溪心法》“痿之不足乃阴血不足也”,根据痿证病机与致病因素,严谨辨证与精当用药。
御方生髓汤诊疗痿症共济失调三大阶段!第一阶段:止痛通络,修复脊髓神经系统活血镇痛,通脉透骨,活化病变部位的缺血缺氧状态,改善脊髓的病态肿胀,减轻对神经根的压迫,修补损伤的神经细胞,兴奋中枢神经细胞,消除并抑制炎性水肿,使触觉逐渐恢复,改善患者的疼痛、麻木、痉挛、僵硬等外在表现!第二阶段:补精养髓,营养神经提高免疫力补肾生髓,滋养督脉,补充受损部位流失的大量营养成份,滋养神经细胞,提高神经细胞的繁殖和再生能力,重建神经网络。
小脑性共济失调的病理知识

脊髓小脑性共济失调的病理知识本病的生化改变尚不明。
有的病人有丙酮酸氧化的缺陷。
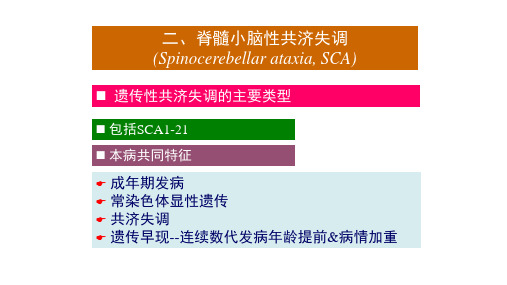
脊髓小脑性共济失调(SCA)是遗传性共济失调的主要类型,其共同特征是中青年发病。
血管病、占位病、常染色体显性遗传和共济失调,经CT扫描证实排除其他累及小脑及脑干的变化。
脊髓小脑性共济失调的患者都会存在着生活上和工作上的问题,当然了一旦患有了脊髓小脑性共济失调这种疾病之后,我们也需要做好患者的心理调适,这是因为患者的心理上往往会存在着一定的落差,不能接受患有这种疾病的现实。
本病的生化改变尚不明。
有的病人有丙酮酸氧化的缺陷。
脊髓小脑性共济失调(SCA)是遗传性共济失调的主要类型,其共同特征是中青年发病。
血管病、占位病、常染色体显性遗传和共济失调,经CT扫描证实排除其他累及小脑及脑干的变化。
常染色体显性遗传的脊髓小脑性共济失调具有遗传异质性,最具特征性的基因缺陷是扩增的CAG三核苷酸重复编码多聚谷氨酰胺通道,该通道在功能不明蛋白和神经末梢上发现的P/Q型钙通道αDA亚单位上;其他类型突变包括CTG三核苷酸(SCA8)和ATTCT四核苷酸(SCA10)重复序列扩增,在许多病例中这种扩增片断的大小与疾病严重性有关,且年龄愈小,病情愈重。
SCAs基因突变改变蛋白的性质,实质无法被正常加工,异常加工的片断与一种参与非溶酶体降解的缺陷蛋白泛素结合,共同以蛋白酶体的符合体形式转运至核内,推测这种核内蛋白聚集可影响细胞核的功能。
每种SCA亚型基因位于不同的染色体,有不同的大小和基因突变部位,例如,SCA1基因位于染色体6q22-23,基因组跨度450kb,cDNA长11Kb,含9个外显子,编码816个氨基酸残基组成ataxia-1蛋白,该蛋白位于细胞核,CAG突变位于8号外显子,,扩增拷贝数为40-83,正常人为6-38。
SCA3(MJD)是我国最常见的SA亚型,基因位于14q24..3-32,至少含4个外显子,编码960个氨基酸残基组成ataxia-3蛋白,分布细胞地中,CAG突变位于4号外显子,扩增拷贝数为61-89,正常人为12-41。
脊髓小脑性共济失调诊疗指南

脊髓小脑性共济失调诊疗指南概述脊髓小脑性共济失调(spinocerebellar ataxia,SCA)是一组由基因突变导致小脑、脑干、脊髓退行性变,以进行性运动协调功能减退、平衡失调为主要临床表现的神经系统遗传性疾病。
最初定义和经典类型SCA为常染色体显性遗传,因相应基因外显子(CAG)三核苷酸拷贝数异常重复扩增产生多谷氨酰胺所致。
后来也发现了常染色体隐性遗传、X连锁遗传和线粒体遗传(NARP、MERRF以及CoQ10缺乏)的类型。
目前已发现数十种SCA致病基因,但还有部分类型未找到明确致病基因。
主要SCA亚型与相应致病基因,见表111-1。
表111-1 主要SCA亚型与相应致病基因主要常染色体显性SCA为SCA1(ATXN1)、SCA2(ATXN2)、SCA3(ATXN3)、SCA6(CACNA1A)、SCA7(ATXN7),主要常染色体隐性共济失调致病基因为FXN、ATM、APTX、SETX。
以上很多致病基因突变后可造成小脑结构功能异常,但病理受损范围并不局限于小脑系统,因此疾病表型还涉及锥体系、锥体外系、高级智能、视觉和听觉等方面。
在疾病分类上也存在一些交集,比如有一类疾病亚型,肢体痉挛性瘫痪(锥体束损害)和共济失调(小脑损害)都较为突出,称为痉挛性共济失调(spastic ataxia,本章节不作详述),在遗传性共济失调和遗传性痉挛性截瘫均被分别纳入,实为两类疾病的交集。
下面我们以经典的常染色体显性遗传SCA中最常见的亚型——SCA3为代表,介绍具有共性的脊髓小脑性共济失调诊疗常规。
病因和流行病学SCA3又称为Machado-Joseph病(MJD),是由于AXTN3基因编码序列中(CAG)三核苷酸重复数异常扩增,从而导致相应编码蛋白近羧基端多聚谷氨酰胺链(PolyQ)异常增多所导致的疾病。
SCA3被认为是最常见的经典型常染色体显性遗传SCA亚型,其具体占比在不同人种中存在差异。
在中国人群中,SCA3占所有常染色体显性遗传SCA的51.1%~72.5%。
脊髓小脑性共济失调
该蛋白位于细胞核 CAG突变在8号外显子 扩增拷贝数40~83(正常为6~38)
病因&发病机制
SCA3(MJD)--我国最常见SCA亚型
基因位于14q24.3-32, 至少含4个外显子, 编码960个 氨基酸残基的ataxia-3蛋白
SCA发病与种族有关 SCA1-2 意大利\英国多见 SCA3 中国\德国\葡萄牙常见
脊髓小脑性共济失调(SCA)
病因&发病机制
常染色体显性遗传, 具有遗传异质性
❖ 特征性基因缺陷— CAG三核苷酸重复编码多聚谷氨酰胺通道
该通道位于功能不明蛋白(ataxins)& 神经末梢P/Q型钙通道α1A亚单位上
脊髓小脑束&后索受损 黑质\基底节\脊髓前角细胞很少受累 SCA2--下橄榄核\脑桥\小脑损害重 SCA3--脑桥\脊髓小脑束损害 SCA7--视网膜神经细胞变性
SCA临床表现
要点提示
脊髓小脑性共济失调是高度遗传异质性疾病 各亚型症状相似, 交替重叠 SCA典型表现遗传早现现象--同一家系发病
临床表现
2. 各亚型特点
SCA1 眼肌 早期大腿肌痉挛
眼球慢扫视运动明显
\下视震颤\复视
\位置性眩晕 SCA3 肌萎缩\面肌&舌肌纤颤
\眼睑退缩形成凸眼
SCA10 纯小脑征
SCA8 发音困难
&癫痫发作
SCA5 进展极慢, 症状轻
SCA7 视力减退&丧失 \视网膜色素变性\心脏损害
CAG突变位于4号外显子, 扩增拷贝数61~89 (正常12~41)
◙ SCA基因突变机制相同 ◙ 各亚型表现雷同, 但有差异 ◙ 伴眼肌麻痹\视网膜色素变性 ◙ 除多聚谷氨酰胺毒性作用, 其他因素可能参与发病
小脑共济失调治愈案例

小脑共济失调治愈案例小脑共济失调(SCA)是一种常见的神经系统疾病,通常表现为姿势不稳、步态不稳、手部不协调等症状。
本文将介绍一个小脑共济失调治愈的真实案例,希望能为患者和家属提供一些启发和帮助。
患者小李,男,45岁,因为长期的手部不协调、步态不稳而前来就诊。
经过详细的检查和诊断,确认为小脑共济失调。
在治疗过程中,患者采取了以下措施:首先,患者积极配合医生进行药物治疗。
医生根据患者的病情开具了一系列的药物治疗方案,患者按时服药,并定期复诊调整治疗方案,有效控制了病情的发展。
其次,患者进行了康复训练。
康复训练是治疗小脑共济失调的重要手段,通过锻炼和训练可以改善患者的姿势和步态,提高身体的协调性。
患者在康复训练中,坚持不懈地进行各项训练,取得了明显的效果。
最后,患者在日常生活中进行了一些自我管理。
患者在饮食、作息等方面进行了调整,避免了一些可能加重病情的因素,保持了良好的生活习惯。
经过长期的治疗和努力,患者的病情得到了明显的改善,手部不协调、步态不稳等症状逐渐减轻,生活质量得到了明显提高。
这个案例告诉我们,小脑共济失调并非不可治愈,通过积极的治疗和努力,患者可以取得良好的疗效。
在治疗小脑共济失调的过程中,患者和家属需要保持乐观的心态,相信医生的治疗方案,积极配合治疗。
同时,患者还需要注意日常生活中的一些细节,如饮食、作息等,避免不良的生活习惯对病情的影响。
总之,小脑共济失调是一种可以治愈的疾病,患者只要积极配合医生的治疗方案,进行康复训练,并注意日常生活中的一些细节,就能够取得良好的疗效。
希望这个案例能够给正在治疗小脑共济失调的患者和家属一些启发和帮助,让他们更加坚定治愈的信心。
sca3基因型

sca3基因型(最新版)目录1.SCA3 基因型概述2.SCA3 基因型的相关疾病3.SCA3 基因型的研究进展4.SCA3 基因型的应用前景正文1.SCA3 基因型概述SCA3 基因型,即脊髓小脑共济失调 3 型基因型,是一种遗传性疾病的基因类型。
脊髓小脑共济失调(Spinocerebellar Ataxia,简称 SCA)是一类遗传性神经系统疾病,主要表现为进行性的共济失调、运动障碍和神经功能减退等症状。
在众多 SCA 亚型中,SCA3 是最常见的一种,占到了所有 SCA 病例的约 15%。
2.SCA3 基因型的相关疾病SCA3 基因型主要与脊髓小脑共济失调 3 型相关。
这种疾病通常在成年后开始出现症状,随着年龄的增长而逐渐恶化。
患者常常出现行走不稳、步态蹒跚、言语不清等症状,严重时可能导致瘫痪和生活不能自理。
除了运动障碍外,SCA3 还可能导致患者出现认知和精神方面的问题,如记忆力减退、抑郁等。
3.SCA3 基因型的研究进展近年来,针对 SCA3 基因型的研究取得了一定的进展。
研究人员已经发现了导致 SCA3 的基因突变,这些突变主要发生在 ATXN3 基因上。
ATXN3 基因编码一种名为 ATXN3 的蛋白质,这种蛋白质在细胞中具有多种生物学功能,包括参与氧化应激反应、调节基因表达等。
通过对 SCA3 基因型的研究,研究人员已经深入了解了该病发病机制和病理生理过程,为治疗和预防 SCA3 提供了理论基础。
4.SCA3 基因型的应用前景随着对 SCA3 基因型的研究不断深入,研究人员已经开始探索针对该病型的治疗方法和预防措施。
目前,针对 SCA3 的治疗主要是对症治疗,旨在缓解患者的症状和减轻病程进展。
未来,随着基因治疗技术的发展,针对 SCA3 基因型的基因治疗可能会成为现实。
此外,通过对 SCA3 基因型的研究,还可以为其他类似遗传性疾病的诊断和治疗提供参考和借鉴。
总之,SCA3 基因型作为一种常见的遗传性疾病基因类型,在相关疾病的研究、诊断和治疗方面具有重要意义。
小鼠共济失调实验报告

共济失调是一种常见的神经退行性疾病,其特征是运动协调性障碍。
脊髓小脑共济失调(SCA)是一组以小脑损害为主的遗传性共济失调,可分为多种亚型,其中SCA1、SCA2、SCA3等亚型主要由基因突变引起。
本研究旨在构建小鼠共济失调模型,并通过脑深部刺激(DBS)治疗探讨其治疗效果。
二、实验材料与方法1. 实验动物:选用健康成年C57BL/6小鼠,雌雄各半,体重20-25g。
2. 实验分组:将小鼠随机分为以下四组:(1)正常对照组:未经任何处理的健康小鼠;(2)模型组:通过基因敲入技术构建SCA1小鼠模型;(3)DBS治疗组:在模型组基础上,采用DBS技术进行脑深部刺激治疗;(4)DBS+运动治疗组:在DBS治疗组基础上,进行运动训练。
3. 实验方法:(1)模型构建:采用基因敲入技术,将SCA1基因敲入C57BL/6小鼠,获得SCA1小鼠模型。
(2)脑深部刺激(DBS)治疗:将DBS电极植入小鼠小脑区域,通过电刺激改善运动功能。
(3)运动训练:对DBS+运动治疗组小鼠进行一定时间的运动训练,观察其对运动功能的影响。
4. 数据采集与处理:(1)运动功能评估:采用旋转杆实验、平衡木实验等方法评估小鼠的运动功能;(2)神经电生理检测:采用脑电图(EEG)等方法检测小鼠的神经电生理变化;(3)组织学观察:采用苏木素-伊红(HE)染色、尼氏染色等方法观察小鼠小脑组织病理变化;(4)统计分析:采用SPSS软件对实验数据进行统计分析,比较各组小鼠运动功能、神经电生理变化和组织病理变化等指标。
1. 运动功能评估:与正常对照组相比,模型组小鼠在旋转杆实验、平衡木实验等运动功能测试中表现明显下降,说明模型构建成功。
DBS治疗组和DBS+运动治疗组小鼠的运动功能较模型组显著改善。
2. 神经电生理检测:与正常对照组相比,模型组小鼠EEG波幅降低,频率降低,表明神经电生理功能受损。
DBS治疗组和DBS+运动治疗组小鼠的EEG波幅和频率较模型组显著提高。
脊髓小脑性共济失调护理查房PPT

处理措施:进行康复训练,如物理治疗、言语治疗等 预防措施:保持良好的生活习惯,避免过度劳累,注意饮食健康,适当运 动,保持良好的心理状态。
语言障碍
• 症状:患者可能出现言语不清、发音困难、理解能力下降等症状
• 原因:脊髓小脑性共济失调可能导致语言中枢受损,影响语言功能
认知障碍
症状:记忆力 减退、注意力 不集中、思维
混乱等
原因:脑部病 变、神经退行 性疾病、药物
副作用等
处理措施:药 物治疗、康复 训练、心理辅
导等
预防措施:保 持良好的生活 习惯、定期体 检、避免过度
劳累等
情绪问题
患者情绪波动:由于病情影响,患者可能出现情绪波动,如焦虑、抑郁等
家属情绪压力:家属可能因照顾患者而产生情绪压力,如焦虑、疲劳等
运动护理:鼓励患者进行适 当的运动,如散步、瑜伽等
心理护理:关注慰
饮食护理
饮食原则:高蛋白、低脂肪、低盐、低糖 食物选择:瘦肉、鱼、蛋、奶、豆制品、蔬菜、水果等 饮食方式:少量多餐,避免暴饮暴食 饮食禁忌:避免刺激性食物,如辛辣、油腻、生冷等 饮食监测:定期监测体重、血糖、血脂等指标,调整饮食方案
效果评价
评估指标:患者生活质量、功能恢复程度、并发症发生率等
评价方法:问卷调查、临床观察、实验室检查等 评价结果:护理措施对患者生活质量、功能恢复程度、并发症发生率等有 显著改善 评价建议:根据评价结果,调整护理措施,提高护理质量
护理问题及处理措施
肢体功能障碍
症状:患者可能出现肢体无力、肌肉萎缩、关节僵硬等症状
患者心理问题:关注患者心 理状态,提供心理支持和疏 导
患者康复问题:制定康复计 划,定期进行康复训练,提
Neurology病例:上视麻痹是脊髓小脑性共济失调3型的体征

Neurology病例:上视麻痹是脊髓小脑性共济失调3型的体
征
32岁女性,表现为共济失调,吞咽困难和构音障碍5年;有脊髓小脑性共济失调3型(SCA3)阳性家族史。
查体可见眼球凸出,向上凝视麻痹伴垂直性眼球震颤,双侧水平眼球震颤,扫视缓慢,辨距不良和步态共济失调(视频)。
神经影像学可见小脑中度萎缩。
神经心理学评估提示记忆和执行功能缺陷。
根据患者的临床表现和相关检查,怀疑SCA3,随后通过基因检测证实。
上视麻痹似乎是SCA3一个有鉴别意义的特征,可能有助于SCA 的临床分型。
(视频:可见明显的步态共济失调,眼球凸出,向上凝视麻痹和相关的垂直性眼球震颤,双侧水平眼球震颤和缓慢的扫视运动)[参考文献]
Nascimento FA, Marques Garcia BC, Teive HAG.Teaching Video NeuroImages: Upward gaze palsy is
a sign of spinocerebellar ataxia type 3.Neurology. 2018 Jul 31;91(5):e494.
温馨提醒请尊重我们的劳动成果,转载不要忘了注明出处哦~~~。
脊髓小脑性共济失调干预护理

家长要与患者一起制定康复计划,督促患者按时完成康复训练,并定期评估康复效果,根据情况调 整康复计划。
家庭支持和社区资源的利用
家庭支持:为患者提供情感支持和日常照顾,帮助其应对疾病带来的挑战。
社区资源:利用社区康复中心、医疗机构和志愿者组织等资源,为患者提供康复训练、 护理指导和心理支持等服务。
并发症处理:针对不同的并发症,如肺部感染、褥疮等,制定相应的饮食计划
食谱推荐和注意事项
推荐食谱:高蛋白、低脂 肪、高纤维的食物,如鱼、 瘦肉、豆类、蔬菜、水果 等
注意事项:避免过度饮酒, 控制糖分摄入,避免食用 高脂肪、高胆固醇的食物, 如动物内脏、肥肉等
饮食护理的实践和效果评估
饮食原则:提 供高蛋白、低 脂肪、高纤维 的食物,保持
添加副标题
脊髓小脑性共济失调干预护 理
汇报人:
目录
CONTENTS
01 汇报人员
02 脊髓小脑性共济失 调的概述
03 脊髓小脑性共济失 调的护理干预
04 脊髓小脑性共济失 调的饮食护理
05 脊髓小脑性共济失调 的并发症预防和处理
06 脊髓小脑性共济失调 患者的自我管理和家 庭教育
汇报人员:XX医院XX
脊髓小脑性共济失调的 饮食护理
营养需求和饮食原则
营养需求:根据 患者的具体情况, 提供足够的热量、 蛋白质、维生素
和矿物质。
饮食原则:选择 易消化、高营养 的食物,避免过 度油腻和刺激性 食物,保持饮食
均衡。
针对不同症状的饮食调整
轻度症状:保持均衡饮食,增加蛋白质和维生素的摄入 中度症状:控制脂肪和糖分的摄入,避免加重病情 重度症状:根据医生的建议,可能需要禁食或进行鼻饲喂养
sca医学名词解释

sca医学名词解释
SCA在医学上可能有以下三种含义:
心脏骤停:SCA是心脏骤停的英文sudden cardiac arrest的缩写,是指心脏突然停止跳动,导致脑及其他重要脏器的血供中断,如未得到及时救治将导致死亡。
脊髓小脑性共济失调:SCA是脊髓小脑性共济失调的英文spinocerebellar ataxia的缩写,是一种神经系统疾病,主要临床表现为小脑性共济失调,还可伴有眼球运动障碍、视神经萎缩、肌萎缩、周围神经病、痴呆等。
视力检查结果:SCA是视力检查结果的三个缩写,s表示球镜度数,即近视或远视的度数;c 表示散光度数;a主要代表散光轴位方向。
脊髓小脑性共济失调的研究进展

脊髓小脑性共济失调的研究进展崔海燕,康龙丽*,李旭光,朱敏霞,戎浩(西藏民族学院医学院,高原环境与疾病相关基因研究省级重点实验室,生命科学基础实验室,咸阳712082)摘要:脊髓小脑性共济失调是一类遗传性神经系统变性疾病,目前已知的脊髓小脑性共济失调亚型有30余种(DRPLA,SCA1-8,10-23, 25-31)。
基因表达异常,谷氨酸及钙离子依赖性细胞信号转导异常是引起小脑功能紊乱的重要原因。
关键词:脊髓小脑性共济失调;神经变性疾病中图分类号: R744. 7文献标识码: AThe study progress of spinocerebellar ataxiaCUI Hai-yan, KANG Long-li, LI Xu-guang, ZHU Min-xia, RONG Hao (Medical Department of Tibet Nationality College,Plateau Environment and Genes Related to Diseases Key Laboratory of Tibet,Life Science Base laboratory, Xianyang 712082, China)Abstract: Spinocerebellar ataxias is a group of hereditary neurodegenerative disorders. To date, more than 30 kinds of subtypes of genetic spinocerebellar have been identified (DRPLA,SCA1-8,10-23,25-31). Both gene expression and glutamate-dependent and calcium-dependent neuronal signaling as important pathways leading to cerebellar dysfunction.Key words:Spinocerebellar ataxias ; Neurodegenerative disorders脊髓小脑性共济失调(spinocerebellar ataxias ,SCAs)是一组以共济失调为特征的常染色体显性遗传性神经系统变性疾病,病变除了累及脊髓、小脑外,其它组织如脊神经、脑神经、交感神经、基底神经节、丘脑、丘脑下部、大脑皮层均可受累,因此,许多患者除了共济失调外还有其它神经系统症状。
小脑性共济失调的鉴别诊断

小脑性共济失调的鉴别诊断小脑性共济失调症状易与其他疾病引起的共济失调相混淆,应认真区别。
(一)脊髓结核性共济失调脊髓结核性共济失调也会出现步态异常,但表现步幅不均,无序,虽然也出现蹒跚步态,但在视力矫正下可改善,因此病人行走时常低头两眼注视地面和脚的动作,闭眼后躯体摇晃增强。
例如病人洗脸时常向脸盆内倾倒(洗脸盆征阳性)。
而小脑性共济失调的病人无论有无视力矫正皆不稳。
此外,脊髓结核尚有深感觉障碍、腱反射丧失和瞳孔异常(Argyll-Robertson瞳孔)等表现。
小脑共济失调合并有辨距不良、肌张力降低及小脑言语等。
细致检查可以鉴别。
(二)前庭迷路性共济失调小脑性共济失调与前庭迷路性共济失调主要鉴别要点如下:1.前庭性共济失调在运动时明显例如体位转动时突出,而单纯做简单随意运动时共济失调并不明显。
2.前庭性共济失调常伴有眩晕呈发作性或持续性,而单纯的小脑性共济失调则无眩晕。
3.前庭性共济失调站立时躯体向侧方倾倒,或摇晃不稳,其发病有一定的潜伏期,摇晃不稳逐渐增强,甚至出现倾倒,倾倒方向以侧方为多。
小脑病变时躯体向各个方向摇晃,但以前后方向为主。
4.前庭性共济失调躯体倾斜与头位有密切关系而小脑共济失调与头位无关。
临床实际工作中由于小脑与前庭束之间有密切联系,两种组织往往同时受累,出现小脑前庭迷路综合征。
即使纯粹的小脑病变(例如小脑肿瘤),由于颅内压增高也可直接或间接地影响到前庭迷路系统。
(三)大脑性共济失调大脑病变伴有痉挛性偏瘫或失语等典型症状时,很容易鉴别,但既有大脑病变又合并小脑病变时鉴别起来就比较困难。
小脑病变时不出现锥体束征,广泛的小脑病变,例如小脑外伤、小脑脓肿呈现肌张力降低,或伴有肌力减低,但较大脑病变的肌力改变轻且短暂,不产生持久性瘫痪。
可通过联合屈曲试验进行鉴别:病人仰卧位,将两臂在胸前交叉,做起身动作,此时大脑病变与小脑病变表现相同,都表现起身时患侧股关节屈曲。
如果检查者将两下肢按住,使之固定不动,观察病人上半身起床的动作。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
• 参考文献
• 7、朴钟源,宋琳,江新梅.脊髓小脑性共济失调的分型 进展[J].中风与神经疾病杂志,2008,25(4):504506 • 8、谢秋幼,梁秀龄,李洵桦.脊髓小脑性共济失调的分 子遗传学诊断与临床应用[J].中华医学遗传学杂志, 2005,22(1):71-73 • 9、邬剑军,蒋雨平.小脑共济失调的过去现在和未来 [J].中国临床神经科学,2006,14(6):638-648
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
• SCAs常见类型:
• SCA3型/MJD:平均起病年龄40岁,病程平均2 1年。主 • 要临床表现为锥体束征、面肌和舌肌肌束震颤、眼球 扫视变慢、眼险退缩、突眼、吞咽困难、肌萎缩等。 致病基因为位于染色体14q24.3一q32的ATXN3基因。巴 西、葡萄牙、日本、德国、荷兰等国常见,也是中国 最常见的类型
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs) • SCA各型之间临床表型存在较大的重 叠,临床分型非常困难,因此诊断SCA 必须依靠基因检测
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
• 临床诊断SCA可遵循以下几点: • 1、确定是否为SCA,主要根据病史、临床和辅助检查 • 2、确定其遗传方式,根据有无家族史确定其是家族 • 性或散发性,家族性根据遗传方式判断是常染色体显 性还是隐性遗传
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
• 参考文献
• 10、江泓,唐北沙.脊髓小脑性共济失调的l晦床及基因 诊断进展[J].2002,29(4):290-293. • 11、王康,王国相.遗传性共济失调临床和基因诊断进 展[J].中华神经科杂志,2001,34(6):378-381 • 12、David A.Greenberg ,Michael J.Aminoff ,Roger P.Simon.Clinical Neurology。(临床神经病学,第 八版,人民卫生出版社,王维治、王化冰主译)2015: 199-234。 • 13、王新德,陈生弟.神经病学第18卷神经变性性疾 病.1版.北京:人民军医出版社,2006:155-218
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
• SCAs常见类型:
• SCA6型:平均发病年龄为52岁,病程一般长达25年以 上。主要临床表现为发作性共济失调,偶伴肌张力障 碍、肢体痉挛等,该型可无家族史,进展极慢。致病 基因为位于染色体19p13.13上的ataxia-6基因。该类 型在德国、日本、美围及荷兰有报道,中国报道较少。
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
• SCAs常见类型:
• SCA12型:平均起病年龄为33岁。主要临床表现为发病 较早者头部和肢体震颤。震颤常作为首发症状。晚发 者表现为轻微帕金森症状、痴呆、认知减退及精神症 状。致病基因位于染色体5q31.q32,由基因PPP2R2B5’ 端非编码区CAG重复扩增致病.是唯一以动作性震颤为 主的SCAs亚型,见于德国和印度家系。
遗传性共济失调(hereditary ataxia,HA)
• 临床上以共济运动障碍为主要特征,可伴有复杂的神 经系统损害,如锥体束、锥体外系、大脑皮质、脊髓、 脑神经、脊神经、自主神经等症状,亦可伴有非神经 系统表现如心脏病变、内分泌代谢异常、骨骼畸形、 皮肤病变等。 • HA的遗传方式以常染色体显性遗传(autosomal dominant,AD)为主,部分可呈常染色体隐性遗传 (autosomalrecessive,AR),极少数为X连锁遗传(Xlinked)和线粒体遗传(mitochondrial);散发病例亦 不少见
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
• SCAs又称为常染色体显性遗传性小脑共济失调 (ADCAs),为遗传性共济失调的主要类型,是一大类 以小脑功能失调或合并其他神经功能异常为特征的神 经系统变性疾病。
• 病变主要累及:脑干、小脑、脊髓 • 主要临床表现:共济失调 • 典型特征:遗传早现现象
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
• 参考文献:
• 4、王希恒,潘学峰,李红权等.(CAG)n·(C TG)n三核苷酸重复序列扩增及相关疾病机制研究 进展[J].国际遗传学杂志,2016,39(5):274-280 • 5、夏明荣,黄月,张杰文等.脊髓小脑共济失调基因 分型、临床及影像学特征分析[J].郑州大学学报(医 学版),2015,50(6):860-863 • 6、俞立强,何晓辉,方琪等.脊髓小脑性共济失调症 状前诊断初探[J].中国神经免疫学和神经病学杂志, 2013,20(3):197-199
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
• SCAs常见类型:
• SCA7型:一般起病年龄在35岁,病程约20年。主要临 床表现是锥体束征、黄斑萎缩、视网膜病变,并导致 视觉丧失、可有听力丧失。致病基因为位于染色体 3p12-p21.1上的ATXN7基因。在北美、欧洲多见,中国 发病率较低。
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
• SCAs常见类型:
• SCA17型:起病年龄在1 8~55岁之问,平均病程8年。 主要临床表现为肌张力障碍、智能障碍、舞蹈样运动、 锥体束征、精神症状。致病基因位于染色体6q27。
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
• SCAs常见类型:
• SCA2型:起病平均年龄为34岁,病程平均1 0年。 • 主要临床表现为帕金森样症状、眼球慢扫视、肌阵挛、 周同神经病、动作性震颤、痴呆等。致病基因为位于 染色体12q24.13上的ataxia-2基因,见于美国、意大 利、印度,是印度发病率最高的类型。
Hale Waihona Puke • 而根据致病基因分型,现SCAs已达到40种
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
• SCAs常见类型:
• SCA1型:平均起病年龄在30~40岁,病程长约15年。 主要临床表现为锥体柬征、眼球扫视和辨距过度、额 叶执行功能障碍、精神症状、痴呆(晚期)等,致病基 因为染色体6q22.3区域的1型致病基因ataxia-1。该类 型多见于意大利、南非、德国和印度。
• 3、推断最可能的SCA亚型,以便基因诊断时确定先后 顺序,节约资源和时间
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
• 参考文献:
• 1、中华医学会神经病学分会神经遗传学组.遗传性共 济失调诊断与治疗专家共识[J].中华神经科杂志, 2015,48(6):459-463 • 2、宋建萍,杨丹,陈涛等.脊髓小脑共济失调各亚型 致病基因及临床表现[J].国际遗传学杂志,2016,39 (2):85-89 • 3、刘钰鹏,田增民.脊髓小脑性共济失调的基因研究 进展[J].医学综述,2013,19(15):2722-2725
• SCAs常见类型:
• 齿状核红核苍白球路易体萎缩症(denta-tonlbralpallidoluysian atrophy,DRPLA : • 少年型:20岁以前起病,主要表现为智力发育迟滞、 癫痫、肌阵挛和共济失调,病情进展迅速 • 成年型:20岁之后起病,主要表现为小脑性共济失调、 手足徐动症、痴呆和精神症状。 • 致病基因位于染色体l2p13.31上。以日本发现居 • 多,其次在欧洲、北美、非洲及高加索人中有报 • 道,中国人群中极为罕见。
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
遗传性共济失调(hereditary ataxia,HA)
• 定义:由遗传因素所致的以共济运动障碍、辨距不良 为主要临床表现的一大类中枢神经系统变性疾病 • 三大特征:世代相传的遗传背景 • 共济失调的临床表现 • 脊髓、小脑、脑干损害为主的病理改变
脊髓小脑性共济失调
(spinocerebellar ataxia,SCAs)
• 根据遗传类型和临床表现至少分为三类:
• ADCAⅠ型:除小脑性共济失调外,还包括眼球运动障碍, 慢眼运动、视神经萎缩、锥体束征、锥体外系征、肌萎缩、 周围神经病和痴呆等,如SCA1、SCA2 、SCA3/MJD、SCA4、 SCA12等 • ADCAⅡ型:小脑性共济失调班视网膜色素变性,如SCA7 • ADCA Ⅲ型:单纯小脑性共济失调,如SCA5、SCA6 、SCA8、 SCA10、SCA11、SCA14等