测序比对表
测序数据分析流程

测序数据分析流程测序数据分析是基因组学研究的重要步骤之一,它可以帮助科研人员了解DNA序列的组成和功能,从而揭示生物体内的遗传信息和生物过程。
测序数据分析流程可以分为以下几个主要步骤:数据预处理、质控、比对、变异检测和功能注释。
1.数据预处理测序数据通常以原始测序片段(short reads)的形式存在,首先需要将这些片段进行预处理。
预处理的主要目的是去除低质量序列、去除引物序列和适配器序列,并且进行去除重复序列。
2.质控质量控制是一个重要的步骤,可以帮助去除测序过程中引入的错误和杂质。
这一步骤通常使用质量分数来评估每个碱基的可靠性,并使用阈值过滤出具有较高质量的片段。
常用的质控工具包括FastQC和Trimmomatic等。
3.比对比对是将测序片段与一个已知的参考序列进行比较的过程。
比对的目的是找到碱基序列与参考序列的相似性和差异,并将测序片段映射到参考序列上。
常用的比对软件包括Bowtie2、BWA和STAR等。
4.变异检测变异检测是为了发现测序样本与参考序列之间的碱基差异。
这些差异可能是单核苷酸多态性(SNP)、插入缺失(INDEL)或结构变异等。
常用的变异检测软件有GATK和SAMtools等。
5.功能注释功能注释是将变异位点与已知的生物信息进行关联,以帮助解释它们的潜在影响和功能。
这一步骤可以帮助研究人员找到与特定疾病相关的功能变异,并进一步研究其机制。
功能注释工具包括ANNOVAR、Variant Effect Predictor (VEP)和SNPEff等。
除了上述的主要步骤外,测序数据分析流程还可以包括其他附加步骤,如数据可视化和统计分析。
数据可视化可以将测序数据和结果以图表或图像的形式展示出来,帮助研究人员更好地理解和解释分析结果。
统计分析可以帮助评估数据的可靠性和统计学意义,并进一步探索数据背后的模式和关联。
测序数据分析是一个复杂且多步骤的过程,需要研究人员具备一定的生物信息学和统计学知识。
BLAST检索和比对

BLAST检索和比对Alignment: 序列比对。
将两个或多个序列排在一起,以达到最大一致性的过程(对于氨基酸序列是比较它们的保守性),这样可以评估序列间的相似性和同源性。
Algorithm: 算法。
在计算机程序中包含的一种固定过程。
Bioinformatics: 生物信息学。
一门结合生物技术和信息技术从而揭示生物学中新原理的科学。
Bit score: 二进制。
二进制值S'源于统计性质被数量化的打分系统中产生的原始比对分数S。
由于二进制值相对于打分系统已经被标准化,它们常用于比较不同搜索之间的比对分数。
BLAST: 基本的局部相似性比对搜索工具。
在序列数据库中快速查找与给定序列具有最优局部对准结果的序列的一种序列比对算法。
初步搜索是对打分至少为T、长度为W的词进行的。
打分的过程是用一个替代矩阵对查询序列和该词作比较。
然后词长可以试着向两端伸长以获得一个超过阈值S的打分。
参数T反映了搜索的速度大小和敏感性。
可以参见BLAST的用户指南和BLAST使用指导来获得更详细的信息。
BLOSUM: 模块替换矩阵。
在替换矩阵中,每个位置的打分是在相关蛋白局部比对模块中观察到的替换的频率而获得的。
每个矩阵被修改成一个特殊的进化距离。
例如,在BLOSUM62矩阵中,是使用一致性不超过62%的序列进行配对来获得打分值的。
一致性大于62%的序列在配对时用单个序列表示,以避免过于强调密切相关的家族成员。
Conservation: 保守。
指氨基酸或DNA(普遍性较小)序列某个特殊位置上的改变,并不影响原始序列的物理化学性质。
Domain: 结构域。
蛋白质在折叠时与其他部分相独立的一个不连续的部分,它有着自己独特的功能。
DUST: 一个低复杂性区段过滤程序。
E value: E值。
期望值。
在一个数据库中所搜索到的打分值等于或大于S的不同比对的个数。
E值越低,表明该打分值的显著性越好。
Filtering: 过滤,也叫掩蔽(masking)。
有参考基因组的转录组生物信息分析模板

v1.0 可编辑可修改一、生物信息分析流程获得原始测序序列(Sequenced Reads)后,在有相关物种参考序列或参考基因组的情况下,通过如下流程进行生物信息分析:二、项目结果说明1 原始序列数据高通量测序(如illumina HiSeq TM2000/MiSeq等测序平台)测序得到的原始图像数据文件经碱基识别(Base Calling)分析转化为原始测序序列(Sequenced Reads),我们称之为Raw Data或Raw Reads,结果以FASTQ(简称为fq)文件格式存储,其中包含测序序列(reads)的序列信息以及其对应的测序质量信息。
FASTQ格式文件中每个read由四行描述,如下:@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACGGCTCTTTGCCCTTCTCGTCGAAAATTGTCTCCTCATTCGAAACTTCTCTGT+@@CFFFDEHHHHFIJJJ@FHGIIIEHIIJBHHHIJJEGIIJJIGHIGHCCF其中第一行以“@”开头,随后为illumina 测序标识符(Sequence Identifiers)和描述文字(选择性部分);第二行是碱基序列;第三行以“+”开头,随后为illumina 测序标识符(选择性部分);第四行是对应序列的测序质量(Cock et al.)。
illumina 测序标识符详细信息如下:第四行中每个字符对应的ASCII值减去33,即为对应第二行碱基的测序质量值。
如果测序错误率用e表示,illumina HiSeq TM2000/MiSeq的碱基质量值用Qphred表示,则有下列关系:公式一:Qphred = -10log10(e)illumina Casava 版本测序错误率与测序质量值简明对应关系如下:2 测序数据质量评估测序错误率分布检查每个碱基测序错误率是通过测序Phred数值(Phred score, Q)通过公式1phred转化得到,而Phred 数值是在碱基识别(Base Calling)过程中通过一种预测碱基判别发生错误概率模型计算得到的,对应关系如下表所显示:illumina Casava 版本碱基识别与Phred分值之间的简明对应关系测序错误率与碱基质量有关,受测序仪本身、测序试剂、样品等多个因素共同影响。
基因测序 序列比对

基因测序序列比对英文回答:Gene sequencing is a technique used to determine the order of nucleotides in a DNA molecule. It is a fundamental tool in genetics and has revolutionized our understanding of the human genome and the genomes of other organisms. Sequencing allows us to identify genetic variations, mutations, and other important information that can help us understand the genetic basis of diseases and develop personalized treatments.One of the key steps in gene sequencing is sequence alignment, which involves comparing the sequences of different DNA molecules to identify similarities and differences. Sequence alignment is important because it allows us to determine the degree of similarity between sequences and infer evolutionary relationships between organisms.There are several methods and algorithms available for sequence alignment, but the most commonly used one is called the Needleman-Wunsch algorithm. This algorithm uses dynamic programming to find the optimal alignment between two sequences by considering all possible alignments and assigning a score to each alignment based on the similarity of the aligned nucleotides.The Needleman-Wunsch algorithm works by creating a matrix that represents all possible alignments between the two sequences. Each cell in the matrix represents aspecific alignment and contains a score that represents the similarity of the aligned nucleotides. The algorithm then fills in the matrix by considering three possible ways to reach each cell: from the cell above, from the cell to the left, or from the cell diagonally above and to the left. The optimal alignment is determined by tracing back through the matrix, starting from the bottom right cell, and selecting the path with the highest score.Sequence alignment is a computationally intensive process, especially when dealing with large genomes. Toaddress this issue, several optimization techniques have been developed, such as the Smith-Waterman algorithm, which is a variant of the Needleman-Wunsch algorithm that allows for local sequence alignment. Local sequence alignment is useful when we are interested in identifying regions of high similarity within a larger sequence.In conclusion, gene sequencing and sequence alignment are critical tools in genetics research. They allow us to decipher the genetic code and understand the complexities of the genome. The Needleman-Wunsch algorithm and its variants, such as the Smith-Waterman algorithm, are widely used for sequence alignment and help us uncover the evolutionary relationships and genetic variations that shape life on Earth.中文回答:基因测序是一种确定DNA分子中核苷酸顺序的技术。
测序比对基因突变位点与修水黄羽乌鸡卷羽性状的关联性

张危红,魏岳,康昭风,等.测序比对基因突变位点与修水黄羽乌鸡卷羽性状的关联性[J ].中南农业科技,2024,45(4):31-34.羽毛是鸟类表皮细胞衍生的角质化产物,具有保暖、信息交流、飞行等作用,且有显著的表型多样性,十分复杂。
鸡是具羽毛形态表型变异的鸟类,不同羽毛形状的变异可作为不同品种的重要表型标记,也可作为标记辅助选择在育种中发挥作用。
羽毛可根据表型划分为平羽、卷羽、丝羽等,其中,卷羽是由羽杆向外卷曲而形成。
鸡的卷羽基因是不完全的常染色体显性遗传,研究者不断对卷羽鸡进行选育与推广,在生产性能等方面做了大量研究[1-5]。
然而,对卷羽性状的分子研究相对欠缺,研究者相继对卷羽鸡系谱进行分析,结果表明在鸡22号染色体上的连锁群E22C19W28_E50C23内角蛋白基因簇中,KRT75基因的内含子5与外显子5间的连接处,存在1个69bp 的缺失突变,该突变导致23个氨基酸的缺失,从而导致羽轴结构的改变,使羽毛尖端扭曲,表现为卷羽表型,这些研究奠定了鸡羽毛性状的遗传基础[6,7]。
但陶林等[8]的研究结果表明,中国现有卷羽鸡的卷羽性状不是由这段69bp 碱基序列的缺失引起的,而可能与KRT75基因CDS 区域中的3个突变位点(954bp :T>C ;967bp :T>C ;978bp :C>T )有关。
修水黄羽乌鸡作为江西省独特的地方家禽品种资源,具有耐粗饲、抗逆性强等优点,全身披黄色羽毛,羽毛紧凑,色泽光亮,羽毛形态以平羽为主。
前期研究发现修水黄羽乌鸡中,也有部分个体自出壳以来,羽毛逐渐表现为外卷形态,为弄清修水黄羽乌鸡羽毛外卷的原因,本研究对基础群进行系统选育,且通过测序寻找与卷羽性状相关的位点,为利用修水黄羽乌鸡卷羽性状奠定分子基础。
1材料与方法将具有羽毛外卷表型的个体从修水黄羽乌鸡群体中挑选出来,与平羽表型个体分开饲养,后期繁殖实行卷羽鸡与卷羽鸡交配,平羽鸡与平羽鸡交配,后代仅留与父母代相同表型的个体,选育3代后,采集后代卷羽鸡和平羽鸡的血液提取DNA ,经PCR 扩增后送南京金斯瑞生物科技有限公司进行Sanger 测序,再进行数据比对,寻找与卷羽变异相关的变异位点。
二代测序基因组拼接和短序列比对

二代测序基因组拼接和短序列比对二代测序技术广泛应用于基因组测序,它可以快速、高效地产生大量短序列。
然而,由于短序列的长度限制,对于较大的基因组,需要进行拼接和短序列比对来重建完整的基因组序列。
基因组拼接是将测序得到的短序列片段按照它们的重叠区域进行拼接,以恢复原始的基因组序列。
这个过程涉及到数据处理、序列比对和重建等步骤。
通常,基因组拼接可以分为两种主要策略:重叠图策略和重铺策略。
重叠图策略是通过将短序列片段之间的重叠关系可视化为一个图形,然后使用图算法来寻找最长的路径,从而确定序列的重叠顺序。
这样,就可以将短序列片段逐步拼接成较长的连续序列,最终得到完整的基因组序列。
另一种常用的方法是短序列比对。
在这种方法中,短序列片段与已知的参考序列进行比对,以确定它们在基因组中的位置和顺序。
通过将多个短序列片段按照参考序列进行比对,可以逐步填补基因组的空白区域,最终重建出完整的基因组序列。
无论是基于重叠图策略还是短序列比对,基因组拼接都需要处理大量的数据和进行复杂的算法计算。
此外,由于测序过程中可能存在错误和噪音,拼接和比对过程中还需要考虑纠错和过滤掉低质量的序列片段。
总结来说,二代测序产生的短序列需要通过基因组拼接和短序列比对的方法来重建完整的基因组序列。
这些方法涉及到数据
处理、序列比对和重建等步骤,以实现对较大基因组的测序和分析。
如何使用NCBI对比测序结果资料

Vector contamination
输入测序结果
运行
Vector contamination
红色:强烈 紫红:中间 绿色:弱 无色:目的序列
成功插入目的序列
• 找出无色ห้องสมุดไป่ตู้域中序列的范围(28~299) • 返回Blast页面
空载体
假阳性,表明目的基因并没有成功插入到载体中,转化到菌株中的只是空 载体。
其他方法——搜索酶切位点或标记
例如使用了pET28a-MS2载体,用到BamHI和HindIII酶切位点,则在测序结 果中搜索这两个酶切位点的序列,插入片段为180bp。那么查看搜索结果, 两个酶切位点中只有十几bp之差,肯定不会是插入了目的序列,那么就是空 载体。如果中间的片段大概有目的片段大小,则把该序列复制到NCBI上搜索
如何使用NCBI对比测序结果
禤淑霞 2013.11.19
内容
• What is NCBI? • How ?(未知序列,已知序列)
What is NCBI?
NCBI (National Center for Biotechnology Information ),是指美国国立生物技术 信息中心 主要用途 1. 查找基因信息/序列 2. 对比 3. 文献
How
打开NCBI的主页:/
• 或直接打开主页/Blast.cgi
basic
Specialized BLAST
未知序列对比——不知道插入片段的序列
• 搜索出 插入载 体中的 目的序 列
balst主页
核酸序列
选择核酸来源, 例如人类,老鼠, 还是其他 others
选择合适的数据库:other(其它)
搜索结果
比对的序列来源
序列比对,构建进化树
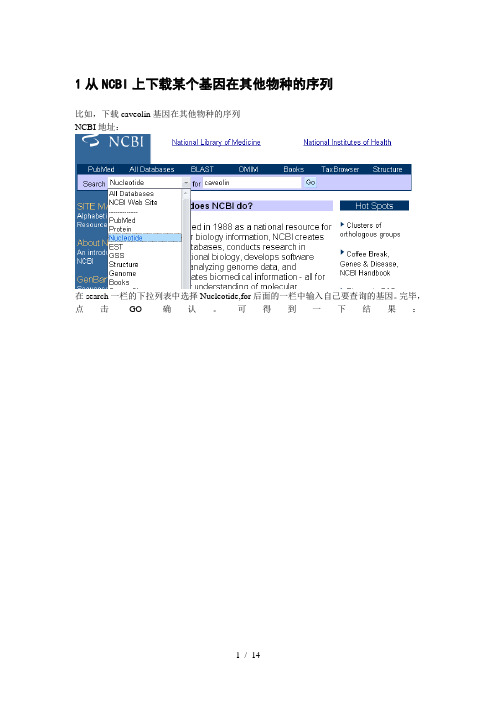
1从NCBI上下载某个基因在其他物种的序列比如,下载caveolin基因在其他物种的序列NCBI地址:在search一栏的下拉列表中选择Nucleotide,for后面的一栏中输入自己要查询的基因。
完毕,点击GO确认。
可得到一下结果:每一条记录分别是某个物种的caveolin的序列,以第10条记录为例,称为GenBank 登录号。
为拉丁文的人类的字母,表示物种,表示基因名称(caveolin基因家族共有3个主要基因,分别称为1,2,3)表示此序列为cDNA,不含含子。
下图中的NEXT表示翻页,查看剩余的记录。
打开第10条记录可看到下图:现在你需要保存下来得就是上面的这一串(碱基)核酸序列。
复制黏贴(包括上面表示顺序的数字)到TXT文本中备用。
打开DNAMAN软件,左上角点击file-new,出现下图:可以把先前从NCBI下载的序列(保存到TXT文本中得)复制到箭头指示处,得到:并按照上图左上角file-save as(注意此文件得保存名称为保存的此物中得名称),已上是DNAMAN软件中seq序列格式的保存方法。
2 序列编辑和比对(DNAMAN软件)你们实验PCR得到的序列只是某个基因上的一部分,所以为了进行不同物种间的比对,要把下载下来的其他物种的某个基因的序列进行删减,以使两段基因是大约相同长度的片段进行比对。
以人类caveolin1基因为例说明一下。
按照1,2,3得顺序依次打开,得到下图:点击上图中的1,你会得到下图,点击2是清楚所有刚才选进比对的序列(为了重新选择序列),3是有选择的删除某个序列。
当然,把你的所有准备的序列保存好以后,从查找围这个下拉列表中寻找你要比对的序列。
可以按住ctrl点击你要比对的几个序列(同时选中)选完点击打开。
再点下图中得确定键。
得到下图:找好这两个物种重合的那个核苷酸的序号(前后两段都是),然后打开你保存的seq格式的序列,数出刚才比对重合部分的后端的碱基数,把这个碱基后面的序列删掉,再用此方法把比对重合部分前段得序列删掉,保存。
2_重测序BSA分析项目结题报告

重测序BSA项目结题报告客户单位:报告单位:联系人:联系电话:传真:报告日期:项目负责人:审核人:目录目录 (1)1 项目概况 (1)1.1 合同关键指标 (1)1.2 项目基本信息 (1)1.3 项目执行情况 (2)1.4项目结果概述 (2)2 项目流程 (3)2.1 实验流程 (3)2.2 信息分析流程 (3)3 生物信息学分析 (5)3.1 测序数据质控 (5)3.1.1 原始数据介绍 (5)3.1.2 碱基测序质量分布 (7)3.1.3碱基类型分布 (9)3.1.4 低质量数据过滤 (10)3.1.5测序数据统计 (10)3.2 与参考基因组比对统计 (11)3.2.1 比对结果统计 (11)3.2.2 插入片段分布统计 (11)3.2.3 深度分布统计 (12)3.3 SNP检测与注释 (14)3.3.1 样品与参考基因组间SNP的检测 (14)3.3.2 样品之间SNP的检测 (17)3.3.3 SNP结果注释 (19)3.4 Small InDel检测与注释 (22)3.4.1 样品与参考基因组间Small InDel的检测 (22)3.4.2样品之间Small InDel检测 (22)3.4.3 Small InDel的注释 (23)3.5关联分析 (26)3.5.1 高质量SNP筛选 (26)3.5.2 SNP-index方法关联结果 (26)3.5.3 ED方法关联结果 (28)3.5.4候选区域筛选 (29)3.6候选区域的功能注释 (30)3.6.1 候选区域的SNP注释 (30)3.6.2 候选区域的基因注释 (30)3.6.2.1候选区域内基因的GO富集分析 (31)3.6.2.2候选区域内基因的KEGG富集分析 (33)3.6.2.3候选区域内基因COG分类统计 (36)3.7结果可视化 (37)4 数据下载 (38)4.1结果文件查看说明 (38)参考文献 (39)1 项目概况1.1 合同关键指标(1) 完成X个样品的重测序,共产生XGbp Clean Data,保证Q30达到80%。
反向引物测序比对结果

反向引物测序比对结果-概述说明以及解释1.引言1.1 概述概述部分的内容:反向引物测序比对结果是生物学和遗传学研究中的一个重要环节。
它通过对DNA序列进行测序,并将其与参考序列进行比对,从而得到反向引物的测序结果。
这项技术的发展促进了基因组学、遗传学以及其他许多领域的研究进展。
在反向引物测序的过程中,研究人员会首先合成反向引物,然后利用该引物与待测序列特异性结合,通过测序仪进行测序。
测序结果会被与已知的参考序列进行比对,以确定相对应的基因或序列信息。
比对结果的分析对于理解反向引物测序的可靠性和准确性至关重要。
通过分析比对结果,我们可以评估测序过程中的潜在错误,并确定反向引物测序的可靠性。
此外,比对结果还可以用来确定测序样本中可能存在的变异、突变或其他遗传变化。
反向引物测序比对结果的意义不仅在于验证反向引物测序的准确性,同时还提供了一种检测和分析基因组中特定区域的方法。
比对结果可以用来研究个体间的遗传差异、基因组的结构和功能等方面。
此外,比对结果还可为医学诊断、药物研发以及基因治疗等领域的研究提供重要参考。
总的来说,反向引物测序比对结果是基因组学和遗传学研究中不可或缺的一部分。
它不仅能够验证反向引物测序的准确性,还为研究人员提供了一种探索基因组和遗传变异的方法。
随着测序技术的不断发展,我们可以期待反向引物测序比对结果的应用范围将进一步拓展,并为更多科学领域带来新的突破。
1.2 文章结构文章结构:本文共分为引言、正文和结论三个部分。
引言部分主要概述了反向引物测序比对结果这一主题的背景和意义,并介绍了本文的结构和目的。
首先,我们会概述反向引物测序的原理和应用,以便读者对这一技术有一个基本的了解。
其次,我们将通过分析反向引物测序比对结果,揭示这些结果的重要性和意义。
最后,我们将总结反向引物测序比对结果,并展望其未来的发展方向。
正文部分是本文的核心内容,具体分为四个小节。
首先,我们会详细介绍反向引物测序的原理,解释其基本原理和操作流程。
三代RNA测序序列的比对和分析工具

三代RNA测序序列的比对和分析工具为了理解人类的遗传机制,以便用于人类疾病的治疗和预防,对于生物序列的研究成为了研究分析遗传的基础。
最基本和最关键的研究就是对于人类遗传物质的序列研究。
随着测序技术的发展,第二代测序技术和第三代测序技术先后引领了一段生物信息学的发展前沿。
随着测序技术的进步,为了适应发展需求,大量的序列比对算法开始逐渐产生和发展。
二代的DNA序列比对技术相比于传统的DNA序列比对技术来讲速度更快,节省更多空间。
二代测序技术同样带动了RNA序列比对技术的发展,为了满足二代测序数据产生的RNA序列数据的比对需要,开发出了很多类型的二代RNA序列比对工具。
对于RNA序列比对的研究,首先要研究RNA的生物学特性,在了解清楚RNA 的生物学背景后,我们才能研究出适应发展需求的RNA序列比对工具。
而三代测序技术的发展则带动了RNA测序技术的更进一步发展,但适用于三代RNA测序序列的算法工具目前几乎没有,目前的RNA序列比对软件在三代RNA序列的比对方面还急需加强。
本文设计了一个针对三代RNA测序序列的比对分析算法,实现了对三代RNA 测序数据的比对和外显子组分析,其主要工作是对三代RNA测序数据进行比对,根据其长读长的特点将其进行分割,对得到的短序列使用BWA方法进行比对,比对到参考基因组上得到比对结果。
根据比对得到的结果对匹配上的位置区域做合并得到全基因组上的外显子岛区域信息,通过构造连通图,使用动态规划等计算最优路径等找出外显子岛的比对信息。
之后则利用三代RNA长序列来进行序列的外显子组成分析,使用上一步得到的外显子岛序列,来对序列的外显子岛组成进行分解,根据各个长序列的组成分析。
对于同一基因的不同剪接,体现为相似序列的外显子岛的组成不同,主要是依据序列的外显子岛是否有重复,得到可变剪接的对比结果。
基因组测序中的序列比对使用教程

基因组测序中的序列比对使用教程序列比对在基因组测序中扮演着重要的角色,它是将测序得到的短序列与已知基因组进行比对,以确定这些短序列在基因组中的位置和功能。
本文将为您提供一份基因组测序中序列比对的详细使用教程。
一、理解序列比对的基本概念序列比对的基本概念是将测序得到的短序列与已知基因组进行匹配。
测序通常会产生大量的短序列,这些短序列需要通过比对才能确定其在基因组中的位置和功能。
在序列比对中,通常会引入一个参考基因组,该参考基因组是一个已知的基因组序列,可以是某个物种的基因组或某个特定区域的基因组。
二、选择合适的序列比对工具选择合适的序列比对工具对于准确地比对测序数据非常重要。
常见的序列比对工具包括Bowtie、BWA、BLAST等。
以下是这些工具的简介:1. Bowtie:Bowtie是一款非常快速的短序列比对工具,适合于比对长度较短的序列。
2. BWA:BWA适用于比对长度较长的序列,比如全基因组测序。
3. BLAST:BLAST是一款广泛应用于序列比对的工具,可以根据序列的相似性进行比对。
根据实际需求和数据类型选择合适的比对工具,以确保比对的准确性和效率。
三、准备比对所需的参考基因组和测序数据在进行序列比对之前,需要准备比对所需的参考基因组和测序数据。
参考基因组可以从公共数据库(如NCBI)下载,也可以使用自己的实验室已有的基因组数据。
测序数据通常是以FASTQ文件格式存储的,包括了测序reads的序列和对应的质量分数。
在比对之前,需要先将FASTQ文件进行质量控制和预处理,例如使用Trimmomatic工具去除低质量reads和适配体序列。
四、进行序列比对选择合适的比对工具后,可以开始进行序列比对。
以下是比对的一般流程:1. 将参考基因组索引化:大部分比对工具都需要将参考基因组进行索引化,以加快比对速度。
通过运行工具提供的索引化命令将参考基因组转换为索引文件。
2. 进行比对:根据选择的比对工具和参数设置,将准备好的测序数据与参考基因组进行比对。
三代转录组测序数据比对构建索引

三代转录组测序数据比对构建索引序号1:概述随着生物学研究的发展,基因组学研究成为当前生物科学领域的热点之一。
而其中,转录组测序数据比对构建索引作为基因组学研究的重要组成部分,一直备受关注。
序号2:转录组测序数据比对构建索引的概念转录组测序数据比对构建索引是指根据RNA序列的原始数据,通过比对和构建索引,将RNA序列与已知的基因组序列进行比对,从而寻找出RNA序列中的基因结构和功能信息的过程。
通过比对分析,可以实现在基因组水平上进行对RNA序列的定位和注释,从而进一步理解RNA序列的生物学功能。
序号3:转录组测序数据比对构建索引的重要性转录组测序数据比对构建索引对于生物学研究具有重要意义。
通过比对构建索引可以帮助科研人员揭示RNA序列的基因组定位及其可能的功能。
转录组测序数据比对构建索引是深入探究基因调控的重要方式,可以帮助揭示基因在不同条件下的表达情况,从而进一步深入了解基因的调控网络。
转录组测序数据比对构建索引也为基因组学研究提供了重要的数据支持,为后续的功能分析和生物信息学研究提供了基础。
序号4:转录组测序数据比对构建索引的方法与技术转录组测序数据比对构建索引的方法与技术涉及到多个方面。
首先是数据预处理,即对原始测序数据进行质量控制、去除低质量序列和接头序列等步骤。
其次是参考基因组的选择和索引的构建,即选择合适的已知基因组序列进行比对,并根据该基因组序列构建索引。
最后是比对算法的选择和优化,根据具体的研究目的选择合适的比对算法,并对算法进行优化,提高比对的准确性和效率。
序号5:转录组测序数据比对构建索引的挑战与解决方案转录组测序数据比对构建索引在实际操作中也存在一些挑战,如测序数据的质量、参考基因组的选择、比对算法的选取等问题。
针对这些挑战,科研人员们通过不断的探索和实践,提出了一些解决方案,如应用质量控制工具处理测序数据、利用最新版本的基因组参考序列、采用多样化的比对算法等,来提高转录组测序数据比对构建索引的准确性和可靠性。
基于比对骨架的第三代测序数据比对与变异检测方法

变异检测
基于比对骨架,使用变异检测 算法识别基因组中的单核苷酸 变异、插入和缺失等变异类型 。
结果输出
将变异检测结果以可视化和表 格形式输出,便于后续分析。
结果分析与讨论
变异注释
对检测到的变异进行功能注释,了解其对基因 和蛋白质结构与功能的影响。
结果比较
将本方法与现有方法进行比较,评估其准确性 和效率。
数据来源
实验数据来自公共数据库和合作实验 室,包括人类基因组、小鼠基因组以 及特定组织或细胞系的测序数据。
实验环境
实验环境包括高性能计算集群、存储 设备和软件工具,用于处理大规模测 序数据。
实验过程与结果
数据预处理
对原始测序数据进行质量控制 、去噪和标准化处理据与参 考基因组进行比对,构建比对 骨架。
变异类型识别
目前该方法主要关注单核苷酸变异,对于结构变异和表观 遗传变异的识别仍需进一步研究和改进。
未来研究方向
1 2 3
算法优化
针对数据处理复杂度问题,未来研究可以进一步 优化算法,提高处理大规模数据的效率。
跨物种比对
针对参考基因组依赖性问题,未来研究可以探索 跨物种比对方法,以适应与参考基因组差异较大 的物种或个体。
临床诊断
第三代测序技术在临床诊断中具有广泛的应用前景 ,如遗传病诊断、肿瘤基因检测等。
生物信息学分析
第三代测序技术产生的数据量巨大,需要借 助高性能计算和生物信息学分析方法进行数 据处理和解读。
02
基于比对骨架的数据比对方法
比对骨架的基本概念
比对骨架
指将测序得到的原始读段(reads)进行预处理和组装,形成较长的序列片段 或超级读段(superreads),再对这些超级读段进行精确比对的一种数据结构 。
基于宏基因组测序技术的非靶向筛查方法检测肉类食品中的动物源性成分

基于宏基因组测序技术的非靶向筛查方法 检测肉类食品中的动物源性成分丁清龙,杨丹婷,谢爱华,韦 云,陈秀芬,周 露*(广东省食品检验所,广东广州 510435)摘 要:目的:建立肉类食品中动物源性成分非靶向筛查方法。
方法:基于宏基因组测序技术建立肉类食品中动物源性成分非靶向筛查方法,将该方法用于模拟样品和实际样品检测,并用现有标准检测方法对实际样品检测结果进行确认。
结果:模拟样品检测结果与模拟情况一致;在实际样品中检出与样品名称不一致或样品标签未标识的动物源性成分,且非靶向筛查方法检测结果与现有标准方法检测结果一致。
结论:该方法可以快速锁定样品中未知的动物源性成分,检测结果准确可靠,可为政府打击肉类食品掺假提供更为有力的技术支撑。
关键词:宏基因组测序;动物源性成分;非靶向;掺假Determination of Animal-Derived Ingredients in Meat Products by Non-Targeted Screening Method Based on MetagenomicSequencing TechnologyDING Qinglong, YANG Danting, XIE Aihua, WEI Yun, CHEN Xiufen, ZHOU Lu*(Guangdong Institute of Food Inspection, Guangzhou 510435, China)Abstract: Objective: To establish the non-targeted screening method for animal-derived ingredients in meat products. Method: The non-targeted screening method for animal-derived ingredients in meat products was established based on metagenomic sequencing technology. The method was applied to the determination of simulated samples and actual samples, and the results of actual samples were confirmed by existing standard determination methods. Result: The determination results of simulated samples were consistent with design of simulated samples. The animal-derived ingredients inconsistent with sample name or label identification were detected in actual samples, and the results of non-targeted screening method and existing standard methods were consistent. Conclusion: This method could quickly test the unknown animal-derived ingredients of samples, and the determination results were accurate and reliable. It could provide more powerful technical support to combat meat adulteration for the government.Keywords: metagenomic sequencing technology; animal-derived ingredients; non-targeted; adulteration肉类食品是人们餐桌必备的主要食品之一,常见的高经济价值的肉类食品有牛肉、羊肉及其制品等。
微生物室室间比对记录表

微生物室实验室室间比对记录表
项目名称: 一般细菌培养+鉴定 细菌血清学凝集
比对单位
参比单位
比对人
参比人
本结果为: 比对单位
参比单位
序 号
样本编号
标本 类型
检验项目
比对结果 参比结果
1
2
3
4
5
6
7
8
9
1 0
结 论
一致 性
评语
专业组组长签字:
比对时间 比对单位
微生物室实验室间比对记录表
项目名称: 涂片革兰染色镜检查细菌 涂片抗酸染色镜检 涂片革兰染色镜检查真菌
参比单位
比对人 本结果为: 比对单位
参比人 参比单位
序 号
样本编号
标本 类型
标本处理方法
比对结果 参比结果
一致 性
评语
不需缩标本 1
需浓缩标本 不需浓缩标本 2 需浓缩标本 不需浓缩标本 3 需浓缩标本 不需浓缩标本 4 需浓缩标本 不需浓缩标本 5 需浓缩标本
结 论
专业组组长签字:
基因组测序中的质控和序列比对

基因组测序中的质控和序列比对基因组测序作为分子生物学研究的重要手段之一,已经成为了各领域生物学之间进行对比与研究的基础。
对于普通人而言,基因测序被广泛应用于宠物、人类的健康管理以及食品安全等领域。
然而,基因组测序中存在繁杂的技术问题,其中最重要的问题就是测序质量的控制和基因序列的比对。
基因组测序的质量控制可以从测序设备、样本质量、操作环境和生物信息学算法等多个方面进行。
首先,测序设备应保证其每次操作的一致性,并在使用前进行正确的校准。
此外,测序的样本质量也是非常关键的。
对于动物和植物而言,应选择优质的DNA或RNA样本,并保证其在取样、储存、运输等各个环节中不会受到污染和损伤。
在操作环境上,应选择干净、无菌、温度适宜且空气流通良好的区域进行。
最后,基因组测序的生物信息学算法也是保证测序质量的一个重要环节。
测序质量控制是基因组测序的关键。
基因组测序的过程中会产生大量的数据,质量较差的数据会给后续的研究带来负面影响。
因此,必须对测序数据进行质量控制。
主要的质量控制指标包括reads长度、reads质量和reads GC含量等。
对于reads长度,在基因组测序的过程中,可以通过合理构建文库、正确的数据处理和质控等方法来确保reads长度的均匀性。
对于reads质量,单序列错误可能会导致大量错误的序列比对和基因注释结果,因此对于reads质量的控制至关重要。
最后GC含量,虽然与测序质量的关系不大,但是如果GC含量的变化比较突出,就必须在数据分析过程中进行一些特殊的处理。
基因组序列比对也是基因组测序的重要部分。
随着DNA测序技术的发展,可以测定的基因组存在高度异质性。
其中,单核苷酸多态性(SNP)和结构变异(copy number variation,CNV和重复序列)等功能和进化最重要的特征,不仅仅决定着基因组水平结构和功能的差异,也是致病基因和新基因等的主要来源。
因此,在深入分析基因组序列之前,必须对基因序列进行比对。
基因序列比对长度

基因序列比对长度主要取决于测序技术的类型。
二代测序序列长度较短,大约为300bp,错误率低,主要错误类型为替换错误。
这种技术可以直接查询每条序列在基因组中的准确比对区域,得到比对结果。
而三代测序序列的错误率较高,且错误类型主要为插入或删除错误,难以通过文本查找直接找到序列在基因组中的比对区域,需要采用针对性的查询策略找到比对区域。
另一方面,在比对阶段,由于三代序列读段长(平均长度超过10kbp),直接采用全局比对(Needleman-Wunsch,NW)或局部比对(Smith-Waterman,SW)比对算法需构建L×L大小的得分矩阵(其中L表示序列长度),计算空间复杂度较高。
体外诊断试剂在进行临床试验时,如采用测序方法作为对比方法,针对测序方法应提供哪些临床资料?

体外诊断试剂在进行临床试验时,如采用测序方法作为对比方法,针对测序方法应提供哪些临床资料?
A :
1. 信息性内容:采用测序方法时,临床试验资料中应提供测序方法的相关信息。
1.1 应提供测序方法原理、测序仪型号、测序试剂及消耗品的相关信息;
1.2 应提供测序方法所用引物相关信息,如基因区段选择,分子量、纯度、功能性实验等资料。
引物设计应合理涵盖考核试剂扩增的靶核酸区段、位点、及所有突变类型。
2. 方法学验证信息
2.1 对所选测序方法的分析性能进行合理验证,尤其是最低检测限的确认,建议将所选测序方法与申报试剂的相关性能进行适当比对分析。
2.2 测序方法应建立合理的阳性质控品和阴性质控品对临床样本的检测结果进行质量控制。
3.测序结果信息
除结果数据表中的测序结果外,应提交有代表性的样本测序图谱及结果分析资料。
审评六部供稿。
DNA测序结果分析比对(实例)

DNA(一)测序结果分析比对(实例)dna测序结果2013-08-22 11:59来源:互联网点击次数:14423从测序公司得到的一份DNA测序结果通常包含.seq格式的测序结果序列文本和.ab1格式的测序图面是一份测序结果的实例:CYP3A4-E1-1-1(E1B).ab1CYP3A4-E1-1-1(E1B).seq .seq文件可以用系统自带的记事本程序件需要用专门的软件打开。
软件名称:Chromas软件Chromas下载 .seq文件打开后如下图:.ab1文件打开后如下图:通常一份测序结果图由红、黑、绿和蓝色测序峰组成,代表不同的碱基序列。
(下图原图的后半段被剪切掉了)大约50个碱基的测序图部分通常杂质的干扰较大,无法判读,象。
这也提醒我们在做引物设计时,要避免将所研究的位点离PCR序列的两端太近(通常要大于50个碱基测序后难以分析比对。
我的课题是研究基因多态性的,因此下面要介绍的内容也主要以判读测序图中的等为主。
实际上,要在一份测序图中找到真正确实的等位基因多态位点并不是一件一般认为等位基因位点假如在测序图上出现像套叠的两个峰,就是杂合子位点。
实际比对后才知道么简单,下面测序图中标出的两个套峰均不是杂合子位点,如图并说明如下:说明:第一组套峰,两峰的轴线并不在同一位置,左侧的T峰是干扰峰;第二组套峰,置相同,但两峰的位置太靠近了,不是杂合子峰,蓝色的C峰是干扰峰通常的杂合子峰由一高一略相同的峰组成,此处的序列被机器误判为“C”,实际的序列应为“A”,通常一个高大碱基峰的前面产生一个相同碱基的干扰峰,峰的高度大约是高大碱基峰的1/2,离得越近受干扰越大。
一个摸索出来的规律是:主峰通常在干扰峰的右侧,干扰峰并不一定比主峰低。
最关键的拿疑似为杂合子峰的测序图位点与测序结果的文本序列和基因库中的比对结果相比较;一个位点的较;你得出的该位点的突变率与权威文献或数据库中的突变率相比较。
通常,对于一个疑似突变位点来说,即使是国际上权威组织大样本的测序结果中话,那么单纯通过测序结果就判定它是突变点,是并不严谨的,因一份 PCR产物中各个碱基的实同,很难避免不产生误差的。