先天性肾上腺皮质增生症研究进展
先天性肾上腺皮质增生症的长期治疗及疗效评价

第 24卷第 8 期 2009年 4 月 J App l C lin P ed ia tr, Vo l. 24 N o. 8, Ap r. 2009
# 569#
先天性肾上腺皮质增生症的长期治疗及疗效评价
孙文鑫
(上海交通大学医学院附属瑞金医院 儿内科, 上海 200025)
Long- Term T rea tm ent and E ffect Eva lua ted in Congenita l Adrenal H yperplas ia
剂量用药可能是造成儿童期及 以后长期生长 受抑制的 一 个潜在原因。在此 阶段 的治 疗应 尽量 使用 小剂 量, 以 避 免对生长的不利影响。
雄激素过多可刺激下丘脑-垂体-性腺轴提早启动, 当 GC治疗控制雄激素不理想时, 骨 成熟加快提 前进入青 春 发动, 患者青春 发动的 年龄 明显 早于 健康 人。青 春期 是 非常重要的生长 阶段, 患者开 始青 春发 动到 出现 生长 激 素 ( GH ) 分泌高峰和出现生长速率高峰的 时间明显缩 短。 生长速率高峰的提早出现使患 儿损失了青春 发动到生 长 速率峰值出现之 间这 一段身 高迅 速增 长的 宝贵 时间, 减 少了最终 身高 [ 9]。 而在 此时 过 量使 用 GC 治疗, 可 抑 制 GH 的分泌及抑制青春期 GH 的峰值 水平, 亦使 身高突 增 减少。青春期启动时皮质激素使 用的剂量与最 终身高 呈 负相关 [ 10] 。 2. 1. 3 GC剂量的影响 未治疗或治疗控制不佳患者的 雄激素分泌过多导致提前成熟而影响身高。过量 GC治疗 不仅抑制 GH 分泌, 而且同样影 响生长。在儿童 和青少年 生理性的可的松分泌 4. 8 ~ 8. 7 mg / ( m2 # d), 大 剂量氢化 可的松对生长有明显的负面影响。 > 20 m g /( m2 # d )就 可 能影响到生长 [ 11] , 早有 报道 15 mg / ( m2 # d ) 氢化 可的 松 明显较 25 m g /( m2 # d) 治疗 的患 者 生长 更 好 [ 。 12] 对 予 ( 17. 6 ? 3. 6 ) m g / ( m2 # d) 氢化可的松治疗 8 a的患者 随 访发现, 达到终身高者均低于靶 身高, 未达到终 身高者 其 71. 5% 预 测身高 低于 靶身高。 根据 17-OHP 和 ACTH 水 平评估控制情况, 控制 好的患 者组 预测 身高 明显 好于 控 制差的患者组 [ 11] 。 GC和终 身高之间 的关 系在不 同的 年 龄段可能有不同 的影 响, 婴儿 期和 青春 期是 生长 速率 最 快的时候, 分组在 6~ 12 个月、8 ~ 10 岁和 12~ 14岁的 年 龄段中剂量和身高呈明显负相关, 高剂量 GC在婴儿期和 青春期对生长潜能有更大的损害 [ 4]。
2024CAH-X综合征研究进展(全文)

CAH-X综合征研究进展(全文)摘要CAH-X综合征是指先天性肾上腺皮质增生症(CAH)患者中,合并肌腱蛋白X(TNX)缺陷而出现埃勒斯-当洛综合征表型的特殊亚群,占CAH 患者的10%~15%。
TNX缺陷可导致一系列结缔组织症状,包括全身性关节活动过度、皮肤过度伸展、反复关节脱位、慢性疼痛、心脏缺陷等,严重影响患者生存质量。
CAH-X 综合征的遗传学病因是CYP21A2和TNXB基因的连续性缺陷,由于致病基因的复杂性,其分子诊断充满挑战。
现对CAH-X综合征研究进展进行综述,以提高临床医师对于这一新发现疾病的认识。
关键词先天性肾上腺皮质增生症;CAH-X综合征;埃勒斯-当洛综合征;TNXB 基因先天性肾上腺皮质增生症(congenital adrenocortical hyperplasia,CAH)是一类肾上腺类固醇合成酶缺乏的常染色体隐性遗传病,由CYP21A2基因缺陷所致的21-羟化酶缺乏症(21-hydroxylase deficiency,21-OHD)是CAH中占比约95%的主要类型,以肾上腺皮质功能不全和高雄激素血症为临床特征[1]。
埃勒斯-当洛综合征(Ehlers-Danlos syndrome,EDS)是一组异质性遗传性结缔组织病,以全身性关节活动过度、皮肤过度伸展和组织脆性为特点[2]。
一部分EDS是由于肌腱蛋白X(tenascin-X,TNX)缺陷所致,编码TNX的TNXB 基因与编码21-羟化酶的CYP21A2基因紧密连锁。
现已发现一种CYP21A2及TNXB基因的连续性缺陷,可同时引起21-OHD和EDS表型,称为CAH-X综合征。
自该综合征于2013年被命名以来,全球范围内累计报道的患者数量已近200例,但鲜有中文文献报道。
近年研究显示,CAH-X综合征在CAH患者中占比10%~15%[3-8]。
为提高临床医师对于这一新发现疾病的认识,现就CAH-X综合征研究进展进行综述。
先天性肾上腺皮质增生症

胆固醇碳链酶缺陷症
✓ StAR缺陷症是CAH中最严重和最少见的一种类型,极罕见,为 常染色体隐性遗传性疾病
✓ StAR缺陷症可致所有类型类固醇激素合成受阻,其特征性病 理改变为明显增生的肾上腺呈脂肪样外观,这是由于肾上腺 皮质细胞中胆固醇和胆固醇酯大量堆积所致
21-羟化酶缺陷症临床表现
轻型表现
非经典型CAH患者21-羟化酶活性保存20%-50%,发病时间较 晚(多见于年长儿童或青春期),因肾上腺过度增生代偿皮 质醇不足,同时出现轻度雄激素分泌过多。非经典型患者症 状较经典型轻微,性毛早现可作为重要的提示症状。
11β-羟化酶缺陷症
11β-羟化酶突变
✓ 人类CYP11B有两种同工酶,即CYP11B1(11β羟化酶) 和CYP11B2
典型女性出生时青春期 提前,多毛,轻型患者 无 月经紊乱
无
男性(46,XY)
无
男性(46,XY)
有
-
各类型CAH的实验室鉴别
CAH类型
CYP21缺 陷症
∆5-17P DHEA 17羟孕酮 正常/↑ 正常/↑ ↑↑↑
3β-HSD缺 陷症
正常/↑
正常/↑
正常/↑
CYP11B缺 陷症
↑↑↑
↑↑
CYP17缺 陷症
• 成年男性CAH患者容易发生睾丸肾上腺残余组织肿瘤和男 性不育症。
• ART主要位于肾上腺附近或睾丸下降的胚胎途径中,如腹 腔血管丛、阔韧带、精索、睾丸或卵巢及肾脏。
• 未经治疗者睾丸ART在高ACTH血症刺激下,容易恶变。
21-羟化酶缺陷症临床表现
中度单纯性男性化型表现
• 婴幼儿期:严重的女性男性化在出生后常被误认为是男婴。 男性患儿在出生时外生殖器一般无异常,少数阴茎稍大, 内生殖器发育正常。
先天性肾上腺皮质增生症

洮南 17 0 3 10 吉林 省 白城 地 区洮 南 市 医 院 ,吉林
【 摘 要 】 本 文对 3 例先天性肾上腺皮 质增生症 ( A )采用强的松及外生殖器畸形矫正手术。外 生殖 器异常按 Pae 的分型 ,其 1 CH r r d 中 I型 5例 , Ⅱ型 9例 ,Ⅲ型 l 7例 ,除 1 例无 闭经外 ,其余均为原发性 闭经 。所有病例乳房均未发育 ,呈男性乳头。治疗 前先作地塞米松 试 验,试验后 1 一K 7 s值均明显下降,根据试验前 1一 K 7 s值选择强 的松用量。全部患者第二性征得 到改善 ,月经来潮 。其 中已婚 1 例 ,6 1 例妊娠,母婴健康 。观察 中发现部分病毒停药后仍能维持月经周期 。本文就此对 C H病人是否需终生服药问题进行 了讨论 。 A 【 关键词 】 肾上腺皮质增生症 ;2 一羟化酶;尿 l 1 7酮 【 中图分类号 】R 5 . 4 8+ 3 【 文献标 识码】A 【 文章编号 】10 — 5 7 (0 0 3 0 0 — 1 0 7 8 1 2 1 )2 — 18 0
~
随访 中发现 ,有 5例 病人 月经规 律后 自行停 药 ,半年 至 1年后 ,2例 仍月经 规律 ,3例 又 出现 闭经 、皮肤 变粗 , 2 h尿 1一K 4 7 S在 8 . 5—14 1 mo之 间。2例 服药 3年 以 67 0. I l  ̄ 上 ,月经规律后不 再坚持 服药 ,每 当 出现月经 紊乱 ,甚 至 停经时再开始服药 。亦 可使月经周 期重新建 立 ,查尿 l一 7 K S亦在 正 常 范 围 。
[ ]俞霭峰 妇产科内分泌学.第 1 1 版.上海 :上海科技 出版社 19 ,2 9 98 5
~
261 .
[ ] 肖风云 男女生殖器 系畸 形 2
先天性肾上腺皮质增生(CAH)基因检测

• 结果1:MLPA检测CYP21A2和POR基因,未发现POR基因异常,发 现CYP21A基因IVS-13位置>G的杂合变பைடு நூலகம்。 • CYP21A2基因IVS-13位 置有两种野生型:C和 A,若该位置等位基因 同时发生>G的变异, 有可能导致21-羟化酶 缺乏症。
• 结果2:检测CYP21A2基因,发现微小变异:
• CAH (OMIM # 201910)按已知缺陷酶的种类,可分为6个型。以21羟 化酶缺陷症(21 hydroxylase deficiency,21-OHD)最常见,占90%95%。国际报道发病率1/10000-1/20000,杂合子发生率高达1:60。
• 21-OHD基因位于人类第6号染色体短臂6p21.3的HLA Ⅲ类基因区, 由无活性的假基因(cytochrome P450, family 21, subfamily A, polypeptide 1 pseudogene;CYP21A1P)和有活性的真基因 (cytochrome P450, family21, subfamily A, polypeptide 2;CYP21A2) 串联排列组成,真假基因之间有高度同源性。
• 当血浆中皮质醇(F)增高时,又对ACTH及CRH具有负反馈调节作 用,以维持动态平衡和钠钾含量的相对稳定。
• 21-OHD由CYP21A2基因突 变引起,它编码21-羟化 酶(P450c21),催化17 羟基孕酮(17-OHP)生 成11-脱氧皮质醇和催化 孕酮(P)为11-脱氧皮质 酮,两者分别为皮质醇 和醛固酮的前体。
目前仍处在动物实验阶段。
谢谢
先天性肾上腺皮质增生(CAH)
• 肾上腺皮质增生(CAH)是一组由于肾上腺皮质激素合成途径中 酶缺陷引起的疾病,属于常染色体隐性遗传病,女孩多见,男女 比例约为1:2。
先天性肾上腺皮质增生症(CAH)

性激素(雄激素) 促进雄性的性器官和第二性征的发育和维持 促进蛋白质合成,使身体肌肉发达
CAH的发病机制
CAH的发病机制
先天性肾上腺皮质增生症
(congenital adrenal hyperplasia, CAH)
胡欣 江苏省中西医结合医院 内分泌科
内容概要
CAH的定义及流行病学 CAH的类型 CAH的发病机制 CAH的临床表现 CAH的诊断 CAH的治疗
内容概要
CAH的定义及流行病学 CAH的类型 CAH的发病机制 CAH的临床表现 CAH的诊断 CAH的治疗
Speiser PW, et al. J Clin Endocrinol Metab. 2010, 95(9):4133-4160.
内容概要
CAH的定义及流行病学 CAH的类型 CAH的发病机制 CAH的临床表现 CAH的诊断 CAH的治疗
CAH的类型
Turcu AF, et al. J Steroid Biochem Mol Biol. 2015, 153:63-71.
CAH的诊断
影像学检查
X片:通过骨龄评估个体的生长发育水平和成熟程度 B超:对肾上腺肿瘤定位有诊断价值,但一般不能检测到增生。 CT/MRI:患者表现为双侧肾上腺影普遍增大,边缘略呈结节状, 但仍保持其大体形态,结构正常。 131I标记胆固醇肾上腺皮质扫描:示肾上腺细胞摄取胆固醇增加, 双侧肾上腺皮质增生, 131I-胆固醇浓集于双侧肾上腺皮质区,呈 双侧对称性增强。
CAH的定义
先天性肾上腺皮质增生症:因肾上腺皮质激素生物合成酶系 中某一或几种酶的先天性缺陷,而致肾上腺皮质激素合成不 足所致的一组疾病。
先天性肾上腺皮质增生症的诊断和治疗进展(精)

先天性肾上腺皮质增生症的诊断和治疗进展先天性肾上腺皮质增生症(congeAnital adrenal hyperplasia,CAH)是一组因肾上腺皮质激素合成途径中酶缺陷引起的疾病,属常染色体隐性遗传病,新生儿发病率为1∶20 000~1∶16 000[1]。
由于类固醇激素合成过程中某种酶的先天缺陷,导致肾上腺皮质束状带合成的皮质醇受阻,经负反馈作用促使下丘脑-垂体分泌的促肾上腺皮质激素释放激素(corticotropin releasing hormone,CRH)-促肾上腺皮质激素(adrenocorticotropin,ACTH)增加,导致肾上腺皮质增生,酶阻断的前质化合物如17-羟孕酮(17-hydroxyprogesterone,17-OHP)增多,某些前体经旁路代谢可转化为雄激素[2],临床上可出现肾上腺皮质功能减退症状,受累女性新生儿可有外生殖器男性化体征,男性则出现假性性早熟;并发的醛固酮缺失可引起以发育停滞、血容量减少及休克为特征的失盐症状。
常见的酶缺陷包括21-羟化酶、11β-羟化酶、3β-类固醇脱氢酶、17α-羟化酶缺陷等,其中21-羟化酶缺陷(21-hydroxylase deficiency,21-OHD)缺乏最常见,90%以上的CAH患儿为该酶缺陷所引起。
新生儿筛查统计,全世界21-OHD发生率为1∶13 000,北美为1∶15 000,欧洲国家为1∶14 000~1∶10 000,日本为1∶15 000[3],我国上海地区发生率为1∶20 000。
1 21-羟化酶缺乏的临床表现与分型根据21-羟化酶缺乏程度不同,可分为失盐型、单纯男性化型和非典型型三种类型。
1.1 失盐型(salt wasting phenotype) 21-羟化酶完全缺乏,占21-OHD患儿总数约75%,临床上除单纯男性化的一系列临床表现外,还可出现因醛固酮严重缺乏导致的失盐症状,同时伴有皮质醇合成障碍,而出现肾上腺皮质功能不全表现:常于新生儿期2~16天发病,表现为呕吐、腹泻、脱水、严重的代谢性酸中毒、低血钠、顽固性高钾血症和低血糖,如不及时治疗可因血容量不足、血压下降、休克、循环衰竭而死亡[4]。
石家庄市新生儿先天性肾上腺皮质增生症筛查及基因突变分析

doi :10.3969/j.issn.1002-7386.2023.17.032·调查研究·石家庄市新生儿先天性肾上腺皮质增生症筛查及基因突变分析封露露 马翠霞 贾立云 马倩倩 封纪珍项目来源:河北省医学科学研究课题计划(编号:20210689)作者单位:050000 河北省石家庄市妇幼保健院遗传科(封露露、马翠霞、贾立云、封纪珍);河北省石家庄市妇产医院(马倩倩)通讯作者:封纪珍 E⁃mail:279406985@ 【摘要】 目的 分析石家庄市新生儿先天性肾上腺皮质增生症(congenital adrenal hyperplasia ,CAH )的发病率及CYP21A2基因突变情况,为患儿的个体化治疗和遗传咨询提供依据。
方法 采用免疫荧光法对石家庄市2018年9月至2021年12月活产新生儿进行筛查,对可疑阳性患儿进行基因检测,分析基因与临床表型的联系。
结果 257751例新生儿中,筛查阳性203例,召回后根据临床表现和生化检测确诊10例患儿,发病率为1/25775。
确诊患儿中,男8例,女2例;失盐型6例,单纯男性化型4例,其中3例进行了基因测序分析,均为21⁃羟化酶缺乏症。
进行基因检测的3例患儿中,纯合突变1例,杂合突变2例,共5种突变类型,突变位点主要包括c.518T>A 、c.293⁃13C>G 、c.955C>T 、c.923dupT 和外显子1~7杂合缺失。
结论 石家庄市新生儿CAH 的患病率为1/25775;男性患病率高于女性,且以失盐型为主;CYP21A2基因突变分析发现5种基因突变类型,补充和完善了石家庄市新生儿CYP21A2基因数据库。
【关键词】 先天性肾上腺皮质增生症;21羟化酶缺乏症;基因测序;CYP21A2基因【中图分类号】 R 586 【文献标识码】 A 【文章编号】 1002-7386(2023)17-2696-04Screening of neonatal congenital adrenal hyperplasia and the gene mutation analysis in Shijiazhuang City FENG Lulu ,MA Cuixia ,JIA Liyun ,et al.Department of Genetics ,Shijiazhuang Maternity &Child Healthcare Hospital ,Hebei ,Shijiazhuang 050000,China【Abstract 】 Objective To analyze the incidence of congenital adrenal hyperplasia (CAH )in neonates of Shijiazhuang City ,and the rate of the CYP21A2gene mutations in them ,thus providing references for individualized treatment and genetic counseling.Methods The living newborns in Shijiazhuang from September 2018to December 2021were screened by immunofluorescence method.Children with suspected CAH were subjected to genetic testing.The correlation between genetic findings and clinical phenotypes of CAH was analyzed.Results Among the 257,751screened neonates ,203cases of suspected CAH were screened.Finally ,10(1/25775)cases were diagnosed as CAH according to clinical and biochemical tests after the recall ,involving 8male infants and 2female infants.There were 6cases of salt⁃wasting phenotype and 4cases of simple virilizing phenotype.Three cases were tested for gene sequencing ,all of whom were 21⁃hydroxylase deficiency.There were 5types of mutations in 1child with homozygous mutation and 2with heterozygous mutations ,including the c.518T >A ,c.293⁃13C>G ,c.955C>T ,c.923dupt and heterozygous deletion of exon 1-7.Conclusion The prevalence of neonatal CAH in Shijiazhuang City is 1/25775,which is higher in males than females.The salt⁃wasting phenotype is the predominant phenotype of CAH.Five gene mutations in the CYP21A2gene are detected in CAH neonates.Our findings have supplemented and improved the profile of the CYP21A2gene mutations in neonatal CAH in Shijiazhuang City.【Key words 】 congenital adrenal hyperplasia ;21⁃hydroxylase deficiency ;gene sequencing ;CYP21A2gene 先天性肾上腺皮质增生症(congenital adrenalhyperplasia,CAH)是一种常染色体隐性遗传病[1],按照酶缺陷的不同,分为6型,21⁃羟化酶缺乏症(21⁃hydroxylase deficiency,21⁃OHD)为最常见的一种类型,患儿常出现肾上腺功能不全等症状,新生儿期表现为厌食、呕吐、体重不增、喂养困难,严重者表现为电解质紊乱等症状;儿童期会出现性早熟、皮肤色素沉着、痤疮等临床表现,根据临床表现分为三种类型:失盐型、单纯男性化型和非典型型[2]。
病例分析:一例先天性肾上腺皮质增生症((CAH))
病例
一例先天性肾上腺皮质增生症(CAH)
患者男性,31岁,结婚3年未避孕,未育入我院生殖中心检查:无精症......
基本资料:患者男性,31岁,结婚3年未避孕,未育入生殖中心检查:无精症;
生殖中心检查结果:ACTH 472 ng/L(0-46),F(皮质醇):199nmol/L,遂转来我科(内分泌科);
实验室检查:
血压正常,电解质正常
FSH:0.45IU/L(1.27-19.26),LH:0.33IU/L(1.84-2.62);
E2:204.9pmol/L(73.4-172.4),T:16.49nmol/L(6.07-27.1);
P:86.14nmol/l(1.27-3.5),PRL:239.3mIU/L(55.9-278.4);
甲状腺功能正常;
ALD:214ng/L,肾素:8.6ug/L/h;
影像学检查:肾上腺CT:两侧肾上腺占位性病变,腺瘤可能;
治疗方案:予地塞米松 7.5mg bid
预后转归:ACTH 7.64 ng/L(0-46)下降,F:3nmol/L;
FSH:6.27IU/L(1.27-19.26),LH:2.92IU/L(1.84-2.62);
E2:25.3pmol/L(73.4-172.4),T:11.08nmol/L(6.07-27.1);
P:12.28nmol/l(1.27-3.5),PRL:218.03mIU/L(55.9-278.4);
肾上腺CT:两侧肾上腺占位性病变,较之前缩小;
基因检测:确认21羟化酶缺失。
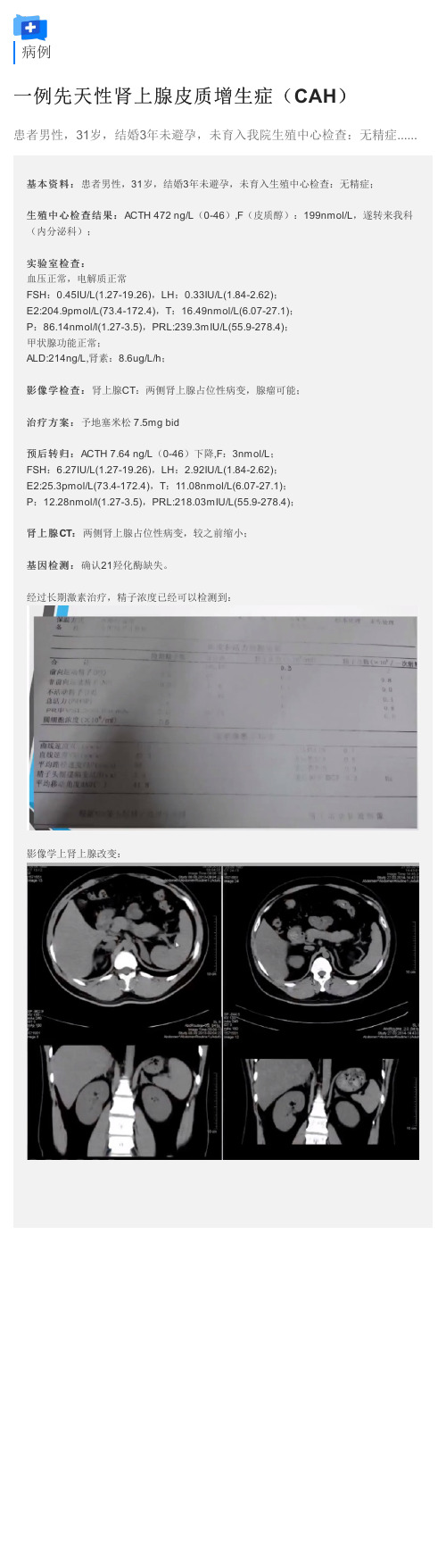
经过长期激素治疗,精子浓度已经可以检测到:
影像学上肾上腺改变:。
先天性肾上腺皮质增生症的诊断及治疗

可产生库欣样特征。 J2 的剂量应维持在能充分抑制雄性激素、 控制男性化症
[ !! ] 状、 保持正常生长的最小剂量 。儿童期治疗时剂量应依据
激素水平及时调整, 通常 !,-羟孕酮控制在部分抑制的水平即 可, 浓度为 !"" / !""" #$ % &’ ( ( / (" #)*+ % ’ ) 。过量的 J2 虽 可使 !,-羟孕酮处于正常水平, 但可能导致医源性库欣综合征 的发生。雄烯二酮及睾酮的水平应维持在与年龄、 性别相适 合的水平。其他能反映疗效的指标还包括骨龄的评定和生长 曲线的监测等。 无症状的非典型 .!-羟化酶缺乏症婴儿或儿童常不需治 疗。新生儿筛查中发现的非典型 234 婴儿应严密检测雄激 素过量的体征, 及时给予治疗。 .1 盐皮质激素( )<#MGO+*F*GH<F*<&I,P2 ) 5 P2 可协同 J2 的作用, 使患儿 3294 分泌进一步减少。失盐型婴儿除 J2 治 疗外, 还应给予 P2 治疗, 通常为氟氢可的松 ( Q+E&G*F*GH<I*#M ) "1 ! / "1 . )$ % &。同时应补充钠盐以纠正水、 电解质紊乱, 每 日补充的量为氯化钠 ! / . $ 或钠 !, / (0 ))*+。 (1 生 长 激 素 ( J4 )及 促 性 腺 激 素 释 放 激 素 类 似 物 ( J#74O) 5 J2 替代治疗一直作为 234 的基础治疗, 但其生 长抑制效应与长期的高雄激素共同作用限制了 234 患儿的 身高增长。尽管患儿在病初时身高常超过正常儿, 但大多数
第 !" 卷第 # 期 !$$% 年 & 月’ ! "##$ %$&’ ()*&+,-, ./$0 !" 1/0 # "#-0 !$$%
先天性肾上腺皮质增生症外科诊治的经验总结

先天性肾上腺皮质增生症外科诊治的经验总结【摘要】先天性肾上腺皮质增生症是一种罕见的遗传性疾病,容易被误诊或漏诊。
本文将就该疾病的临床表现、影像学特征、手术指征、手术方式和术后管理进行详细讨论。
通过总结多年来的诊治经验,为临床医生提供参考。
本文也展望未来在该疾病诊治方面的挑战和发展方向。
经验总结的目的是为了更好地指导临床实践,提高患者的治疗效果。
也要感谢所有在这一领域做出贡献的专家和患者的支持与信任。
希望本文的内容能够为相关领域的医生和患者提供帮助,促进先天性肾上腺皮质增生症的治疗水平和患者生活质量的提高。
【关键词】先天性肾上腺皮质增生症, 外科诊治, 经验总结, 临床表现, 影像学特征, 手术指征, 手术方式, 术后管理, 诊治经验, 展望未来, 致谢.1. 引言1.1 疾病背景先天性肾上腺皮质增生症(Congenital Adrenal Hyperplasia,CAH)是一种常见的遗传性内分泌疾病,主要由于11β-羟化酶缺乏或其他酶缺失导致肾上腺皮质激素合成代谢障碍所致。
CAH的发病率在不同族群和地区有所不同,但全球范围内的患病率大约为1/10000-1/20000。
其中以盐皮质醇缺乏型(传统型)最为常见。
临床上,CAH患者常出现生长迟缓、早熟、骨龄提前、男性外生殖器发育不全、女性外生殖器异常等,严重者可出现低血糖、低钠血症、高钾血症、原发性肾上腺皮质功能减退等危及生命的并发症。
CAH的早期诊断和治疗非常重要,可有效避免患者出现恶性高血压、肾盂积水、青春期障碍、性腺功能坏死等严重后果。
及早发现CAH,进行规范化治疗,对于患者的生长发育和生活质量具有重要意义。
在本文中,我们将探讨CAH的外科诊治经验,以期对相关疾病的临床工作有所帮助。
1.2 治疗意义先天性肾上腺皮质增生症是一种罕见的遗传疾病,患者常常出现肾上腺皮质增生导致的激素异常,严重影响其生活质量。
针对这种情况,外科手术治疗具有重要的意义。
先天性肾上腺皮质增生症诊治进展

多巴
苯丙酮酸
甲状腺素
黑色素
苯乳酸 苯乙酸
多巴胺
肾上腺素
合肥市妇幼保健所
新筛中心
四氢生物蝶呤合成代谢图
三磷酸鸟苷(GTP)
GTPch
p35 +
新蝶呤
三磷酸二氢新蝶呤
PTPS
(N)
6-丙酮酰四氢蝶呤
SR
苯丙氨酸
PAH
酪氨酸
TH TPH
色氨酸
四氢生物蝶呤(BH4)
DHPR
PCD
二氢生物蝶呤(BH2)
生物蝶呤(B)
传统Guthrie细菌抑制法(半定量),简便、重复性好 全定量法: 荧光法、HPLC、串联质谱技术(MS/MS)等
方法
对象 出生72小时(哺乳6-8次以上)的新生儿 判断血Phe>120 mol/L为阳性,召回 确诊血苯丙氨酸(Phe)及酪氨酸测定(全定量法),排除其他原因所致HPA
随着年龄增长, 智能发育明显落后 神经系统体征
肌张力增高 腱反射亢进 严重者可有脑性瘫痪 伴有点头样或婴儿痉挛样抽痉 约1/4患儿伴有癫痫发作,80%患儿脑电图异常
合肥市妇幼保健所 新筛中心
新生儿HPA筛查
意义
新生儿期患儿无临床表现,生化异常已存在,早期诊 治,预防智能障碍
完善 BH4鉴别诊断试验 2005年开展BH4缺乏症基因诊断及产前诊断
合肥市妇幼保健所 新筛中心
HPA PKU Phe PAH BH4
高苯丙氨酸血症 苯丙酮尿症 苯丙氨酸 苯丙氨酸羟化酶 四氢生物蝶呤缺乏症
合肥市妇幼保健所
新筛中心
先天性肾上腺增生症1例

论著·临床论坛CHINESE COMMUNITY DOCTORS 中国社区医师2019年第35卷第6期肾上腺生殖器官综合征又称先天性肾上腺皮质增生症(CAH),为常染色体隐性遗传病,是由于肾上腺皮质激素合成过程中所需酶的先天性缺陷所导致的一组疾病。
由于肾上腺类固醇的21-羟化酶(CYP21)的CYP21A2的编码基因(P450c21)突变是CAH 最常见的类型,约90%~95%[1,2]。
其中失盐型CYP21完全缺乏约占21-羟化酶缺乏(21-OH D)总数75%[3]。
我们收集了1例CAH(21-OH D 失盐型)患儿,采集其家系父母亲及两位姐姐外周血DNA,进行PCR扩增采用DNA聚合酶,直接测序扩增产物,检测CYP21A2点突变,现整理病例,报告如下。
资料与方法患儿,男,26d,因“少吃、少动伴气促1d,发现肾功能异常0.5d”入院。
患儿系G3P3,母孕期经过顺利,无口服药物及毒物接触史,孕39周足月顺产出生,出生时无窒息及抢救史,Apgar评分正常,羊水及脐带无异常,出生体重3500g。
生后4d 曾因新生儿高胆红素血症予光疗退黄处理,查电解质未见异常,黄疸消退后出院。
生后31d 临床初步诊断:CAH(21-OH D),予琥珀酸氢化可的松针50mg/m 2,静脉输注1次/d,治疗1周后复查,即38日龄上午8:00血ACTH 176.40pg/mL,同步皮质醇94.2nmol/L,复查K +6.36mmol/L,钠离子113.24mmol/L,血氯85.1mmol/L。
继续琥珀酸氢化可的松针q 12h,治疗1周再次复查,即45日龄上午8:00血ACTH 46.27pg/mL ,同步皮质醇18.3nmol/L,复查血K +5.86mmol/L ,血钠121.87mmol/L,血氯94.9mmol/L ,改为口服泼尼松2mg/kg,2次/d,连续2d,再次出现频繁呕吐、尿少,复查K +6.61mmol/L,钠离子122.24mmol/L,血氯92.1mmol/L,将泼尼松改为1次/d,加氢化可的松针50mg/m 2静脉输注,呕吐缓解,尿量增加,3d 后复查K +7.44mmol/L,钠离子108.42mmol/L,血氯81.2mmol/L。
单纯男性化先天性肾上腺皮质增生症合并正常生育1例并文献复习

单纯男性化先天性肾上腺皮质增生症合并正常生育1例并文献复习目的:提高对先天性肾上腺皮质增生症(CAH)的认知,探讨其诊治方法,了解最近研究进展。
方法:对1例单纯男性化CAH合并正常生育患者临床资料进行分析,并结合文献复习。
结果:患者行激素替代联合外阴整形术、阴道扩大术。
术后18个月自然受孕,术后28个月顺产1正常男婴。
结论:CAH的早期、规律、系统治疗对于改善患者生活质量和成年后生育能力尤为关键,治疗目前多以激素替代联合外生殖器整形术为主。
标签:先天性肾上腺皮质增生症;女性假两性畸形;诊断;治疗先天性肾上腺皮质增生症(Congenital adrenal hyperplasia,CAH)是一组肾上腺皮质类固醇激素合成障碍导致的疾病,属常染色体隐性遗传病,其中约90%为21-羟化酶缺乏症(21-hydroxylase deficiency,21-OHD)[1]。
此疾病与以新生儿发病、重度男性化及失盐(salt wasting)为特点的失盐型CAH不同,也不像非典型CAH发病隐匿,单纯男性化CAH表现为外生殖器男性化,但不伴失盐等电解质紊乱[2]。
一般认为CAH女性患者受孕率较低,特别是失盐型CAH最低,而且受孕后容易流产[3]。
国内关于CAH合并妊娠的报道较少,而单纯男性化CAH合并妊娠,产出正常婴儿的报道则更少[4]。
现报道1例如下。
1 资料与方法1.1 一般资料患者女性,23岁,因“外生殖器发育异常”入院,自诉青春期至今无月经来潮及周期性下腹疼痛。
体格检查:颈部有喉结,乳房未发育,呈男性乳房表现;阴毛浓密呈男性分布,可见一条约3 cm阴茎组织,可勃起,龟头未见尿道口,未见阴囊,可见一对翼状小阴唇样皱襞,与阴茎包皮融合,两翼中间可见阴道口较狭窄和较浅,约容1指(图1);阴道难以探入;肛查发现子宫较小,活动度欠佳,无压痛;其余未见明显异常。
实验室检查:尿17-酮皮质类固醇(17-KS)73.2 mg/24 h(6.0~14.0 mg/24 h),睾酮(TES)806.1 ng/dL(9.8~75.2 ng/dL),17α-羟孕酮(17-OHP)89 ng/mL(0.2~4.5ng/mL),电解质未见明显异常,外周血染色体核型报告46,XX。
先天性肾上腺皮质增生症的科普知识

目录 介绍先天性肾上腺皮质增生症 先天性肾上腺皮质增生症的治疗方法 如何预防先天性肾上腺皮质增生症 先天性肾上腺皮质增生症的生活护理 先天性肾上腺皮质增生症的影响 如何提高先天性肾上腺皮质增生症患者的 生活质量 先天性肾上腺皮质增生症的研究进展 结论
介绍先天性肾上腺皮质增生 症
结论
提高患者生活质量的关键在于知识普及 和心理支持。 研究进展将为患者提供更好的治疗方案 。
谢谢您的观赏 聆听
先天性肾上腺皮质增生症的 影响
先天性肾上腺皮质增生症的影响
对生活的影响:可能导致生活质量下降 、体格发育受限等。 对健康的影响:可能引发骨质疏松、代 谢异常等健康问题。
如何提高先天性肾上腺皮质 增生症患者的生活质量
如何提高先天性肾上腺皮质增生症患者的生活质量
知识普及:加强对患者和其家人的知识 普及,使其能够更好地理解和应对疾病 。 心理支持:提供心理咨询和支持,帮助 患者积极面对疾病。
如何预防先天性肾上腺皮质增生症
基因咨询:若家族有先天性肾上腺皮质 增生症病历,应进行基因咨询以了解携 带基因的风险。 prenatal screening:产前筛查可以帮 助早期发现胎儿是否存在先天性肾上腺 皮质增生症的风险。
先天性肾上腺皮质增生症的 生医生的建议 进行治疗。 生活方式管理:合理饮食、适当锻炼、 保持规律的生活作息等。
介绍先天性肾上腺皮质增生症
定义:先天性肾上腺皮质增生症是一种 遗传性疾病,导致肾上腺皮质功能异常 。 病因:由于基因突变导致肾上腺皮质细 胞生长异常。
介绍先天性肾上腺皮质增生症
症状:体格发育异常、从早年开始出现 的肾上腺皮质功能异常症状,如骨质疏 松等。 分类:根据发病时间及病因分为多种类 型。
先天性肾上腺皮质增生症的临床分析与分子遗传学研究

先天性肾上腺皮质增生症的临床分析与分子遗传学探究专业品质权威编制人:______________审核人:______________审批人:______________编制单位:____________编制时间:____________序言下载提示:该文档是本团队精心编制而成,期望大家下载或复制使用后,能够解决实际问题。
文档全文可编辑,以便您下载后可定制修改,请依据实际需要进行调整和使用,感谢!同时,本团队为大家提供各种类型的经典资料,如办公资料、职场资料、生活资料、进修资料、教室资料、阅读资料、知识资料、党建资料、教育资料、其他资料等等,想进修、参考、使用不同格式和写法的资料,敬请关注!Download tips: This document is carefully compiled by this editor. I hope that after you download it, it can help you solve practical problems. The document can be customized and modified after downloading, please adjust and use it according to actual needs, thank you!And, this store provides various types of classic materials for everyone, such as office materials, workplace materials, lifestyle materials, learning materials, classroom materials, reading materials, knowledge materials, party building materials, educational materials, other materials, etc. If you want to learn about different data formats and writing methods, please pay attention!先天性肾上腺皮质增生症的临床分析与分子遗传学探究先天性肾上腺皮质增生症(Congenital Adrenal Hyperplasia,CAH)是一种常见的遗传代谢紊乱性疾病,主要表现为肾上腺皮质增生和激素分泌异常。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
黔
南 民
族 医 专 学
报
第 2期
先 天 性 肾上 腺 皮 质 增 生症 研 究进 展
赵 茜
( 黔南民族医学高等专科学校附属医院内分泌科 ,贵州 都匀 580 ) 500
【 关键词】先天性 肾上腺皮质增生症; 1 羟化酶;糖皮质激素 ;盐皮质激素 ;诊断;治疗 2一
【 中图分类号】 R 2. 【 758 文献标识码】 A 【 文章编号1 O — 9321) — 1 — 3 l 8 48(000 05 0 O 2 0
由于过量的雄激素使 阴唇体皱襞发生不同程 度融合 ,女性患 者表现为阴蒂增大 , 似隐睾 、 类 尿道下 裂 而性 别难 辨 ,但 内生殖 器仍 为女性 ,有 输卵管 、卵巢 、子宫、阴道。阴茎发育而睾丸未 同步增大 , 强烈提示为周 围性早熟 ,常伴多毛、 肌 肉发 达 ,皮 肤 呈 不 同程 度 的色 素 沉着 ( 铜 古
一
素合成过程中酶的缺陷所引起的疾病,为常染色 体隐陛遗传病。该病 的发病率低 ,新 生儿 C H A 的发 病 率 约 1 1 0 / 600~12 0 ,典 型 C H /000 A 发病率约为 1/0万 ,非典型 C H的发病率约 01 A 为典型 的 1 ,女 性多 于男性 ,种族 间差 异大 , 0倍 犹太人多见 。由于此病 在 临床 上 较为 少见 , J
・
10・ 5
ห้องสมุดไป่ตู้ 第 2 第 2期 3卷
21 0 0年 O 6月
黔 南 民 族 医 专 学 报 Junl f i nnMei l oeefr ai aie ora o a a dc l g tnli Qn aC o N o ts
素合成减少 ,旁路途径活跃 ,雄性激素增加 ,引 起一系列水 、电解质及物质代谢紊乱和雄性激素 过多症候群。因糖皮质激素 (l ooi i)是 g ccrc d u to
维持 生命所必 须 ,它 的减 少 ,负反馈 不足 ,继发 促 肾上 腺 皮 质 激 素 释 放 激 素 ( R C H) 增 加 、促 肾上腺 皮质 激素 ( C H) 增 加 ,导致 肾上 腺皮 AT
2 2 一羟化 酶缺 乏症 I l 临床表现 和分 型
且有部分症状不典型 ,很多临床医师对该病 了解 不够 , 临床上容易漏诊。常见 的酶 缺陷包括 在
2 一羟 化 酶 、1 l 1 B一羟 化 酶 、3 1 3一类 固醇 脱 氢 酶 、1o一羟化 酶缺 陷等 ,其 中类 固醇 2 7L l一羟化 酶缺 乏最 常见 ,9 % 以上 的 C H 患者 为 该酶 缺 0 A
乏所引起 。所 以我们主要对 2 羟化酶缺乏 J 1一 所导致的 C H发病机制、临床表现、诊断及治 A
疗 作一综 述 。 1 C H的发病 机理 及病 因 A
C H是指 由于 编码 肾 上 腺 皮质 激 素合 成 过 A 程 中某 种 酶基 因突变 或 缺失 而 导致 糖/ 盐皮 质 激
质增生 ( 达 1 7 ) 。 可 O一 5g
正常肾上腺 以胆 固醇为原料合 成糖皮质激
素 、盐 皮 质 激 素 、性 激 素 ( 、雌 激 素 和 孕 激 雄
素)三类主要激素 ,都是胆 固醇的衍 生物。其 过程极 为 复 杂 ,每 一 步 骤 都 要 经 过 一 系 列 酶 催 化 ,有 些酶是合 成这 一类 激素 或其 中两类 激素 的
体) ,孕 酮转 化 为 去 氧皮 质 酮 (醛 固 酮 的前 体) 1 羟化酶活性的减少或缺失将阻碍皮质 。2 一 醇的合成 ,皮质醇合成不足,血中浓度降低 ,由 于负反馈作用刺激垂体 ,分泌促肾上腺皮质激素 (CH A T )增多 ,刺激性 肾上腺皮质增生并分泌 过多皮质醇前身物 ( 1 脱 氧皮质 醇和肾上腺 1一 雄 酮等 ) ,某些 前体 可转 化 为 雄 激素 ,而发 生 一 系列临床症状 。糖皮质激素减少时 ,对黑色素 细胞刺激素 ( S M H)和 ( C H)分泌的反馈抑 AT 制减弱,A T C H增多的同时伴 随 M H分 泌的增 S 多 ,患者会有不同程度的皮肤黏膜色素沉着。
过程 中所共 同需 要 的 。常 见 的 酶 缺 陷 包 括 2 l一
羟化酶、lp一羟化 酶、3 1 p一类 固醇脱 氢 酶、
形 ,男性出生时可无症状 , 出生后 6 在 个月左右
作者简介 :赵 茜 (9 6一) 17 ,女 ,贵州罗句县人 ,内科 学硕士 ,住 院 医师 ,主要从 事内分泌与代 谢临床及研 究工作 。
先天性 肾上腺 皮质增 生症 (Cneil de ognt r- aa nl yepai A a hprls C H) 是 一 组 由于 肾上 腺 皮 质 激 a
1a一 7 羟化酶缺陷等 , 中类 固醇 2 一 其 1 羟化酶缺 乏最常见 ,类 固醇 2 一羟化 酶 由 C Y 1 2编 l P 2A 码 ,也称为 C P 1 P 5 c1 Y 2 或 4 02 ,是位于肾上腺皮 质内质网的一种细胞色素 P 5 4 0酶 ,它能催化 1 7 羟孕酮转 化 1 1一脱 氧皮 质 醇 (皮 质 醇 的前
色) 。女 婴/ 则 因 阴 蒂 增 大 ,类 似 隐 睾 、尿 道 孩 下裂而 性别难 辨 ,至青 春期则 表现 为原发 性闭经 伴 外生 殖器 发 育 异 常 的男 性化 表 现 。根 据 2 1一 羟化酶 缺乏程 度不 同 ,可 分为失 盐型 、单纯 男性 化 型和非 典型 型三种 类型 【 。 3 J 2 1 单纯男 性化 型 (ipevrin p ) 为 . s l ili t e m iz g y 2 一羟化 酶不 完 全 缺乏 所 致 ,醛 固 酮合 成 正 常 。 1 该 型发病 年龄 早 ,女 性在 出生 时即发病 ,呈 现程 度 不 同的男性 化体征 ,临床表现 为女性 假两性 畸