发酵液还原糖的测定方法
国内最准确的生产实用发酵液糖检测方法大公开
国内最准确的生产实用发酵液糖检测方法大公开4.1 总糖的测定4.1.1 试剂和溶液4.1.1.1斐林储备液A液:称取硫酸铜120克,加水溶解稀释至2000毫升B液:称取氢氧化钠250克,溶于部分水中,加375克酒石酸钾钠,加水溶解稀释至2000毫升。
C液:称取碘化钾300克,于1000毫升棕色容量瓶中,稀释至刻度。
依照B、A、C的顺序依次将上述溶液倒入干燥洁净的棕色试剂瓶中备用。
4.1.1.2 淀粉指示剂(1%)称取可溶性淀粉1克于200毫升烧杯中加水10毫升,调制浆,倒入沸腾的90毫升水,煮沸1-2分钟,冷却即可。
(有效期一周)4.2标准曲线制备及回归方程的确定。
4.2.1准确称取预先以105℃干燥恒重的无水葡萄糖1.0克(精确至0.0002克),以0.01mol/L盐酸溶解,用水稀释至100毫升容量瓶中摇匀。
按上表用吸量管准确吸取溶液于250毫升三角瓶中,各分别加入定量蒸馏水,使溶液呈5毫升,置电热板加热,从沸腾计时3分钟,取下循环水冷却至室温。
同时以5毫升水做空白。
4.2.3 加入3滴酚酞,用20%NaOH调制为淡粉色;加入20毫升费林混合试剂,置于电热板加热,沸腾计时3分钟,循环水冷却至室温。
4.2.4加入6mol/L盐酸10毫升,摇匀,立即用0.1mol/L硫代硫酸钠滴定,溶液呈淡黄色时加入1毫升淀粉指示剂,继续滴定至刚好蓝色消失为终点。
4.2.5 回归方程的确定。
4.3总糖测定4.3.1样品预处理4.3.2稀释液:准确吸取5毫升离心后发酵液于50毫升容量瓶中,用蒸馏水稀释至刻度。
精密吸取上述稀释液2.5毫升(根据发酵液含糖量可增减)于三角瓶中,加蒸馏水使溶液成5毫升,加入10毫升蒸馏水、5毫升6mol/L盐酸,置电热板加热至沸,煮沸3分钟,取下循环水冷却至室温,加入1%酚酞指示剂三滴,用20%氢氧化钠滴定中和至淡粉色。
准确加入混合液20毫升,置电热板加热至沸,从沸腾计时3分钟,取下自然冷却至室温。
发酵液中还原糖含量测定
发酵液中还原糖含量测定(3,5-二硝基水杨酸比色法)一、实验目的1、掌握还原糖测定的基本原理2、学习比色法测定还原糖的操作方法和分光光度计的使用二、实验原理还原糖的测定是糖定量测定的基本方法。
还原糖是指含有自由醛基或酮基的糖类。
单糖都是还原糖,双糖和多糖不一定是还原糖,例如乳糖和麦芽糖是还原糖,蔗糖和淀粉是非还原糖。
利用各种糖的溶解度不同,可将植物样品中的单糖、双糖和多糖分别提取出来。
对非还原性的双糖和多糖,可用酸水解法使其降解成还原性单糖进行测定,再分别求出样品中还原糖和总糖的含量(常以葡萄糖含量计)。
还原糖在碱性条件下加热可被氧化成糖酸及其它产物,而氧化剂3,5-二硝基水杨酸则被还原为棕红色的3-氨基-5-硝基水杨酸。
在一定范围内,还原糖的量与棕红色物质颜色的深浅成正比关系,利用分光光度计在540nm波长下测定光密度值,查对标准曲线并计算,便可求出样品中还原糖和总糖的含量。
由于多糖水解为单糖时,每断裂一个糖苷键需加入一分子水,所以在计算多糖含量时应乘以系数0.9。
三、实验材料和试剂1、实验材料发酵液2、实验试剂①1mg/ml葡萄糖标准液准确称取80℃烘至恒重的分析纯葡萄糖100mg,置于小烧杯中,加少量蒸馏水溶解后,转移到100ml容量瓶中,用蒸馏水定容至100ml,混匀,4℃冰箱中保存备用。
②3,5-二硝基水杨酸(DNS)试剂将6.3g DNS和262ml 2M NaOH溶液,加到500ml含有185g酒石酸钾钠的热水溶液中,再加5g结晶酚和5g亚硫酸钠,搅拌溶解,冷却后加蒸馏水定容至1000ml,贮于棕色瓶中备用。
四、实验器材具塞璃玻刻度试管:20ml×8移液管:2ml×2;10ml×1 微量移液器:1000μL×1容量瓶:100ml恒温水浴锅沸水浴可见光分光光度计五、操作步骤1、制作葡萄糖标准曲线取7支具塞刻度试管编号,按表1分别加入浓度为1mg/ml的葡萄糖标准液、蒸馏水和DNS试剂,配成不同浓度的葡萄糖反应液。
葡萄酒酿制过程中糖酸和酒精度的检测

葡萄酒酿制过程中还原糖、总酸及酒精度的测定方法一、还原糖的测定在葡萄酒发酵前,测定葡萄的还原糖含量,以确定要添加的糖含量;发酵之后测定酒中的残糖含量。
1、测定方法:裴林试剂热滴定法(1)裴林氏A 、B 液标定预备试验:取裴林氏A 、B 液各5.00mL 于250mL 三角瓶中,加50mL 水,摇匀,在电炉上加热沸,在沸腾状态下用制备好的葡萄糖标准溶液滴定,当溶液的蓝色将消失呈红色时,加2滴甲基蓝指示液,继续滴至蓝色消失,记录消耗的葡萄糖标准溶液的体积(V1ml)。
正式试验:取裴林氏A 、B 液各5.00mL 于250mL 三角瓶中,加50mL 水和比预备试验少1ml 葡萄糖标准溶液,加热至沸,并保持2min ,加2滴次甲基蓝指示液,在沸腾态下于1min 内用葡萄糖标准溶液滴至终点(消耗葡萄糖标准液,记录消耗的葡萄糖标准溶液的总体积。
(2)样品的测定预备试验:裴林试剂A 、B 液各5ml 250ml 三角瓶中 加水50ml 加入7.5ml 试样 在沸腾状态下用5g/l 的葡萄糖标液滴定至 蓝色消失成红色时 加2滴亚甲基兰指示剂 继续滴定至蓝色消失,记录体积。
正式试验:裴林试剂A 、B 液各5ml 250ml 三角瓶中 加水50ml 加入7.5ml 试样+比预备试验少1ml 的葡标液,在沸腾状态下用5g/l 的葡萄糖标液滴定至 蓝色消失成红色时 加2滴亚甲基兰指示剂 继续滴定至蓝色消失,记录体积。
结果计算:总糖或还原糖含量g/l =稀释倍数)(取样体积测定试样消耗的葡标液标定消耗的葡标液葡标液⨯-⨯V V V C 2、试剂的配制及仪器需求(1)斐林试剂配制方法将36.4g CuSO4.5H2O 溶于200mL 水中,用0.5mL 浓硫酸酸化,再用水稀释到500mL 待用;取173g 酒石酸钾钠KNaC4H4O6.4H2O ,71g NaOH 固体溶于400mL 水中,再稀释到500mL .使用时取等体积两溶液混合。
05-03-010发酵液中还原糖含量的测定——案例(精)

案例
案例1ቤተ መጻሕፍቲ ባይዱ
处理发酵液中杂质很多,其中对菲林试剂法测定影响最大的是杂蛋白,所以应做预处理时,应尽量除去,就需要加入一些沉淀剂,如乙酸锌,亚氰化钾等
问题
如果这些沉淀剂中掺入了硫酸铜,结果会怎样?如何处理?
案例解析
本法是根据一定量的碱性酒石酸铜溶液(Cu2+量固定)消耗的样液量来计算样液中还原糖含量,反应体系中Cu2+的含量是定量的基础,所以在样品处理时,不能用铜盐作为澄清剂,以免样液中引入Cu2+,得到错误的结果。所以需要更换不含Cu2+的沉淀剂。
为了加快滴定速度,可以进行样品溶液预测,其目的:一是本法对样品溶液中还原糖浓度有一定要求(0.1%左右),测定时样品溶液的消耗体积应与标定葡萄糖标准溶液时消耗的体积相近,通过预测可了解样品溶液浓度是否合适,浓度过大或过小应加以调整,使预测时消耗样液量在10ml左右;二是通过预测可以知道样液大概消耗量,以便在正式测定时,预先加入比实际用量少1ml左右的样液,只留下1ml左右样液在继续滴定时加入,以保证在1分钟之内完成继续滴定工作,提高测定的准确度。
案例2
斐林试剂是新配制的溶液,它在加热条件下与醛基反应,被还原成砖红色的沉淀,可用于鉴定可溶性还原糖的存在。菲林试剂法测定还原糖所用指示剂次甲基蓝容易被空气中的氧气氧化,所用需要注意避免空气中氧气进入到溶液中,并且滴定速度不宜过慢。
问题
如何快速准确滴定还原糖的含量?
案例解析
为了避免空气中氧气进入到溶液中,滴定必须是在沸腾条件下进行,其原因一是加快还原糖与Cu2+的反应速度;二是亚甲基蓝的变色反应是可逆的,还原型的亚甲基蓝遇空气中的氧时会再被氧化为氧化型。此外,氧化亚铜也极不稳定,易被空气中的氧所氧化。保持反应液沸腾可防止空气进入,避免亚甲基蓝和氧化亚铜被氧化而增加消耗量。如果不沸腾就进行滴定,结果会偏大。另外,滴定时不能随意摇动锥形瓶,更不能把锥形瓶从热源上取下来滴定,以防止空气进入反应溶液中。
DNS法测定发酵液中总糖含量

DNS法测定发酵液中总糖含量3,5-二硝基水杨酸与还原糖共热后被还原成棕红色的氨基化合物,在一定范围内,还原糖的量和反应液的颜色强度呈现比例关系,利用比色法可测知样品的含糖量。
DNS试剂的配方(3,5-二硝基水杨酸试剂):称取酒石酸钾钠18.2g,溶于50ml蒸馏水中,加热(不超过50℃),于热溶液中依次加入3,5-二硝基水杨酸0.63g,NaOH 2.1g(先配成溶液),苯酚0.5g,无水亚硫酸钠0.5 g, 搅拌至溶解完全,冷却后用蒸馏水定容至100ml,贮于棕色瓶中,室温保存。
0.1%葡萄糖标准液:准确称取100 mg分析纯的葡萄糖(预先在105℃干燥至恒重),用少量蒸馏水溶解后定容至100 ml,冰箱保存备用。
注意事项:1. 在配制的过程中,如果操作不合理,会出现溶液变黑,或者有鸡蛋花一样的絮状沉淀出现。
2.称取DNS(具体重量,按照你的配方来,下同),加水溶解,水浴45℃;3.逐步加入氢氧化钠溶液,同时不断搅拌,直到溶液清澈透明;(a.药品中的氢氧化钠要配制成溶液;直接加颗粒,可能产生鸡蛋花;b.加入氢氧化钠溶液时,溶液的温度会上升,所以要慢慢加,不停地搅拌,同时溶液的温度不能超过48度;温度高了,溶液颜色变黑。
)4. 逐步加入四水酒石酸钾钠、苯酚和无水亚硫酸钠;(顺序最好不要更改!)5.继续45度水浴,同时补水,不断搅拌,直到加入的物质完全溶解;(一定要有耐心地搅拌!)6. 停止加热,冷却至室温,用水定容。
7. 储存在棕色瓶中,避光保存。
室温下存放7天后使用。
有效期为6个月。
(时间不忙的话,最好按照时间来操作,时间紧迫了,时间提前个几天,推后几天,也可以用的)【操作方法】一、葡萄糖标准曲线的绘制取9支大试管,分别按下表顺序加入各种试剂:将上述各管溶液混匀后,用72型分光光度计(520nm)进行比色测定,用空白管溶液调零点,记录光密度值。
以葡萄糖浓度为横坐标,光密度值为纵坐标绘制出标准曲线。
还原糖测定

还原糖含量的测定(斐林试剂法)一、实验目的:学习用斐林试剂测还原糖的方法,二、实验原理:斐林试剂由甲、乙液组成,甲液为硫酸铜溶液,乙液为氢氧化钠与酒石酸钾钠溶液。
平时甲、乙溶液分开贮存,测定时才等体积混合,混合后,硫酸铜与氢氧化钠反应,生成氢氧化铜沉淀:2Na0H十CuS04——→Cu(OH)2↓十Na2SO4氢氧化铜因能与酒石酸钾钠反应形成络合物而使沉淀溶解。
酒石酸钾钠铜络合物中的二价铜是一个氧化剂,在氧化醛糖和酮糖(合称总还原糖)的同时,自身被还原成一价的红色氧化亚铜沉淀:反应终点用次甲基蓝来指示。
三、实验器材与试剂:电炉,滴定管等(1)斐林溶液甲液:称取30g结晶硫酸铜(CuS04·5H20),0.1g次甲基蓝,定容于1000mL水中,如有不溶物须过滤。
乙液:称取100g酒石酸钾钠,108g NaOH,定容1000mL水中,若有沉淀过滤即可。
(2)0.1%标准葡萄糖液:精确称取于105℃烘至恒重的分析纯葡萄糖 1.000g,用水溶解后,定容至1000mL。
四、实验步骤1. 空白滴定准确吸取斐林甲、乙液各5mL,放入250mL锥形瓶中,加水约15mL,并从滴定管加入约20mL 0.1%标准葡萄糖溶液,严格控制不要摇晃三角瓶,将锥形瓶置电炉上加热煮沸,并准确沸腾30s,在沸腾状态下,以每两秒1滴的速度滴入0.1%标准葡萄糖溶液,至溶液刚由蓝色退去,生成砖红色沉淀为止。
后滴定操作应在1分钟内完成,整个煮沸时间应控制在3分钟之内。
记下总耗糖量V0。
2. 预滴定稀释:将样品除气后,进行适当稀释,以期用15~50mL(最好20~30mL)稀释液使滴定完成。
一般将发酵液稀释100倍(吸取1mL发酵液定容到100mL容量瓶)。
准确吸取斐林甲、乙液各5mL,放入250mL锥形瓶中,加水约15mL,吸取稀释样液1mL,放入锥形瓶中,并从滴定管加入约18mL 0.1%标准葡萄糖溶液,其他与空白滴定基本相同,最后在1分钟内滴定完成。
发酵过程主要生化指标的测定氨基氮测定——甲醛滴定法

• 水溶液中的氨基酸并不是以游离的羧基或 氨基形式存在,而是兼性离子,既是质子 供体(酸),也是质子受体(碱)。
COOH
COO-
COO-
H3N+ C H
H3N+ C H
H2N C H
R
2p左K右1
R
9p左K右2
R
甲醛可与氨基酸上的NH3+结合,生成羟甲基衍生物,释放出H+ R-NH2+2HCHO→R-N(CH2OH)2
• 为何两种显色剂?
• 6-7.6 溴酚兰变色范围(黄 蓝)
• 7.4-10 酚酞变色范围(无色 粉红)
• 酚酞终点淡红,不易观察,现在变色范 围是从黄色变为紫色,终点为紫色,易 观察。
• 用标准碱液滴定羧基,按标准碱的消耗 量求出氨基酸的含量。碱完全中和羧基 时的pH值约为8.5~9.5。
滴定注意事项:
• 沙伦逊甲醛滴定法(Sorensenformaltitration) 一种测定氨基或氨基氮的方法。氨基酸 分子中含有碱性氨基和酸性羧基,在固 态或水溶液中以双性离子(内盐)存在, 因此不能直接用酸滴定氨基,也不能用 碱滴定羧基。
• 沙伦逊提出了将氨基酸(水溶液)用中 性甲醛水溶液处理,将氨基“隐蔽”后 滴定羧基的方法。
三、大肠杆菌发酵种子的制备
菌种
接种量1-2环
接种量1%
斜面 一级种子 二级种子
30℃,18h , 4℃长期保存
14h,32℃,
8h,32℃
按学号,4人一组,3组共用一支斜面种子 摇瓶装液量50ml 一级种子种龄在14h±2h内,二级种子7-8h内
时间安排
每一格必须由操作人签名!
一级种子
二级种子
• 甲醛有毒,滴定要快,必要时可戴口罩。 • 现配现滴,滴完盖紧瓶盖。 • 中性甲醛若长时间放置会变酸,滴定前要
应用不同方法测定发酵液中的糖含量
0.5
1
0.359
1
1
0.364
1.5
1
0.372
2
1
0.385
2.5
1
0.377
3
1
0.378
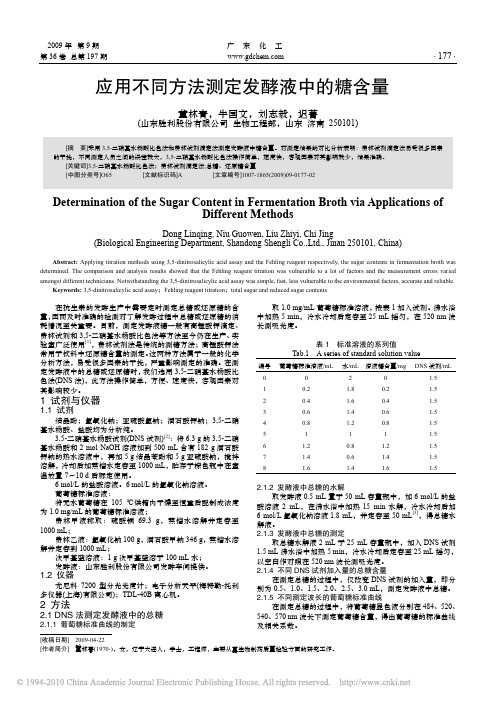
从表 3 看出,在测定发酵液总糖含量时,当 DNS 添加量 大于 1.5 mL 时,对结果的影响虽然不大,但浪费大,当小于 1.5 mL 时,还原糖过量,使得测定结果偏低,所以应选择 DNS 添加量为 1.5 mL。
5
1
1
1
1.5
6
1.2
0.8
1.2
1.5
7
1.4
0.6
1.4
1.5
8
1.6
1.4
1.6
1.5
2.1.2 发酵液中总糖的水解
取发酵液 0.5 mL 置于 50 mL 容量瓶中,加 6 mol/L 的盐 酸溶液 2 mL,在沸水浴中加热 15 min 水解,冷水冷却后加 6 mol/L 氢氧化钠溶液 1.8 mL,并定容至 50 mL[3],得总糖水 解液。 2.1.3 发酵液中总糖的测定
还原糖测定方法的规范_管斌

第18卷第3期1999年9月 无锡轻工大学学报Journal of Wuxi University of Light IndustryV ol.18,No.3 Sep.,1999收稿日期:1998-10-16;修订日期:1999-05-07作者简介:管斌(1957年11月生),男,山东济南人,工学博士,副教授. 文章编号:1001-7453(1999)03-0074-06还原糖测定方法的规范管 斌1,丁友日方2,谢来苏2,隆言泉2(1.山东轻工业学院,山东济南250100, 2.天津轻工业学院,天津300222)摘要:探讨了3,5-二硝基水扬酸(DN S)添加量、显色时间、氧的溶解量、离子强度以及DN S显色后存放时间对纤维素酶酶活力测定的影响.氧的溶解量对测定结果影响显著;测定波长的变化对检测样品光吸收值的灵敏度和线性范围影响很大,一般选定540nm为进行纤维素酶酶活力测定的波长.另外,当以葡萄糖为标准来测定还原糖时,不同的测定方法会使得纤维二糖的测定值不同.因此,在测定纤维素酶酶解产物时,用DN S方法来测定还原糖,使测定结果更加准确.关 键 词:纤维素酶;酶活力;测定条件;还原糖;3,5-二硝基水扬酸中图分类号:T S207.3 文献标识码:A纤维素酶酶活力测定方法的多样性、不准确性以及测定中存在着的难以克服的困难,给纤维素酶的开发和利用带来了很多不利的影响.其主要原因为:1)纤维素酶是由内切β-1,4-葡聚糖酶、外切β-1,4-纤维二糖分解酶和纤维二糖酶(或者β-葡萄糖苷酶)组成的复合酶系[1,2].各组分的协同作用促使结晶纤维素转化为纤维二糖和葡萄糖.2)纤维素酶的作用底物纤维素是不溶于水的高聚糖[3].在纤维素与水组成的多相反应体系中,酶总处于底物不饱和状态,因此,难以用测酶反应初速度的方法来测定酶活力[4].3)酶与纤维素的反应是在固体界面上进行的,其酶解反应速率受到底物对酶蛋白的吸附速率和产物扩散速率的影响[5].现今,在纤维素酶活力测定中,主要是采用测定纤维素酶解反应平均速率的方法.这主要包括测定酶解前后纤维素的溶解量,如同以CM C作底物来测定粘度的方法;而多数方法是测定底物酶解后还原糖的生成量,如以葡萄糖为标准,用3,5-二硝基水杨酸(DN S)法[6]或Som og yi法[7]等.但是,由于酶解产生还原糖的种类和氧化能力有所不同、底物不饱和程度以及酶对纤维素结晶区域和过渡区域水解能力存在差异,因此会给纤维素酶活力测定带来误差.所以说,采用的纤维素酶活力测定方法以及测定条件,都对测定结果有较大影响.本文对纤维素酶活力测定的影响因素进行了分析,并提出了建议.1 材料与方法1.1 材料酶样A 由作者所在实验室制备,它是由菌种T .reesei G B 发酵后,经过滤、浓缩制成的酶液.酶样B 由Nov o 公司生产并提供.1.2 分析方法1.2.1 还原糖测定方法 取上清液,按照Miller [6]的方法,使用DNS 试剂,通过测定540nm 波长下的光吸收值来确定还原糖的含量,并以葡萄糖为标准来表示.另外,也可用Som og yi [7]方法来测定还原糖的生成量.1.2.2 纤维素酶活力测定方法 采用Ma ndels 等[8]的测定方法.1)滤纸酶活(Filter Paper Activity ,简称F P A):取25m L 的刻度试管,加入经稀释的0.5m L 酶液和1m L 浓度为0.05mol /L 的柠檬酸缓冲溶液,并加入一条1cm ×6cm 的滤纸,50℃保温1h .用DN S 试剂测定所形成的还原糖量,并扣去空白.其酶活力单位用国际单位(1μmo l /(minm L ))来表示.2)CM C 酶活(CM Case):取25m L 的刻度试管,加入经适当稀释的酶液0.5m L 和1m L 质量分数为1%的CM C 缓冲溶液,50℃保温30min.用DN S 试剂来测定所形成的还原糖量,并扣去空白.其酶活力单位用国际单位(1μmol /(minm L ))来表示.纤维素酶酶活力单位的定义:在酶的催化下,每分钟形成1μmo l 葡萄糖时所用该酶的量为1个酶活国际单位(1μmol /(min m L)).2 结果与分析2.1 DN S 测定条件对纤维素酶活力(还原糖)测定的影响2.1.1 DN S 与还原糖的显色时间对还原糖测定的影响 DN S 在碱性、沸腾的条件下与糖图1 显色时间对还原糖测定的影响类化合物的还原基团发生反应并显色.DN S 与还原糖显色后,其颜色深浅的比例可作为还原糖测定的依据.另外,该反应对还原糖种类没有选择性[9].在试验条件下,DN S 与还原糖进行反应并显色,显色(煮沸)时间对还原糖的测定有很大影响.图1为显色时间对还原糖测定的影响.可以看出,显色时间在3min 内显色液的吸光度随显色时间的延长而增加.3min 之后,吸光度随时间变化不大.尤其在5min 之后,显色后的吸光度已基本趋于稳定.所以,一般取DN S 与还原糖反应的显色时间为5min.2.1.2 DN S 添加量对还原糖测定的影响 在试验条件下,DN S 添加量对还原糖测定的影响如图2所示.可以看出,随着DN S 添加量的增加,显色液的吸光度变化不大,但有逐渐增加的趋势.这可能是由于DN S 添加量的增多使其吸光度增大的缘故.DN S 添加量主要与待测液的还原糖含量有关.一般在纤维素酶活力测定时,DN S 添加量为3m L 较适宜,且还原糖含量与吸光度之间的线性范围较宽.2.1.3 氧的溶解量(溶液的氧化能力)对还原糖测定的影响 在试验条件下,待测液或者蒸馏水的氧化性高低直接影响到DN S 与还原糖显色的比例性,进而影响到对糖测定的准确性.图3为氧的溶解量(用H 2O 2代替溶解氧添加到待测液中)对还原糖测定的影响.可以看出,随着氧在待测液中溶解量的增加,显色液的吸光度呈现明显的降低趋势.其原因可能是由于DNS 与还原糖反应实际上是一种氧化还原反应.因此,待测液中氧溶解量的变化对该反应产生显著的影响.图2 DN S 添加量对还原糖测定的影响 图3 氧的溶解量对还原糖测定的影响图4 离子强度对还原糖测定的影响2.1.4 离子强度对还原糖测定的影响 进行纤维素酶的酶活力测定时,必须在一定缓冲液存在时完成.缓冲液和待测液中的离子强度对还原糖测定的影响如图4所示.通过在0.05mol /L 柠檬酸缓冲液中改变氯化钠浓度来改变溶液的离子强度.可以看出,随着溶液的离子强度增加,其吸光度变化不大,但有缓慢降低的趋势.2.2 测定波长对纤维素酶酶活力测定的影响在分光比色分析测定时,一般选定最大光吸收值作为测定波长,这样做可获得最大的灵敏度.但在纤维素酶酶活力测定过程中,不同的研究人员[11,10]所选择的波长有所不同.作者用紫外可见分光光度仪比较了波长对葡萄糖的光吸收的影响,如图5所示.发现在可见光范围内,显色液在486nm 处有较大吸收.作者分别在486,510,540,570nm 波长下测得葡萄糖的标准曲线以及相关系数,如表1和图5所示.从表1可看出,不同波长下所得的标准曲线和相图5 测定波长对纤维素酶活力测定的影响关系数存在着差异.波长越短,其标准曲线的相关系数偏离1的数值越大;随着波长的增加,这种偏离程度变小.究其原因可能是在486nm 波长下测定时,存在最大光吸收,所以灵敏度最高;但在此波长下测定,所测得的数据稳定性差,相关系数偏离1的数值趋大.在570nm 波长下测得的数据,虽然数据稳定性好,但灵敏度偏低.从图5可以看出,为兼顾两者,540nm 是在最大光吸收的波长和灵敏度最低的波长中间,既能获得较高的灵敏度,且所测得的数据稳定性又较好.因此,在纤维素酶的酶活力测定过程中,一般取540nm 为测定波长.另外,在光比色分析过程中,一般取透光度20%~80%,吸光度在0.1~0.7范围内时,测定的数据较准确.从表1可看出,波长越长,标准曲线的线性范围越宽,这对纤维素酶的酶活力测定非常有利;另一方面它又使测定的灵敏度降低,给测定带来不利影响.从上面分析可看出,兼顾两者,又照顾到纤维素酶酶活力测定的实际需要,选540nm 为还原糖比色测定的波长较为适宜.表1 不同波长下葡萄糖曲线的回归方程相关系数和线性范围测定波长/nm葡萄糖曲线的回归方程 相关系数线性范围486y =0.3467x 0.97960.28~ 2.02510y =0.5852x -0.12380.99730.38~ 1.41540y =0.4306x -0.095240.99910.45~ 1.85570y =0.2583x -0.057140.99920.61~ 2.932.3 还原糖的种类对纤维素酶酶活力测定的影响图6 还原糖的种类对纤维素酶活力测定的影响Miller 等人[12]曾经指出,用DN S 等分析试剂对还原糖进行测定时,其纤维素酶酶解产物不同(如葡萄糖、纤维二糖和其它纤维寡糖).不同的还原糖氧化还原能力存在差异,这种差异不可避免地给测定带来误差,而这些却一直被人们所忽略.图6为还原糖的种类对还原糖测定的影响.从图中可看出,以葡萄糖为标准,纤维二糖测定值较之低一些,而木糖测定值则高一些.在进行纤维素酶酶活力测定时,酶解液中主要含有葡萄糖和纤维二糖以及少量纤维寡糖[13].由此可以看出,以葡萄糖为标准测定纤维素酶酶活力,所得到的结果比实际酶活偏低.2.4 不同测定方法对纤维素酶酶活力测定的影响常用的还原糖分析方法有两种,即DNS 法和Somo gyi 法,能够分析测定葡萄糖和纤维二糖.以葡萄糖为基准,分别用两种方法分析相同含量的纤维二糖,会得出不同的测定值,其结果见表2.在还原糖测定中,Som ogyi 方法会得到更准确的值.可是,纤维素酶酶解产物,表2 不同测定方法对纤维素酶酶活力测定的影响测定方法葡萄糖纤维二糖DNS10085~88Somogyi 10055~58通常不是形成某一类还原糖,而是形成以葡萄糖和纤维二糖为主的、并含有寡聚糖的混合物.在这种情况下,DNS 方法仅有最小误差[13].3 讨 论3.1 DN S 测定方法的原理和适用性DNS 方法适于碱性条件,DNS 与还原糖发生氧化还原反应,生成了3-氨基-5-硝基水扬酸,其产物在煮沸条件下显色,其颜色深浅与还原糖含量有比例关系,利用这一原理测定还原糖的含量.DNS 与还原糖显色后,其颜色的深浅与糖类游离出还原基团的数量有关,而对还原糖的种类没有选择性.DNS 方法适合用在多糖(如纤维素、半纤维素和淀粉等)水解产生多种还原糖的体系中.对于测定纯的、单一的外切β-纤维二糖分解酶或者内切β-葡聚糖酶,用Som og yi 方法为宜.如果测定一个纤维素酶系对不溶性纤维素的酶解能力,用DNS 方法较适宜.3.2 DN S试剂的组成及其作用DNS试剂是由DNS、氢氧化钠、酒石酸钾钠、苯酚和亚硫酸氢钠加蒸馏水配制而成.配制方法是把DN S加入一定量的氢氧化钠溶液中,待完全溶解后,加入已溶解的酒石酸钾钠溶液,混合均匀,再加入苯酚和亚硫酸氢钠,贮存于棕色试剂瓶中.在室温下,放置7~10d 后即可使用.酒石酸钾钠在DN S试剂中的作用是防止溶解氧的侵入;苯酚的作用是增加DN S显色后的颜色深度,以及平衡脲形成时对糖测定的影响;亚硫酸氢钠与苯酚配合使显色后颜色更加稳定;碱性环境是DNS与还原糖作用的条件.3.3 进行纤维素酶酶活力测定时DN S方法的规范在25m L刻度试管中盛有进行纤维素酶酶活力测定的溶液,加入3m L DN S试剂,在沸水中煮沸5min进行显色,然后在流水中急速冷却.用蒸馏水定容到25m L.1h内,在540 nm波长下比色测定光吸收值,对照葡萄糖标准曲线,查出还原糖的含量.4 结 语 纤维素酶系作用于水不溶性纤维素,其分解能力可分别用糖化型纤维素酶活力或者纤维素溶解的纤维素酶活力来表述.前者指分解产生还原糖的能力,通常用滤纸酶活和CM C 酶活来表示;后者指纤维素降解为低分子糖的能力,常用CM C粘度降低法[14]和滤纸崩溃法[15]来表示.由于微生物分泌纤维素酶酶系的复杂性,底物纤维素又是处于多相复杂体系,天然纤维素除含有纤维素之外还有半纤维素和木素等,这些都给纤维素酶活力和纤维素酶作用机制的分析和纤维素酶的分离精制带来困难.这就要求进一步探讨和改进现有的研究和分析手段,使纤维素酶的研究和利用有所突破,开发和利用丰富的可再生纤维素资源,使之造福于人类.参考文献:[1] G ERRI T B,PET T ERSSO N B.Ex tracelllar enzy me sy stem utilized by the fung us for the breakdow no f cellulo se[J].Eur J Bio ch em,1985,146:301~308[2] W O OD T M.Purificatio n a nd some pro perties of a(1,4)-β-D-g lucan gluco hydro la se a ssociated withth e cellulase[J].Biochem J,1968,109:217~227[3] G HO SE T K.Studies o n the mecha nism of enzyma tic h ydro ly sis of cellulosic[J].Bio techno l Bio eng,1979,21:131~146[4] O K AK I M,M O O-YO U N G M.Kinetic o f enzymatic hydr olysis of cellulo se:Ana ly tical desc riptio n o fa mecha nistic model[J].B io tech no l B io eng,1978,20:637~663[5] ERIK SSO N K E.Proper ties and mide o f actio n o f cellulase[J].FEBS Lett,1974,49(2):282[6] M I LL ER G e o f dinitr osalicylic acid r eag ent fo r deter mination o f reducing suga r[J].AnalChem,1959,31(3):426~428[7] SOM O GY I M.A new r eage nt fo r the deter minatio n o f reducing suga r[J].J B io l Chem,1952,195:119[8] M AN DEL S M.M easurement o f saccharifying cellula se[J].Bio techno l Bio eng,1976,(6):21~33[9] 福田作藏.还原糖の定量法[M].东京:学会出版センタ-,1989.[10] 北京大学生物化学教研室著.生物化学实验指导[M].北京:人民教育出版社,1979.[11] G HO SE G T.M easurem ent o f cellulase activ ities[J].Pur e Applied Chem,1968,59:257~268[12] M IL L ER G L.M ea sur ement o f ca rbox yme th ylcellulase activ ity[J].Anal B iochem,1961,2:521~528[13] 高培基,刘垂王干.研究纤维素酶活时测定还原糖方法的选择[J].植物生理通讯,1985,2:51~54.[14] A LM IN K E.V isco metric dete rmination of car bo x ymethy lcellulase in sta ndard inter na tio nal[J].B io chem Bio phy s Acta,1967,39:248~253[15] N U M M I M,FOX C P.N ephelo metric and turidomet ric assays o f cellulase[J].Anal Biochem,1981,116:133~136The Modification of the D N S Method for theDetermination of Reducing SugarGU AN Bin1,DIN G You-fang2,X IE Lai-su2,LON G Yan-quan2(1.Sha ndong Institute o f Light Industry,Jinan250100;2.Tianjin Institute o f Ligh t Industr y,Tia njin 300222)Abstract:In this paper,the procedure and co ndition(fo r exa mple,the dosag e of DN S reag ent,heating time,dissolv ing ox ygen and ionic streng th)on the m easurem ent o f reducing suga r w ere studied.Results sho w tha t the choice o f m easuring wav elength w as affected by hy dro lysis products.Therefo re,the measuring wav elength at540nm w as chosen to ex pand the liner range of g lucose a nd obtain proper sensitivity and stabler da ta. On the o ther hand,when g lucose w as used as sta nda rd,v arious m ethods fo r reducing sug ar w o uld giv e different v alues.When determining the saccharifying effect o f a cellulase com plex,the result sho uld be independent o f the ty pe of suga r fo rmed.Thus,the erro r is actually smallest w ith th e DN S m ethod in the case.Key words:saccharifying cellulase;activity;testing co ndition;DNS;reducing sug ar。
发酵液还原糖含量测定法

标准管理规程STANDARD OPERATING PROCEDURE发酵液还原糖含量检查法版本:2005版生效日期:2005年月日编号:SOP—QC—3002—00制定:审核:QA审核:签发:日期:日期:日期:日期:颁发部门:质量部部门审核:QA审核:签发:1.目的:还原糖的测定也是为了控制抗生素新陈代谢过程。
2.范围:车间化验室3.责任:QC化验室化验人员和技术人员。
4.原理:单糖中醛基在碱性溶液中与铜盐作用,铜盐则还原成亚铜盐,此亚铜盐之量即代表糖的多少,本法测定之糖含量为100ml远洋中含由糖的克数。
5.程序5.1 样品测试:5.1.1吸取发酵过滤液0.5ml于100ml碘量瓶中,加2-3ml蒸馏水,再加入10ml菲林试剂,摇匀。
5.1.2将碘量瓶置于电炉上煮沸水解3分钟,取下冷却。
5.1.3待冷后,加入4mol/L硫酸3.5ml密塞振摇,然后用0.1N硫代硫酸钠进行滴定,滴定至颜色呈淡米色时,加入1%淀粉指示剂1ml,摇匀,再用0.1N硫代硫酸钠滴至蓝色刚好消失,记下滴定毫升数。
5.1.4用蒸馏水代样品作一份空白,以空白滴定的毫升数减去样品滴定的毫升数,查糖标准曲线表,取得还原糖含量。
5.2标准曲线制备:5.2.1取适量的无水葡萄糖置于1050C恒温烘箱中干燥3小时,然后取出于干燥罐中冷却。
5.2.2取出冷却后的葡萄糖5.0g溶于蒸馏水中,加水定容至100ml,摇匀,制成5%葡萄糖溶液。
标准管理规程STANDARD OPERATING PROCEDURE版本:2005版生效日期:2005年月日编号:SOP—QC—3002—005.2.3别吸取上述标准溶液0.1、0.2、0.3、0.4、0.5ml各分别加水0.9、0.8、0.7、0.6、0.5ml至100ml碘量瓶中,将总量为1ml,浓度为0.5、1.0、1.5、2.0、2.5g/100ml的各点标准样品。
5.2.4于碘量瓶中各加入10ml菲林试剂,然后置于电炉上煮沸水解3分钟,取下冷却。
糖测定实验实验报告

一、实验目的1. 掌握DNS比色法测定还原糖的原理和方法。
2. 学会利用DNS比色法测定发酵液中还原糖含量。
3. 了解不同食品中总糖和还原糖的含量。
二、实验原理DNS比色法是一种测定还原糖含量的常用方法。
在碱性条件下,还原糖与3,5-二硝基水杨酸(DNS)发生反应,生成棕红色的化合物。
在一定浓度范围内,还原糖的量与光吸收值呈线性关系,通过比色法可以测定样品中的还原糖含量。
三、实验材料1. 样品:发酵液、不同食品(如苹果、香蕉、橙子等)。
2. 试剂:DNS试剂、NaOH溶液、苯酚、亚硫酸钠、酒石酸钾钠、葡萄糖、蒸馏水。
3. 仪器:可见分光光度计、电子天平、真空干燥箱、数显恒温水浴锅、离心机、移液器、枪头、容量瓶、试管、烧杯。
四、实验步骤1. DNS试剂的配制:准确称取DNS试剂1.0g,溶解于100ml的蒸馏水中,加入NaOH溶液5ml,混合均匀,备用。
2. 样品处理:取发酵液或食品样品适量,加入蒸馏水稀释至一定浓度。
3. 标准曲线的绘制:准确移取不同浓度的葡萄糖标准溶液,加入DNS试剂,混匀,置于水浴锅中加热5分钟,取出后冷却至室温,在540nm波长下测定光吸收值。
以葡萄糖浓度为横坐标,光吸收值为纵坐标,绘制标准曲线。
4. 样品测定:准确移取处理后的样品溶液,加入DNS试剂,混匀,置于水浴锅中加热5分钟,取出后冷却至室温,在540nm波长下测定光吸收值。
5. 计算还原糖含量:根据样品的光吸收值,从标准曲线上查得相应的葡萄糖浓度,计算样品中还原糖的含量。
五、实验结果与分析1. 标准曲线的绘制:根据实验数据,绘制标准曲线,线性关系良好。
2. 样品测定:对不同食品和发酵液进行测定,得到还原糖含量如下:(1)苹果:还原糖含量为5.0g/100g;(2)香蕉:还原糖含量为11.0g/100g;(3)橙子:还原糖含量为8.5g/100g;(4)发酵液:还原糖含量为3.5g/100ml。
六、实验讨论1. 实验过程中,DNS试剂的配制和样品处理对实验结果有较大影响。
DNS法测定还原糖

DNS法测还原糖方法一原理DNS即二硝基水杨酸法是利用碱性条件下,二硝基水杨酸(DNS)与还原糖发生氧化还原反应,生成3—氨基—5—硝基水杨酸,该产物在煮沸条件下显棕红色,540nm处有吸收,且在一定浓度范围内颜色深浅与还原糖含量成比例关系的原理,用比色法测定还原糖含量的。
二试剂及方法2。
1 试剂1. 1 mg/ml的葡萄糖溶液2. 3,5-二硝基水杨酸用 400mL 蒸馏水溶解 6.3g3,5—二硝基水杨酸,逐步加入 21g 氢氧化钠,再加入182g 四水合酒石酸钾钠、5.0g 苯酚、5。
0g 无水亚硫酸钠,温水浴(不超过 48℃),不断搅拌,直至溶液清澈透明.用蒸馏水定容至 1000mL,保存在棕色瓶中,与二氧化碳隔绝,静置 5~7 天后使用,贮存期为 6 个月。
2.2 葡萄糖标曲的绘制表1 葡萄糖标曲的绘制方法试管试剂012345葡萄糖标准液(ml)00.10.20。
30.40.5蒸馏水(ml)0.50。
40.30.20。
103,5-二硝基水杨酸0。
50。
50。
50。
50.50。
5(ml)糖含量(mg)00.10.20.30.40。
5混合均匀,沸水浴5min,冷却后加入4mL蒸馏水,混匀,在540nm下测OD值2.3 发酵液样品测定将发酵液稀释一定倍数后,取3只比色管标号1,2,3,加入稀释和的发酵液0.5 ml ,加入 DNS 试剂0。
5 ml 混匀,沸水浴5min,冷却后加入4 ml 蒸馏水混匀,测OD 540表 2 样品的测定方法管号0 1 2还原糖待测样品 (mL ) 0。
0 0。
5 0。
5 蒸馏水 (mL ) 0。
5 0。
0 0。
0 DNS 试剂 ( mL ) 0.5 0.5 0.5 蒸馏水 (mL ) 444A 540 nm计算所得还原糖含量 (mg )三 结果计算 3.1 标曲3.2样品的还原糖含量稀释倍数)加人样品的体积()计算得到的还原糖量()还原糖含量(⨯=ml 5.0mg /g L。
直接滴定法测定还原糖的原理与主要影响因素(严选内容)

直接滴定法测定还原糖的原理与主要影响因素日常食品检测中,还原糖是一个常规理化检验项目,涉及的样品种类很广,如乳制品、肉制品、发酵酒及果蔬制品叶等。
目前还原糖的检测分二大类,一类是具体检测某一还原糖含量,如葡萄糖、果糖等;一类是测定还原糖总量,还原糖总量目前应用较多的是化学滴定法,食品检测中最常用的是国标GB/T 5009.7-2008中的第一法:直接滴定法。
本文讨论的是后者。
检测原理(1)碱性酒石酸铜甲液与乙液混合后,生成蓝色氢氧化铜沉淀,此沉淀立即与酒石酸钾钠反应生成深蓝色的酒石酸钾钠铜络合物。
(2)当碱性酒石酸铜甲、乙液与还原糖共热时,酒石酸钾钠铜被还原生成红色的氧化亚铜沉淀物,而还原糖的醛基或酮基则被氧化为羧基,生成还原糖酸。
(3)当还原糖将溶液中酒石酸钾钠铜耗尽时,稍微过量的还原糖可将亚甲基蓝还原而呈无色,指示滴定终点的到来。
(4)为消除氧化亚铜沉淀对滴定终点观察的干扰,在碱性酒石酸铜乙液中加入少量亚铁氰化钾,与红色的氧化亚铜发生络合反应,生成可溶性无色络合物,利于终点的判定。
检测原理的补充性解释酒石酸钾钠作用。
既然实验须在碱性条件下进行,那么硫酸铜遇碱生成氢氧化铜沉淀后,不利于实验正常进行,必须使其(铜离子)在可溶状态下才行,酒石酸钾钠与铜离子络合物酒石酸钾钠铜是可溶的,从而达到了目的。
亚铁氰化钾作用。
当样品中存在大量的如铁、锰、钴等金属离子,或样品处理时,沉淀剂乙酸锌过量了,都会消耗碱性酒石酸铜乙液中的亚铁氰化钾,当亚铁氰化钾被过量消耗时,不能有效络合氧化亚铜,致使滴定终点不显无色而显暗红色。
可另配制亚铁氰化钾溶液,滴定时往锥形瓶中适量添加,标准与样液添加量一样。
定量标准物质。
碱性酒石酸铜甲液中的硫酸铜的铜离子(Cu2+)为此滴定反应的定量标准物质,碱性酒石酸铜乙液中氢氧化钠提供了强碱性环境,故国标GB/T 5009.7-2008中5.2:“吸取5.0mL碱性酒石酸铜甲液”的体积量取精度应改为“5.00mL”,否?t检测结果的有效位数达不到该标准的要求:“还原糖含量≥10g/100g时计算结果保留三位有效数字”。
发酵测定还原糖意义

发酵测定还原糖意义摘要:一、引言二、发酵测定还原糖的原理1.费林试剂的作用2.还原糖与费林试剂的反应3.反应结果的判定三、发酵测定还原糖的意义1.食品中糖类含量的检测2.生物发酵过程中的监测3.还原糖在医药领域的应用四、发酵测定还原糖的实验方法1.样品处理2.费林试剂的配制与使用3.实验操作步骤4.结果分析与计算五、总结与展望正文:发酵测定还原糖的意义在于对食品、生物发酵过程以及医药领域中糖类物质的检测与研究。
本文将从发酵测定还原糖的原理、意义以及实验方法等方面进行详细阐述。
一、引言发酵现象在生活中无处不在,发酵过程中的糖类物质发生变化,对食品的品质、口感等方面产生影响。
发酵测定还原糖作为一种糖类物质检测方法,对于研究发酵过程、提高食品品质等方面具有重要意义。
二、发酵测定还原糖的原理发酵测定还原糖的原理主要基于费林试剂与还原糖的反应。
费林试剂由氢氧化钠和铜离子组成,当与还原糖反应时,还原糖将被氧化,同时使铜离子还原成红色沉淀。
根据红色沉淀的生成情况,可以判断样品中还原糖的存在与否。
1.费林试剂的作用费林试剂具有很强的氧化性,能与还原糖发生氧化还原反应。
在这个过程中,费林试剂被还原,还原糖被氧化。
2.还原糖与费林试剂的反应将费林试剂加入待测样品中,如果样品中含有还原糖,则还原糖与费林试剂发生反应,生成红色沉淀。
3.反应结果的判定观察反应液的颜色变化,如果由蓝色变为红色,说明样品中含有还原糖。
颜色变化的程度可以用来判断还原糖的含量。
三、发酵测定还原糖的意义1.食品中糖类含量的检测发酵测定还原糖方法被广泛应用于食品行业,对糖类物质进行检测。
如在葡萄酒、果汁、蜜饯等产品中,可以通过发酵测定还原糖的方法检测糖度,以确保产品质量和口感。
2.生物发酵过程中的监测在生物发酵过程中,糖类物质是重要的原料。
通过发酵测定还原糖的方法,可以实时监测发酵过程中糖类物质的消耗情况,优化发酵条件,提高产率和产品质量。
3.还原糖在医药领域的应用某些药物中含有还原糖成分,通过发酵测定还原糖的方法,可以对药物中的糖类物质进行含量测定,以确保药物的安全性和有效性。
发酵液还原能力测定

发酵液还原能力测定
发酵液的还原能力是指其抗氧化能力,通常通过测定其对氧化
剂的还原作用来评估。
常见的测定方法包括抗坏血酸法、铁离子还
原法和DPPH自由基清除法等。
抗坏血酸法是一种常用的测定发酵液还原能力的方法。
该方法
通过将发酵液与含有氧化剂的试剂(如二碘化钾溶液)反应,然后
用碘液滴定未反应的氧化剂,从而计算发酵液中还原抗坏血酸的含量,间接反映其还原能力。
铁离子还原法是另一种常见的测定方法,利用发酵液中的还原
物质与铁离子生成的蓝色络合物的形成来测定还原能力。
还原能力
强的发酵液会生成更多的蓝色络合物,从而可以通过比色法或者光
度计来测定其还原能力。
DPPH自由基清除法是一种常用的体外抗氧化能力测定方法。
该
方法利用DPPH自由基的颜色变化来评估发酵液的抗氧化能力,还原
能力强的发酵液会导致DPPH自由基的减少,从紫色变为黄色或无色,通过测定吸光度的变化来评估其抗氧化能力。
除了上述方法,还有其他一些测定发酵液还原能力的方法,如Folin-Ciocalteu法、FRAP法等。
这些方法可以从不同角度全面地评估发酵液的抗氧化能力,为发酵液的质量评价提供了重要的参考依据。
总的来说,测定发酵液的还原能力是评估其抗氧化能力的重要手段,可以帮助我们了解发酵液的营养价值和保健功效。
发酵液还原糖含量测定

溶液蓝色刚好褪去为终点 记录耗用的葡萄糖标液的体积V1
10ml斐林试剂相当于葡萄糖的质量按下式计算:
m mV1 1 1000
式中: m1—10ml斐林试剂相当于葡萄糖的质量,单位为(g); m—称取葡萄糖的质量,单位为(g); V1—正式标定时,消耗葡萄糖标准溶液的总体积,单位为(ml)。
3 样品的预滴定
吸取甲液、乙液各5.0ml,加入10ml处理过的样 品,摇匀,于电炉上加热,控制在2min内沸腾。
趁沸以每2秒1滴的速度滴加葡萄糖标液
溶液蓝色刚好褪去为终点
记录耗用的葡萄糖标液的体积
4 样品的正式滴定
吸取甲液、乙液各5.0ml,加10ml处理过发酵液 (样品),从滴定管滴加比预测体积少0.5-1mL 的葡萄糖标液,摇匀,控制在2min内加热至沸腾。
思考题
发酵液中的蔗糖、总糖该如何测定??
感谢观看 THANK YOU
02 PART TWO
仪器、器皿
仪器
万分之一天平、电炉、烘箱
器皿
滴定管(25ml)、铁架台、容量瓶、 吸量管(5ml、10ml)、锥形瓶 (150ml)、烧杯、试剂瓶等
03 PART THREE 试剂准备
01斐Leabharlann 试剂甲液硫酸铜15g、亚甲蓝0.05g 溶于水中,并稀释至1000mL
乙液
酒石酸钾钠50g、氢氧化钠75g, 溶解于水中,再加亚铁氰化钾4g, 完全溶解后,用水定容至1000mL。
02
葡萄糖 标准溶液
准确称取经过98℃~100℃烘箱中干燥 2h后的葡萄糖1g,加水溶解后加入盐 酸溶液5mL,并用水定容至1000mL。
此溶液每毫升相当于1.0mg葡萄糖
发酵液还原糖的测定方法

中间体化验操作法发酵液中糖含量的测定:1.原理:利用还原性糖类的自由配合基在碱性溶液中能将高价铜还原成为低价铜,过量的高价铜在酸性溶液中与碘化钾作用生成Cu2I2,同时析出碘,析出的碘以标准硫代硫酸钠(Na2S2O3)溶液滴定,同时做一空白对照,从两者之间求出糖的含量。
反应式如下:酸△(C6H10O6)n n(C6H10O6)水解CuSO4+2NaOHCu(OH)2+Na2SO4CHOH—COONa O—CH--COONaCu(OH)2+Cu +2H2OCHOH—COOK O—CH--COOKO-CHCOOK CHOCu +CHOHO4O-CHCOOK CHOHO--CHCOOK COONa HO--CHCOOK2Cu +NaOH+H2O (CHOH)4+Cu2O+O--CHCOOK CH2OH HO--CHCOONaO—CHCOOK CHOHOOK过量的Cu +2H2SO4O—CHCOOK CHOHOONa+CuSO4+Na++K++SO42-2CuSO4+4KI+2Cu↓2K2SO4+I2(在酸化条件下)I2+2Na2S2O32NaI+Na2S4O62.试剂:1)斐林试剂直接配制法:将酒石酸钾钠800g溶于水中,再溶入硫酸铜160g,加入1N的NaOH溶液1318ml (或固体氢氧化钠580g),最后加入KI400g溶后,加入纯化水至总体积为10000ml,摇匀即得。
2)斐林试剂贮备液的制备a.硫酸铜贮备液:称取硫酸铜(CuSO4·5H2O)800g加纯化水溶解(如浊可用玻璃棉过滤),使总体积成10000ml,摇匀即可。
b.酒石酸钾钠贮备液:称取酒石酸钾钠400g,加纯化水使总体积为1000ml摇匀即得。
c.斐林试剂混合液:取硫酸铜贮备液1000ml,加酒石酸钾钠贮备液1000ml,摇匀加11mol/L氢氧化钠(NaOH)659.5ml或固体NaOH290g,最后加入碘化钾(KI)200g,溶后加入水,使总体积为5000ml。
发酵生产中还原糖和葡萄糖检测指标的分析

发酵生产中还原糖和葡萄糖检测指标的分析史建国杨俊慧孟庆军杨艳马耀宏张利群(山东省科学院生物研究所,济南,250014)摘要采用还原糖测定法和葡萄糖酶电极测定法,对谷氨酸发酵生产上淀粉糖原料和发酵液中还原糖和葡萄糖进行了测定,对其变化的特点和意义进行了研究。
结果表明:淀粉糖液中葡萄糖/还原糖比值变化从80% - 94.2%;发酵过程中,谷氨酸生产菌首先消耗葡萄糖,发酵28h,葡萄糖含量接近零,而还原糖含量为 1.0%;还原糖测定仪用于发酵后期还原糖测定,精密度(RSD%)为2.21,对发酵后期的精确控制具有一定的应用价值。
关键词还原糖,葡萄糖,发酵过程控制微生物发酵生产中常以淀粉为基本原料,经水解生成还原糖或葡萄糖,供发酵使用。
糖的检测是生产过程控制的常规生化指标[1]。
近年来,还原糖测定仪和葡萄糖测定仪已在发酵生产中应用,并逐步取代传统的手工滴定法,实现了还原糖和葡萄糖快速、准确的仪器化分析,大大减少了人为测定的误差[2、3]。
但由于测定原理和方法的不同,使测定结果出现了差异。
本文对多年来在谷氨酸发酵生产中测定的还原糖和葡萄糖结果进行了总结,对还原糖和葡萄糖检测指标进行了比较和分析,对发酵生产中仪器分析方法的应用特点和意义进行了探讨。
1材料和方法1.1实验材料淀粉水解液糖化液为莲花集团不同的糖化车间的送检样品;谷氨酸发酵液为菱花集团三分厂送检样品;化学试剂均为分析纯,用蒸馏水配制。
1.2实验方法还原糖测定(1)斐林试剂滴定法采用国家标准测定法(GB/T 5009.7—1985)(2)还原糖测定仪采用山东科学院生物中心提供的SGD-Ⅲ型还原糖测定仪。
该仪器测定原理同还原糖斐林试剂滴定法。
测定方法如下:接通电源(220V),按“开/关”键,自动启动准备程序;用微量注射器将标准品注入反应池,完成后自动定标;测定时,用微量注射器将被测样品注入反应池,仪器自动完成测定过程,并显示和打印测定值。
葡萄糖测定采用山东省科学院生物研究所提供的SBA-40型谷氨酸-葡萄糖双功能分析仪。
糟醅取样及还原糖测定的基本过程、注意事项。

糟醅取样及还原糖测定的基本过程、注意事项。
糟醅取样基本过程:
1.入池酒酪试样采集入池酒酪应在堆积发酵完毕,即将入池时取样,从堆的四周及中间采集试样10kg左右。
最好在入池过程中,每次相隔一定时间,分次采集相同数量的试样,迅速混匀后,以四分法取出0.5~1kg装入广口瓶中,注明取样时间、班次、池号。
2.出池酒酷试样采集,在酒酷上、中、下各层的3~4处,采集试样10kg 左右,迅速混合均匀,用四分法取出0.5~lkg,装入广口瓶中。
注意事项:
为防止水分等成分改变,须将塞子盖紧,尽快进行检验。
还原糖测定基本过程:
1.样品经除去蛋白质后,在加热条件下,直接滴定已标定过的费林氏液,费林氏液被还原析出氧化亚铜后,过量的还原糖立即将次甲基蓝还原,使蓝色褪色。
2.根据样品消耗体积,计算还原糖量。
注意事项:
(1)本方法测定的是一类具有还原性质的糖,包括葡萄糖、果糖、乳糖、麦芽糖等,只是结果用葡萄糖或其他转化糖的方式表示,所以不能误解为还原糖=葡萄糖或其他糖。
但如果已知样品中只含有
某一种糖,如乳制品中的乳糖,则可以认为还原糖=某糖。
(2)分别用葡萄糖、果糖、乳糖、麦芽糖标准品配制标准溶液分别滴定等量已标定的费林氏液,所消耗标准溶液的体积有所不同。
证明即便同是还原糖,在物化性质上仍有所差别,所以还原糖的结果只是反映样品整体情况,并不完全等于各还原糖含量之和。
如果已知样品只含有某种还原糖,则应以该还原糖做标准品,结果为该还原糖的含量。
如果样品中还原糖的成分未知,或为多种还原糖的混合物,则以某种还原糖做标准品,结果以该还原糖计,但不代表该糖的真实含量。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
中间体化验操作法
发酵液中糖含量的测定:
1.原理:利用还原性糖类的自由配合基在碱性溶液中能将高价铜还原成为
低价铜,过量的高价铜在酸性溶液中与碘化钾作用生成Cu2I2,同时析出碘,析出的碘以标准硫代硫酸钠(Na2S2O3)溶液滴定,同时做一空白对照,从两者之间求出糖的含量。
反应式如下:
酸△
(C6H10O6)n n(C6H10O6)
水解
CuSO4+2NaOH
Cu(OH)2+Na2SO4
CHOH—COONa O—CH--COONa
Cu(OH)2+Cu +2H2O
CHOH—COOK O—CH--COOK
O-CHCOOK CHO
Cu +CHOHO4
O-CHCOOK CHOH
O--CHCOOK COONa HO--CHCOOK
2Cu +NaOH+H2O (CHOH)4+Cu2O+
O--CHCOOK CH2OH HO--CHCOONa
O—CHCOOK CHOHOOK
过量的Cu +2H2SO4
O—CHCOOK CHOHOONa
+CuSO4+Na++K++SO42-
2CuSO4+4KI+2Cu↓2K2SO4+I2(在酸化条件下)
I2+2Na2S2O32NaI+Na2S4O6
2.试剂:
1)斐林试剂直接配制法:将酒石酸钾钠800g溶于水中,再溶入硫酸铜160g,加入1N的NaOH溶液1318ml (或固体氢氧化钠580g),最后加入KI400g溶后,加入纯化水至总体积为10000ml,摇匀即得。
2)斐林试剂贮备液的制备
a.硫酸铜贮备液:称取硫酸铜(CuSO4·5H2O)800g加纯化水溶解(如
浊可用玻璃棉过滤),使总体积成10000ml,摇匀即可。
b.酒石酸钾钠贮备液:称取酒石酸钾钠400g,加纯化水使总体积为1000ml
摇匀即得。
c.斐林试剂混合液:取硫酸铜贮备液1000ml,加酒石酸钾钠贮备液1000ml,
摇匀加11mol/L氢氧化钠(NaOH)659.5ml或固体NaOH290g,最后加入碘化钾(KI)200g,溶后加入水,使总体积为5000ml。
3)8N的硫酸:取36mol/ml 的试剂H2SO4(密度为1.84g/ml)222ml,缓缓注入778ml水中,放冷摇匀,即得。
4)0.5%淀粉指示剂:
a.称取可溶性淀粉5g,溶于约50ml纯化水中。
b.量取950ml纯化水于烧杯中,加热煮沸,将1)液倒入搅拌,使其溶解,冷却至室温倒入1000ml试剂瓶中,置冰箱备用。
5)20%碘化钾溶液:称取c.p碘化钾(KI)100克,用纯化水溶解至500ml,
摇匀即得。
6)4N盐酸(HCl):量取12N的C.P盐酸(比重1.19)334ml,加纯化水
至1000ml,摇匀即得。
7)2N盐酸(HCl):量取上述4N盐酸500ml,加纯化水500ml,摇匀即得。
8)0.4N碘酸钾(KIO3)基准溶液:精确称取经研细,并在105℃干燥至恒重的KIO3(AR)基准试剂3.5670g,用纯化水溶入250ml容量瓶中,并稀释至刻度,摇匀即得0.4N的碘酸钾溶液,称量时误差不得超0.02‰。
W
KIO3的当量浓度=
250×0.0367(E/1000)
3.5670
= =0.4N
250×0.03567
214.02 35.67
E =
=35.67,=0.03567(KIO3的毫克当量)
6 1000
9)1N硫代硫酸钠(Na2S2O3)贮备液:
称取碳酸钠(Na2CO3)20g,溶于约6000ml 水中,加入五水亚硫酸钠(Na2SO3·5H2O)2000g,溶解完全后,稀释至1000ml,摇匀过滤继续放置一周后标化备用。
标化原理:
KIO3+5KI+6HCl 3I2+6KC l+3H2O
I2+2Na2S2O32NaI+Na2S2O6
标化操作:精确吸取已配制1mol/L的硫代硫酸钠溶液100ml于1000ml容量瓶中,稀释至刻度,摇匀,注入已洗净并用待标定溶液冲2~3次的25ml一等滴定管中,另精确吸取5ml0.4N碘酸钾(HIO3)标准溶液于250ml碘瓶中,加5ml20%碘化钾(KI)溶液,加纯化水10ml,再加2mol/L盐酸(HCl)10ml,立即加盖并用纯化水水封在暗处放置3分钟,然后用上述硫代硫酸钠(Na2S2O3)溶液滴定,用淀粉做指示剂至兰色刚刚消失为终点。
要求:0.1mol/ml硫代硫酸钠(Na2S2O3)的结果允许误差在0.0002之间。
10)0.1mol/ml硫代硫酸钠(Na2S2O3):经计算后,量取约1mol/ml的硫代硫酸钠贮备液一定量,稀释至
0.1mol/L,摇匀即可。
标化操作与1mol/L的Na2S2O3相同。
3.测定方法:
1)溶糖测定法:吸取发酵滤液2.5ml于50ml具塞刻度试管中,加5ml4mol/L盐酸(HCl)在沸水中水解10分钟,取出后冷却,用纯化水稀释至25ml,
取5ml稀释于150ml三角瓶中,加入斐林试剂25ml,摇匀后在电炉上加热沸腾3分钟,用冷水冷却后加入8N硫酸(H2SO4)10ml,摇匀立即以0.1mol/L硫代硫酸钠(Na2S2O3)的滴定液滴定,以淀粉做指示剂,滴定至指示剂由兰变白为止,记下消耗的滴定液数。
空白:直接取斐林试剂25ml于150ml三角瓶中,在电炉上沸腾3分钟,其他操作同上。
计算:
(V空白消耗Na2S2O3-V样品消耗Na2S2O3)×N Na2S2O3
V 0.1 Na2S2O3
0.1
由0.1mol/ml的硫代硫酸钠(Na2S2O3)的体积即可计算出溶糖的百分含量。
注:如所用的标准Na2S2O3的摩尔浓度正好是0.1mol/ml,则不需要再计算。
以空白所消耗的Na2S2O3体积减去样品Na2S2O3的体积即为0. 1000mol/ml硫代硫酸钠(Na2S2O3)的体
积,用此数直接查表即可。
2)还原糖测定法:取发酵滤液1ml(如含量在0.4%以上取0.5ml)于150ml
三角瓶中,加斐林试剂25ml,其它操作同溶糖做法。
空白操作同溶糖测定。
计算:以新消耗的Na2S2O3体积数查还原糖表。
4.标准曲线的制作:
精确称取(先60℃烤1小时,80℃烤1小时,105℃再烤2小时)烤过恒重的基准葡萄糖(AR含量在99%以上)0.5g于100ml容量瓶中,加
纯化水稀释至刻度,摇匀,分别吸取0.5ml、1ml、1.5ml、2ml、2.5ml、5ml、10ml,按糖法测之,以糖的百分含量为横坐标,以0.1mol/L 硫代硫酸钠(Na2S2O3)滴定数为纵坐标,做一曲线,再将滴定体积数与糖含量关系列表备用。
1)操作注意事项:斐林溶液加量要准确,因铜量是计算的依据。
2)要保证电炉上加热沸腾3分钟的准确性,加热后必须冷却才能加8mol/L硫酸(H2SO4),以免碘挥发。
3)加入8mol/L硫酸(H2SO4)后开始滴定时要振摇,以免I2挥发。
淀粉指示剂在滴定将近终点时加入为宜,否则I2和淀粉形成复合物,碘淀粉影响滴定的准确度。
4)Na2S2O3的水溶液遇酸和空气中的氧及水中细菌极易起分解反应。
Na2S2O3+H2CO3NaHCO3+S2↓+NaHSO3
Na2S2O3+H2SO4Na2SO4++SO2+H2O
Na2S2O3+1/2O2Na2SO4+S↓
水中如含铜离子也能促其分解。
2Cu2++2S2O32-2Cu++S4O62-
由此在配制时要用煮沸冷却的纯化水,同时加入少量的碳酸钠,放置数日后,使其稳定,使用溶液时间不可太长,否则浓度易变化。
5)KIO3标准液应放在冰箱内,新配制的标准液与旧标准液要核对。
消耗Na2S2O3体积的差应在±0.02ml 以内。
6)标定硫代硫酸钠时个人平行两个结果之差应该小于0.03ml,二人核对结果之差小于0.05ml,否则均应重作。