Enhanced Design Review
第三期 美欧GMP设备规范(印刷版)

美欧GMP专题现场培训全面地科学地理解与实施美欧GMP规范(浙江海正专场)★美欧GMP设备规范(本资料仅供企业内部培训使用)Shanghai NovoScience上海加中生物技术有限公司1美欧GMP 设备规范上海加中生物技术有限公司上海国际药政高级培训中心执行经理程毓渡博士2程毓渡博士(Dr. Frank Cheng)在北美制药领域拥有十几年学习、研究与工作经历,先后在美国JOHNS HOPKINS大学、加拿大国家科学院生物技术研究所、加拿大新科药业、加拿大生化制药、日本第一制药等著名大学、研究所、制药企业工作、合作与交流,在原料药、仿制药、创新药开发、生产与市场准入规范方面具有广泛的专业知识与国际人脉资源。
作为一个在中国工作的加籍华人专家,程毓渡博士目前担任上海加中生物技术有限公司执行总裁,专注于为国内外药企客户提供美欧GMP(ICH Q7A/cGMP)符合性培训与审计服务、为美欧公司和中国药企承担独立第三方GMP审计工作,亦提供面向美欧市场药品合同制造方面的项目管理。
程毓渡博士上海中生物技术有限公司执行经理上海国际药政高级培训中心执行经理3美欧GMP 规范专题现场培训全面地科学地理解与实施美欧GMP 规范•美欧GMP设施规范•美欧GMP物料规范•美欧GMP设备规范•美欧GMP生产规范•美欧GMP质保规范•美欧GMP质控规范•美欧GMP文件规范•美欧GMP审计规范4美欧GMP 设备规范培训序言根据美国FDA 对04/05年的原料药和制剂生产厂家GMP 审计结果统计,涉及违反设施与设备规范方面的缺陷占总缺陷的17%,这些缺陷来自设施设备与工艺设备的设计、调试、确认、验证、清洁、维保、变更等。
美欧GMP 设备规范还包括工艺设备的计算机系统,而这部分涉及的缺陷包括控制系统未经确认或验证、控制程序未经验证、密码管理系统与示踪系统未能建立等。
设施设备和工艺设备对于原料药和制剂的质量控制具有直接作用,本课程将对设施中的空调设备、水设备、空气压缩设备和生产中的主要工艺设备以及计算机系统的GMP 规范和实施进行科学解读,促进贵药厂遵循美欧GMP 在设施和工艺设备方面的管理规范,为通过美欧GMP 认证水平奠定坚实的基础!5美欧GMP 设备规范培训提纲美欧GMP设备规范解读ICHQ7A 第5节(工艺设备)与第12节(验证)中关于设备确认和验证方面的规范条例总共有28条(不包括计算机系统的10条规范),为药厂在设备管理方面实施GMP 提供了原则和依据,科学地和恰当地解读这些规范条例以建立这些规则与管理实施之间的有机联系是药厂的责任……美欧GMP设备规范实施美欧GMP 设备规范的实施涉及设备的设计、构造、调试、确认、操作、清洁等,通过对HVAC 系统、工艺水系统、工艺气体系统、反应罐、离心过滤机、干燥机、制粉机等设施系统和设备的确认以掌握规范实施方法……6美欧GMP 设备规范解读ICHQ7A第5节(工艺设备)与第12节(验证)中关于设备方面的规范条例总共有28条,为药厂在设备管理方面实施GMP提供了依据和规则,科学地和恰当地解读这些规范条例以建立这些规则与管理实施之间的有机联系是药厂的责任……7美欧GMP设备规范解读ICH Q7A 5 工艺设备–ICH Q7A 5.1 设计与构造–ICH Q7A 5.2 设备维护与清洁–ICH Q7A 5.3 校验ICH Q7A 12 验证–ICH Q7A 12.3 确认–ICH Q7A 12.7清洁验证ICH Q7A官方网站:ICH FDA EMEA85.1 设计与构造:5.10Equipment used in the manufacture ofintermediates and APIs should be of appropriate design and adequate size, and suitably located for its intended use, cleaning, sanitization (where appropriate), and maintenance.用于中间体和原料药生产的设备应做到设计合理、体积适中、放置恰当,以符合其使用、清洁、卫生(如有必要)和维护的要求。
ISPE汉译本

项目调试和确认指南1.简介2.本指南的目的、关键概念及定义3.影响评估4.GEP (工程质量管理规范)MISSIONING(项目调试)6.QULIFICATION PRACTICES(确认操作管理规范)7.ENHANCED DESIGN REVIEW(进一步设计审核)8.安装确认9.运行确认10.性能确认11.相关程序1、简介和以前出版的指南一样,Commissioning(项目调试)和Qualification(项目确认)由国际制药工程协会的医药咨询理事会发布,在Commissioning(项目调试)和Qualification(项目确认)指南中所有的方案、指南及技术指导由指导委员会提供。
本指南应用范围本规程适用于药厂建造符合FDA或其他卫生部门规范的生产设施的设计、建造、调试移交及确认,该指南既不是一种标准也不是GMP的规定,它不能取代法定标准、准则、规程。
该指南只是有助于建造符合cGMP要求的公共设施、设备等。
该指南需要谨慎应用。
本指南重点在于工程方法和工程施使,以及及时地提供满足预计要求的经济实用的生产设施,具体来说,使设施、设备和公用设施的设计、建造、调试及确认符合FDA或其他卫生部门要求。
本指南不是为了解决工艺或产品验证的问题,本指南得到FDA和其他权威部门的认定。
Commissioning(项目调试)和Qualification(项目确认)是工序验证的基础,进一步讲,这些操作在提供有效、安全、高效率的设施、设备和公用设施运作起着至关重要作用。
因此,在Commissioning(项目调试)和Qualification(项目确认)操作中实施综合、全面的方案是非常重要的,一套较好的构思和实施Commissioning(项目调试)和Qualification(项目确认)防范能达到及时地、经济实用的验证效果。
本指南只提出了一些对Commissioning(项目调试)和Qualification(项目确认)程序有影响的非工程方面的软件问题,例如,系统支持,文档问题,决定程序等,这些软件对于项目工程师来说比较重要。
制药系统英文简写

GMP 常见英文缩写AQAI(Automated Quality Assurance Inspection Equipment):在线自动质量保证检查设备API(Active Pharmaceutical Ingredient):活性药物物质,即原料药ANDA (Abbreviated New Drug Application):简化新药申请ADR(Adverse Drug Reaction):不良反应BSE(Bovine Spongiform Encephalopathy):疯牛病BPCS(Business Planning and Control System):业务计划及控制系统BIA(Business impact assessment):商业影响评估cGMP(current Good Manufacturing Practice):现行药品生产质量管理规范CCCD(China Certification Committee for Drugs):中国药品认证委员会CIP(Cleaning In Place):在线清洁SIP(Steam In Place):在线灭菌CV(Concurrent Validation):同步验证CDER( Center for Drug Evaluation and Research):药品研究与评价中心COA(Certificate Of Analysis):分析报告单CFR(Code of Federal Regulation):美国联邦法规CDC(Centers for Disease Control and Prevention):疾病预防控制中心COS/ CEP( Certificate of Suitability for European Pharmacopeia):欧洲药典适用性证书CCD(Certification Committee for Drugs):药品认证管理中心CPMP(Committee for Proprietary Medicinal Products):欧洲专利药品委员会CTD(Common Technical Document): 通用技术文件CDC( Centers for Disease Control and Prevention):疾病预防控制中心GMP(Good Manufacturing Practice): 药品生产质量管理规范TQC(Total Quality Control),TQM(Total Quality Management):全面质量管理EU(European Union):欧洲联盟EFPIA(European Federation of Pharmaceutical Industries Associations):欧洲制药工业协会联合会MHW(Ministry of Health and Welfare,Japan):日本厚生省JPMA(Japan Pharmaceutical Manufacturers Association):日本制药工业协会FDA(US Food and Drug Administration):美国食品与药品管理局PRMA(Pharmaceutical Research and Manufacturers of America):美国药物研究和生产联合会WHO(World Health Organization):世界卫生组织IFPMA(International Federation of Pharmaceutical Manufacturers Associations): 国际制药工业协会联合会PDCA(Plan,Do,Check,Action):计划,执行,检查,处理QA(Quality Assurance):质量保证QC(Quality Control):质量控制QS(Quality System):质量体系QM(Quality Management):质量管理SOP(Standard Operating Procedure):标准操作规程SMP(Standard Management Procedure):标准管理程序SOR(Standard Operating Record):标准操作记录GEP(Good Engineering Practice):工程设计规范HVAC(Heating Ventilation and Air Conditioning):空调净化系统HEPA(High Efficiency Particulate air Filter):高效空气过滤器DQ(Design Qualification):设计确认IQ(Installation Qualification):安装确认OQ(Operational Qualification):运行确认PQ(Performance Qualification):性能确认OOS(Out-Of-Specification):检验不合格;超标PFDS(Process Flow Diagrams):工艺流程图MRA(cMutual Reognition Agreements):现场检查多边认同协议DMF( Drug Master File): EDMF(European Drug Master File)欧盟药物主文件EDQM(European Directorate for Quality Medicines): 欧洲药品质量管理局ORA(Office of Regulatory Affairs):药政事务办公室GGPs( Good Guidance Practices):优良指南规范MOA(Method Of Analysis):分析方法VMP(Validation Master Plan):验证主计划VP(Validation Protocol):验证方案MSDS(Material Safety Data Sheet):物料安全技术说明书NDA (New Drug Application):新药申请OTC(Over-the-counter):非处方INN(International Nonproprietary Name):国际非专有名称USP(the united state pharmacopeia):美国药典NF(National Formulary):美国国家药品集GAP(Good Agricultural Practice):中药材种植管理规范GCP(Good Clinical Practice):药物临床试验质量管理规范GLP(Good Laboratory Practice):药物实验室管理规范GSP(Good Supply Practice):药品经营质量管理规范GUP(Good Use Practice):药品使用质量管理规范PMF(Plant Master File); SMF(Site Master File):工厂主文件EDL(List of Essential Drugs ):基本药物目录PI(Package Insert):说明书PCT( Patent Cooperation Treaty):专利合作条约PPAC(Patent Protection Association of China):中国专利保护协会PIC( Person In Charge):负责人PDS(Pharmaceutical Development Services):整体新药研发机构SPC(Summary of Product Characteristics):产品特性摘要SAL(Sterility Assurance Level)无菌保证水平原料药 API,Active Pharmaceutical Ingredient,美国焊接学会 AWS,American Welding Society生物耗氧量 BOD,Biochemical Oxygen Demand菌落数 CFU,Colony Forming Units关键运行参数 COD,Critical Operating Data化学耗氧量 COD设计确认 DQ,Design Qualification增强设计审核 EDR,Enhanced Design Review工厂验收测试 FAT,Factory Acceptance Test良好自动化质量规范 GAMP,Good Automated Manufacturing Practice 良好工程规范 GEP ,Good Engineering Practice生产质量规范 GMP,Good Manufacturing Practice输入输出 IO,Input/Output安装确认 IQ,Installation Qualification运行确认 OQ ,Operational Qualification氧化还原电势 ORP,Oxidation-Reduction Potential管道和仪表工艺流程图 P&ID,Piping and Instrumentation Diagrams可编程序逻辑控制器 PLC ,Programmable Logic Controller性能确认 PQ ,Performance Qualification在系统或系统主要元件交付到现场之前的检查和测试 PDI,Pre-Delivery Inspection 工艺流程图 PFD,Process Flow Diagrams聚偏氟乙稀 PVDF,Polyvinylidene Fluoride聚四氟乙烯 PTFE,Polytetrafluoroethylene聚丙烯 PP,Polypropylene反渗透 RO,Reverse Osmosis电去离子 EDI,ElectroDeIonization标准操作程序 SOP,Standard Operating Procedure现场验收测试 SAT ,Site Acceptance Test用户技术要求说明 URS,User Requirement Specification 总有机碳 TOC,Total Organic Carbon注射用水 WFI ,Water For Injection。
工程量清单计价模式下招投标阶段的工程造价控制英文翻译

英文翻译Abstract :In this paper, according to relevant provisions, approaches to the specific content and spiritual essence of learning and understanding, and combined with the current bidding characteristics, how to do a good job under the model of BOQ valuation bidding phase of the project cost control are discussed.Key words: project quantity list valuation pattern, bidding phase, construction cost controlUnder the model of BOQ valuation bidding in bidding direction, it has unified project to measure, reasonable low wins the bid, bid the unit according to the characteristics of enterprise quota quote independently. Engineering quantity list valuation model on engineering cost control target and specific work has profound influence, the new pricing mode to the bidding phase of project cost control work should have a good grasp of the preparation of BOQ, review, preparation of bid enquiry work, reasonable low wins the bid, determine the type of contract, the terms of the contract agreed to pay attention to reasonable allocation of risk and other key.Carry out a project to measure detailed list valuation method is the embodiment of "control quantity, price, quote independently by the enterprise" project cost reform is an important measure, is also China's accession to the WTO and international standards requirement. The Ministry of construction in 1998 put forward " the conditions of the areas and projects, in accordance with the administrative department in charge of Construction issued a unified engineering calculation rules and project division rules, to bidding of engineering quantity list, reasonable low wins the bid the pilot", and in the Guangdong Shunde , Tianjin and other places of the pilot.Promulgated 2001 carry out" construction project contract and contract valuation management approach" eighth presents a bidding project can use a project to measure detailed list method for the preparation of tendering and bidding price. In 2002 summing up relevant provinces and municipalities to carry out engineering quantity list valuation based on experimental work, construction ministry organized the formulation of" construction of the public bidding of engineering quantity list valuation Interim Measures". Therefore the quota of the separation of quantity and price, market price and norm of construction company interior to determine the bidding price and bid price of the bidding of engineering quantity list tender but英文礦封represent the general trend, but also the future direction of the bidding.In this paper, according to relevant provisions, approaches to the specific content and spiritual essence of learning and understanding, and combined with the current bidding characteristics, how to do a good job under the model of BOQ valuation bidding phase of the project cost control are discussed.a00d上bb上n3?ds丁s?才9丁上2才上?2Under the model of BOQ valuation bidding: engineering quantity list by the owner or entrust a bidding agency organization unit, as part of the bidding documents, the bidder according to their enterprise quota autonomous bidding, bid according to the budget quota, market prices, administration published rates determined in the bid evaluation, only for reference. Compared with the traditional quota + uniform base price fee pricing model of bidding, the bidding work has the following characteristics.Content of tender documents including bill of Engineering quantity. Project volume detailed list is in accordance with the national ( GB50500-2003 ) or local enactment of calculation rules ( unified project code, unified project name, uniform units of measurement, unified project to measure computational regulation ) according to the design drawings, design, drawing review records, consider the person of invite public bidding requirements, the engineering project characteristic calculation and statistics, arrangement, in order to get a list of. It is as tender offer reference file important component provided to bidder, aimed at unifying all bidders bidding in engineering quantity, and price competition to the bidder.Bid price and bidding to objective law to weave. Under the new mode of valuation, bid the unit to the project volume detailed list of engineering quantity examination, according to the enterprise's own construction methods, construction measures and artificial, material, machinery consumption level and material procurement channels and management to determine the level of prices, the indirect rate, profit rate, combined with the market factors in autonomous quotation. It is conducive to play independent valuation of enterprise competence manifests its advantage and experience, to realize the government to fix a price to the market change.Bid according to the budget quota, the project cost administration published rates, market information and price determination. Reflect the social average level.Bid in bid only for reference. Bidding of engineering quantity list of bid price only as market reference price or the owners of the block bid price in bid, only for reference. As a result of project volume detailed list is open to the public, the partiesinvolved from the base price of the bondage, unit of invite public bidding bid for no longer confidential tax one's ingenuity, saving a lot of manpower, material resources. To avoid in project invite public bidding resort to deceit, the black-box operation irregularities.The evaluation methods of comprehensive scoring method or by the lowest evaluated bid method. Review the tender offer not only focus on the price, pay more attention to the price, the same price bidding of engineering quantity, form a clear comparable, facilitate the bid response, quoted price is reasonable evaluation, the advantage is reflected in the price, make the price with quality and construction period of comprehensive evaluation of reasonable low price are effectively guaranteed, at the same time the degree to eliminate the undesirable phenomenon such as string, carry the mark, to avoid malicious distortions of Engineering cost. It will effectively change the person of invite public bidding blindly demand a low price behavior, help to regulate the person of invite public bidding in the bidding behavior, thus truly reflect open, fair, just principle.Bidding phase of project cost control goal is twofold: to determine the reasonable contract price and strict engineering contract price to realize safely. New valuation pattern to these two points will produce profound effect.The contract price formation that makes project cost is more close to the practical value of. First, determine the contract price are two important factors -- the bidding and bid price to objective law to weave, the consumption, price, rate is the market fluctuations in the value of the contract price, therefore can better reflect the nature and characteristics of engineering cost, closer to the market value. Secondly, easy to undertake to project cost dynamic control. In the original pricing mode, no matter the contract with fixed price or adjustable price, no matter how much quantity change, no matter how long construction period, both parties as long as agreed by the national norm cost management department, national adjustment and material price and issued by the price to adjust coefficient, then applied to contract, project settlement. Under the new mode of valuation, engineering quantity by tender offer, quotation competitive quotation based on engineering quantity list value, tender people in order to avoid due to the different understanding of the problems caused by the drawings, generally do not require quotation for engineering quantity puts forward opinion or judgment. But the project will change the situation of construction site, construction organization change, causing the construction cost, profit, management fee rate changes, thus bring the project unit price changes. New valuation mode can realize the real meaning of construction dynamic cost control.In the terms of the contract agreement, both risk and responsibility consciousness strengthens. In the original pricing mode, because the valuation method of single, contract both sides about risks and responsibility consciousness is not strong. New valuation mode, bidding on both sides of the contract price of shared responsibility. The tenderer to provide engineering, engineering quantity change or calculation errors responsibility, bid the unit only to its own quoted price cost, charge. Engineering settlement, according to the actual completion of engineering quantity, according to the agreed measures adjustment. Both sides of the project the understanding to manifest in different ways in the contract price, the tenderer to BOQ, tender side reflected in pricing. In addition, engineering project cost through the detailed list quoted price is clear down, in the future construction process, construction enterprises to obtain the maximum benefit, make use of construction alteration and claim means to pursue additional costs. Therefore, two sides of the contract management consciousness will be greatly enhanced, the terms of the contract agreement will be more careful.Bidding phase of project cost control in three aspects: access to competitive bidding, effective evaluation of the most reasonable quotation, contract to control cost variation. New valuation mode with three aspects of cost control works new content and new emphasis. First engineering quantity list quotation of unified foundation to become competitive bidding guarantee, reasonable low price bid evaluation method without mark bottom so that the selection of more reasonably priced, the terms of the contract and pay more attention to risk the reasonable apportion, pay more attention to the cost dynamic control, pay more attention to the price adjustment and engineering change, claim etc. the promise.The bidding work stage of engineering cost control keyThe author believes that: compared with the traditional quota + uniform base price fee pricing model of bidding, the engineering quantity list bidding phase of project cost control work should hold good the following:1.focus on engineering quantity list preparationProject volume detailed list is the important part of the bidding documents, bidding units to bid and the foundation of fair competition. Therefore the engineering quantity list must be scientific and reasonable, clear content, objective and fair. The preparation of project quantity list should pay attention to the following points:(1)compilation basis. compilation basis must be a comprehensive understanding of project information, the owners to understand intentions, technical specification, site investigation at the scene, understand the actual conditions of construction ( construction of the site, housing, transportation and other environmental conditions,geological, hydrological, meteorological condition, for ) calculating the engineering quantities to lay a good foundation for the future, to minimize engineering change.(2)The project division. requirement project project division between clear boundaries, project content, process and quality standard of clear, convenient measurement, also easy to quote; project division should be fine, avoiding the uneven quotation.(3)concise and comprehensive list description. Including work content added, specific construction technology requirements, the main material specifications and quality requirements, the construction site conditions, natural conditions and so on. Especially the construction site conditions, natural conditions, should accurate representations for bidders, and their understanding of the situation control. Supporting form should design reasonable, practical and intuitive, have operation sex. That is to make the bidding operation without cumbersome, but also conducive to the evaluation of convenient and fast operation.2 .pay attention to mark the establishment of auditAlthough the bid price in bid evaluation is only a reference, but is not essential. Bid is still under evaluation bidder cast unit price and total price rationality important reference, is the production of building products consumed by the socially necessary labor valuation, accounting cost basis, contract management is identified in the contract, price adjustment, claims and additional works at the rates and prices of reference. Therefore, the correct calculation of bidding for project cost control has important significance.The pre-tender price levels should be determined in detail many artificial, material, machinery market situation investigation, and master the more the area with similar conditions of similar project cost information, after careful research, analysis, comparison and calculation, as far as possible the project control in similar engineering social average level.3 .attaches the en quiry work, reasonable low price bidAfter the opening of the tender, bid before the construction project bidding management department under the supervision of the bidders, enquiry. The enquiry, can rule out the possibility of the immaterial, deceptive, excessively extreme and was significantly less than the cost of unreasonable price, avoid error, omission, for bidding on both sides of the issues related to explain to each other, to clarify, to determine a reasonable price of the contract to lay a good foundation to provide a prerequisite.When the evaluation, first to each bid according to file of invite publicbidding "performance, time limit for a project, offer" and other specific requirements specified in the bid invitation documents, and the bidding price adjustment method, to all bidders offer necessary price adjustment, calculates the tender bid price, then according to the bid, the bid evaluation, analysis, further selection of classics evaluation, price reasonable candidates for winning the bid.4 .carefully to determine type of contractIn engineering quantity list in bidding of engineering quantity, due to uniform, the type of contract price bidding strategy become the choice of the more important factors. Because the quantities is the expected value, quantities and the actual required quantity difference greatly bring the contract the two sides of the main interests of risk. So the appropriate selection of contract types in favor of a competitive price, can also be reasonable partake in the course of the contract risk.For large and medium-sized construction is usually adopted to unit price contract, total prices contract for contract form. In the contract that the main project is the unit price contract, other parts of the project uses the total price contract. For small scale, technology is not complicated, short period of the project, can adopt lump-sum contract.5 .signed a contract noticeOn the contract document sections in the bidding process to form supplement, modify, answering questions, enquiry, written summary of the various agreements shall be used as part of the contract documents. Special attention to as payment and settlement based on the engineering quantity list price, should be based on the bid evaluation stage makes a revised rearranged, validation. And should show, which will be completed the engineering calculation for payment, which shall be in the payment.6.on contract termsIn the preparation of the terms of the contract, should pay attention to the risks and responsibilities of the contract, the project management into the terms of the contract, as far as possible the risk quantification, responsibility is clear, fair to safeguard the interests of both sides. The main attention to the following provisions:(1)Procedural provisions. The purpose of regulating the settlement of project cost based on the form, the prevention of unnecessary disputes. Procedural provision throughout the contract behavior always. Information exchange program, including the measurement procedures, engineering change procedure, claims processing procedures, payment procedures, dispute settlement procedures. Written note specific steps, the agreed time period.(2)The engineering measurement terms. Focus on the calculation method of contract: ( general according to the net metering ) strictly define measurement content, enhance concealment engineering measurement agreement. Method of measurement by the general engineering parts and engineering characteristics determining, in order to approved project amount, convenient in the calculation of wages payment principle.(3)The price terms, with special attention to the price adjustment clause. A: the contract does not indicate the price, with or without a separate price valuation method. Or the pricing formula, or the principle of valuation. B: project quantity price adjustment. Contract project quantity exceeding the prescribed range of comprehensive unit price adjustment formula. C : the material price greatly changes caused by factors such as price adjustment, agreed to share principle or price adjustment formula. In short, the adjustment of contract price clause must have clear and detailed content, minimize ready to accept either course controversial terms appear, less left alive, for subsequent engineering change settlement development direction, strong operability terms.(4)The duties of both parties in terms of. In order to further define the responsibility, risk quantification, respond to both the proper description of duties. For those who have been foreseeable future ( such as the now often blackout ) and may influence the cost of the events and circumstances make clear each square responsibility, minimize claims and disputes.(5)Claim clause: clear claim procedures, payment of claims, disputes settlement etc..(6)The general terms and specific terms. Any construction contract should have the general terms and specific terms, terms are in accordance with the laws, administrative regulations and the construction of the need to develop, common to all construction projects construction terms. Special terms are the employer and the contractor in accordance with the laws, administrative regulations, combined with practical engineering, the reach an agreement on the terms, to the general provisions of the specific changes, additions or modifications. In fact there are many construction contract special terms are often written "in general terms first paragraph a few", so completely lost the special terms of meaning, also cannot be reflected to the engineering characteristics, the project contract management and project settlement brings very big hidden trouble, in order to eradicate the hidden trouble for contract management, must quantiation, refinement, deepening the special conditions of contract.In addition the bidding work and prepare the bidding process of the organization work, such as: to determine the means of invite public bidding, tender schedule, the investigation at the scene, held the bidding documents will answer and英文翻译so on, do the work also helps people get reasonable quoted price for bidding, bidding phase of project cost control is also very important. In short, in the bidding process, take seriously every job, to ensure the smooth progress of the work, to control engineering cost at a reasonable level.Bidding of engineering quantity list in our country at present in the pilot phase, the comprehensive promotion but also by some restricted, such as: enterprises have not enough time is accumulated to form enterprise own quotes norm, business level is not high, the text of the contract not complete, construction market information has not adapted the cost management needs, effects of on the bid price is lower than the cost evaluation. So to do the bidding phase of project cost control work, we must continue to learn relevant knowledge, change idea, get used to project cost system and construction of system reform, along with the project construction cost management reform of constantly to explore, to try.Reference.:[1]" The bidding project cost control in the phase of" city construction theory research in 2022 fourth issue[2] Guo Shurong" project cost management" Science Press2022[3] He Zengqin" engineering project bidding strategy" Tianjin University press2004。
fast fashion

• •
• • •
The Model
• • • • • • • • • • • • N 市场容量(消费者总人数),分布函数F(.),均值为μ υ 消费者对产品的估计(所有消费者估价一致) q 零售商采购数量 ,变量 C 采购成本,CQ快速响应附加成本 P 零售价格 , 变量 S 清仓价格,s<c δ ∈[0,1] the level of strategic behavior or patience of the customer population Ф 商品满足率 probability of obtaining a unit υ -p 消费者立即购买的收入 δФ (υ -s) 消费者延迟购买的收入 (x)+ =max(x,0) ,L(q)=E(N-q)+ 期望缺货数量,I(q)= E(q-N)+ 期望剩余 T,Q,D,F 分别是四种系统的缩写
•
• • • • •
Literature Review
• 快速响应技术方面的研究:Fisher and Raman (1996), Eppen and Iyer (1997), Iyer and Bergen (1997), Fisher et al.(2001), and the Sport Obermeyer case study byHammond and Raman (1994),描述了快速响应技术通过降低供给需求之间的偏差给企业带来的好处。 Lee and Tang (1997), Feitzinger and Lee(1997), Goyal and Netessine (2007), and Anand and Girotra (2007).做了有关延迟制造方面的研究。 消费者策略行为的研究:The Coase conjecture, which was described informally by Coase (1972) and formalized by Stokey (1981) and Bulow (1982) ,面对策略消费者,垄断厂商会降 低价格甚至接近边际成本。 关于消费者策略行为下的决策问题研究:Liu and van Ryzin (2008);Aviv and Pazgal (2008); Aviv and Pazgal (2008) ;Su and Zhang (2008),考虑库存策略和消费者策略行为; 消费者策略行为与快速响应技术研究:Cachon and Swinney (2009) and Swinney (2010) ;Cachon and Swinney (2009)。 现有文献都没有涉及到在快速时尚领域,高级设计对消费者策略行为的影响,以及高级设计与快速 响应技术之间的相互影响
02 顾客特定要求评审表-ford
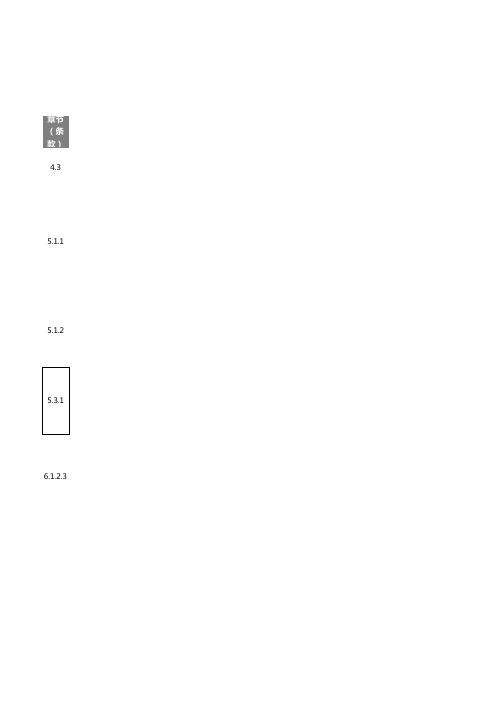
章节(条款)4.35.1.1 5.1.26.1.2.37.1.3.17.1.5.3 .2 7.2.17.5.3.2 .18.18.2.1 8.2.3.1 .18.2.3.1 .28.2.3.1 .3 8.3.1.18.3.2.18.3.48.3.4.48.4.1.3 8.4.2.18.4.2.2 8.4.2.38.4.2.4 8.5.1.2 8.5.2 8.6.18.6.2 8.6.3 8.6.6 8.7.1.19.1 9.1.1.19.1.2 9.1.2.19.2.2.39.3.1.1 9.3.210.2.1 10.2.2 10.2.5更多免费质量管理相关资料,请微信搜索公众号“体系君”,或扫码关注。
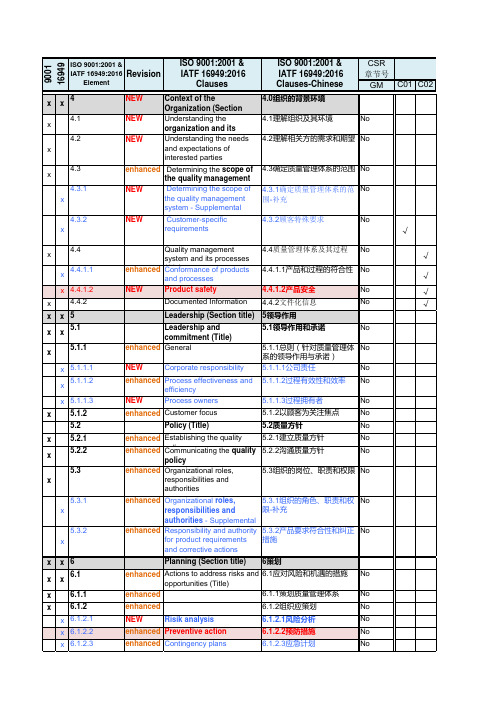
要求(具体描述)Determining the scope of the quality management systemCorporate Responsibility:The organization shall comply with Basic Working Conditions in the Global Terms and Conditions and the related Supplier Social Responsibility and Anti-CorruptionRequirements Web-Guide .组织应遵守基本的全球条款和工作条件条件,相关供应商的社会责任和反腐败要求。
The organization is also encouraged to adopt and enforce a similar code with Ford’s Policy组织还应鼓励采取和实施类似ford的代码与政策.Customer focus:The organization shall demonstrate enhanced customer satisfaction by meeting the continuous improvement requirements of Q1, as demonstrated in the organization’s QOS (Quality Operating System).组织必须通过满足Q1持续改善的要求来证实顾客满意的提高,并在组织的QOS(质量运行系统)中被证实。
质量管理体系过程与IATF 条款 顾客特殊要求 CSR 对照表
7.1.4过程操作环境
No
operation of processes
Environment for the
7.1.4.1过程操作环境-补充 No
operation of processes -
sMuopnpilteomrienngtaalnd measuring 7.1.5监视和测量资源
No
resources (title)
x
7.1.6
x
7.2
x 7.2.1
7.2.2 x
x 7.2.3 7.2.4
x
x
7.3
x
7.3.1
7.3.2 x
x
7.4
x
7.5
x
7.5.1
7.5.1.1
x
7.5.2
7.5.3 x
x
7.5.3.1
enhanced Quality objectives and planning to achieve them (Title)
7.1资源
No
modified General
7.1.1总则
No
NEW
People
7.1.2人员
No
Infrastructure
7.1.3基础设施
No
enhanced Plant, facility, and equipment planning
7.1.3.1工厂、设施和设备策 No 划
enhanced Environment for the
No
x
5.1.2
enhanced Customer focus
5.1.2以顾客为关注焦点
No
5.2
Policy (Title)
项目验证策略

Detailed
Design
详细设计
FAT & SAT
Construction
施工
Commissioning &
Qualification
试车与确认
工厂验收试车与 现场接受测试试车
Startup & Operation Support
启动与运转支持
PQ PV
性能确认与工艺验证
Life Cycle Activity
IQ Protocols 安装确认草案 OQ Protocols运行确认草案 Commissioning Protocols试车草案
IQ Reports 安装确认报告 OQ Reports运行确认报告 Commissioning Reports试车报告
Concept
Design
设计概念
Basic
Design
• 每个部件的建立都应该遵循相关人员批准或授权的计划和规范
• Each component should be inspected, tested, and documented by qualified individuals
• 每个部件都应该检查,测试,并由专业人员记录备案
• A minimum level of documentation should be provided for all systems and equipment
– 70’s to 2001: 确认在调试结束后开始. – 2001 to 现在: ISPE引入 C&Q, 确认和调试相结合. (ISPE
Guide Volume 5). – 未来: 新的基于风险评估的模式.
国内目前验证的整体水平与欧洲企业相差太多, 采用C&Q的策略风险较大,需要从设计阶段开始就要入手
药厂设备英文缩写一览表

序号 英文缩写
6 7 8 9 10 11 12 13 EHS EMA EMS EP EPBD EU EU-GMP EU-OSHA (EASHW)
英文全称
Environment, Health and Safety European Medicines Agency Environment Monitoring System European Pharmacopoeia Energy Performance of Buildings Directive(EU) European Union European Good Manufacturing Practice European Union-Occupational Safety and Health Adminstration(European Safety and Health at Work)
E 1 2 3 4 5 EC EDI EDL EDQM EDR European Commission Electro-de-ionization List of Essential Drugs European Directorate for the Quality of Medicines Enhanced Design Review 欧洲委员会 电去离子 基本药物目录 欧洲药品理事会 增加的设计审核
第3页,共7页
序号 英文缩写
2 3 4 5 6 7 8 9 10 11 IAQ ICH IEST IND INN IPC IQ IR ISO ISPE
英文全称
Indoor Air Quality
中文全称
室内空气质量
International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human 人用药品注册技术要求国际协调会 Use Institute of Environmental Sciences and Technology(US) Investigational New Drug International Non-proprietary Name In-process Controls Installation Qualification Infrared Spectrophotometer International Standard Organization International Society for Pharmaceutical Engineering 美国环境科学与技术学会 临床研究申请 国家非专利名 过程控制 安装确认 红外分光光度计 国际标准化组织 国际制药工程协会
国际制药协会ISPE基本指南05卷

A GUIDE FOR NEW FACILITIES VOLUME 5: COMMISSIONING AND QUALIFICATIONEXECUTIVE SUMMARYJUNE 2000A DOCUMENT DEVELOPED IN PARTNERSHIP BY:23ISPE PHARMACEUTICAL ENGINEERING GUIDECOMMISSIONING AND QUALIFICATIONFOREWORDAs noted in the Baseline® Guides, Volume 1, the pharmaceutical industry has experienced a ratcheting effect in the cost of new facilities. This increase in cost has been driven in part by uncertainty about the requirements for regulatory compliance. Some significant areas of concern are validation, particularly related to automation systems, and the trend to validate back to source utilities, architectural and HVAC. The absence of a consistent and widely accepted interpretation of regulatory requirements has led to one-upmanship. This practice of building increasingly technically advanced facilities has led to increased cost, longer lead times and, in some cases, delays in bringing new products to market.In May 1994, engineering representatives from the pharmaceutical industry engaged in a discussion with the International Society for Pharmaceutical Engineering (ISPE) and the Food and Drug Administration (FDA). That first discussion allowed for the creation of 10 facility engineering guides, now known as the Baseline® Pharmaceutical Engineering Guides. These guides are intended to assist pharmaceutical manufacturers in the design, construction and commissioning of facilities that comply with the requirements of the FDA. Volume 1, covering Bulk Pharmaceutical Chemicals (BPC), was published in June of 1996. This Guide, for Commissioning and Qualification, is the fifth volume in the series.As with the BPC Guide, the Commissioning and Qualification Guide, has been sponsored by ISPE’s Pharmaceutical Advisory Council, made up of senior pharmaceutical engineering executives from owner companies, the FDA and ISPE senior management. Overall planning, direction and technical guidance in the preparation of the Commissioning and Qualification Guide was provided by a Steering Committee most of whom were involved in the BPC Guide. The Commissioning and Qualification Guide itself was produced by a task force of around 60 individuals who expended a great deal of their own time in its preparation and development.Editors’ Disclaimer:This guide is meant to assist pharmaceutical manufacturers in the design and construction of newfacilities that comply with the requirements of the Food and Drug Administration (FDA). TheInternational Society for Pharmaceutical Engineering (ISPE) cannot ensure, and does not warrant,that a facility built in accordance with this guide will be acceptable to FDA.4ISPE PHARMACEUTICAL ENGINEERING GUIDECOMMISIONING AND QUALIFICATIONACKNOWLEDGEMENTSThis guide was developed by an integrated US-European team under the co-leadership of Alan Philips of Pfizer and Christopher Wood of Glaxo Wellcome.The Core Team on the guide was comprised:Alan Philips PfizerChristopher Wood Glaxo WellcomeBob Myers KvaernerGeorgia Keresty, Ph.D. Bristol Myers SquibbThe Extended Review Team was comprised the Core Team plusJan Gustafsson Novo NordiskGraham Shewell SmithKline BeechamTodd Troutman KvaernerSimon Shelley Glaxo WellcomeGene Yuan Hoffman LaRocheThe Chapter Credits are as follows:Introduction Alan PhilipsGeorgia Keresty, Ph.D. PfizerBristol-Myers SquibbKey Concepts & Philosophy Chris WoodGeorgia Keresty, Ph.D. Glaxo Wellcome Bristol-Myers SquibbImpact Assessment Bob MyersSimon ShelleyTodd Troutman KvaernerGlaxo WellcomeKvaerner5Good Engineering Practice John FadoolGraham Shewell Glaxo Wellcome SmithKline BeechamCommissioning Mark E. Miller1Chris WoodGraham Shewell GenentechGlaxo Wellcome SmithKline BeechamQualification Practices Jan GustafssonGene YuanSue Bacso Novo Nordisk Hoffman LaRoche MerckEnhanced Design Review Graham ShewellChris Wood SmithKline Beecham Glaxo WellcomeInstallation Qualification Bob MyersBob AdamsonTodd Troutman Kvaerner Foster Wheeler KvaernerOperational Qualification Bob MyersTodd Troutman Kvaerner KvaernerPerformance Qualification Chris Dell Cioppia KvaernerRelated Programs Georgia Keresty, Ph.D.Todd Troutman Bristol-Myers Squibb KvaernerIllustrative Examples Bob Myers KvaernerThe guide co-team leaders would also like to acknowledge the contributions made by the following part-time members of the guide team:Flemming Steen Jensen (then of) Novo NordiskSteve Heidel MerckCecilia Luna Novartis1 With the support of John Hughes (TVS Inc,), Jon Sheh (Alza Inc.) and Gary Schoenhouse (Genentech)6Tony deClaire APDC ConsultingFDA Reviewers:We would like to thank the following FDA review team for their input to this guide:Robert Sharpnack InvestigatorEric S. Weilage NDA/ANDA Pre-approval Inspection ManagerRobert Coleman National Drug Expert, DEIOBrian Nadel Compliance Officer, CDERWe also appreciate FDA support from the following representatives:Sharon Smith-Holston Deputy Commissioner for External AffairsSusan Setterberg Regional Director, Mid-Atlantic RegionJoe Phillips Deputy Regional Manager, Mid-Atlantic RegionIn addition, we would like to acknowledge the support and contribution of the ISPE Technical Documents Steering Committee, in particular the following members:Paul D’Eramo Committee Chairman, Johnson & JohnsonMel Crichton Eli LillyBruce Davis Astra ZenecaPaul Lorenzo (Retired)71. INTRODUCTION1.1 BACKGROUNDThe design, construction, commissioning and qualification of manufacturing facilities regulated by FDA or other regulatory authorities pose significant challenges to manufacturers, engineering professionals and equipment suppliers. These facilities are required to meet cGMP regulations while remaining in compliance with all other governing codes, laws, and regulations.The cost and time required to bring such facilities on line has been increasing, in many cases due to inconsistent interpretation of regulatory requirements. The ISPE and engineering representatives from a broad base of healthcare companies (e.g. pharmaceutical, device, biotechnology, etc.) have entered into a partnership with the Food and Drug Administration (FDA) to enhance understanding of Baseline cGMP requirements for facilities. This Guide is intended to define key terms and offer a consistent interpretation, while still allowing a flexible and innovative approach to facility design, construction, commissioning and qualification. A fundamental goal of the Guide is to provide value added guidance to industry that will facilitate timely and cost effective commissioning and qualification of facilities.This guide is one in a series of Baseline® Guides being planned and produced by ISPE. The majority of these are specific to one functional area (e.g. Oral Solid Dosage Forms). However, this guide provides advice and guidance that may be applied to all types of facilities, utilities and equipment found in the healthcare industry.This Guide was prepared by the ISPE, and has incorporated comments from:Industry representatives from all areas and disciplinesFDA Field Investigators and personnel from The Center for Drug Evaluation and ResearchIt is recognized that industry standards evolve and this document reflects the understanding of these standards, as of publication date.1.2 SCOPE OF THIS GUIDEThis is a Guide to be used by industry for the design, construction, commissioning and qualification of new or newly renovated manufacturing facilities that are regulated by FDA or other health authorities. It is neither a standard nor a GMP. It is not intended to replace governing laws, codes, standards or regulations that apply to facilities of this type. These are mentioned only for completeness and where their impact affects facility, equipment and utility design relative to cGMP’s. The use of this document for new or existing facilities, equipment or utilities is at the discretion of the owner or operator.This Guide focuses on the engineering approaches and practices involved in providing cost effective manufacturing facilities in a timely manner that meet their intended purposes. Specifically, the Guide addresses the process of designing, constructing, commissioning and qualifying the facilities, utilities and equipment regulated by FDA or other health authorities.This Guide is not intended to address any aspect of process/product validation. This is a subject that has been well defined by FDA and other authorities and for which substantial guidance documentation exists.It must be recognized, however, that Commissioning and Qualification activities are the foundation upon which Process Validation is built. Furthermore, these activities play a crucial role in delivering operationally effective, safe and efficient facilities, utilities and equipment. Therefore, it is important to ensure that a comprehensive approach is 8undertaken during the commissioning and qualification process. A well conceived and executed commissioning and qualification plan can greatly facilitate a timely and cost effective validation effort.Where non-engineering issues are covered (e.g. support systems, documentation, decision processes), the guidance is provided to show engineers the importance of such topics and the impact they have on the commissioning and qualification process. Consequently, non-engineering topics are not covered comprehensively. Specialist advice from QA Departments should be sought where additional information is required.The Guide is intended primarily for facilities, equipment and utilities meeting regulatory requirements to supply the United States (US) market and is aligned with US standards and references. The Guide may also be helpful to manufacturers needing to meet European requirements.91.3 KEY FEATURES AND CHAPTERS OF THIS GUIDEThe following key concepts are defined and used as a basis for guidance:• Direct Impact Systems• Indirect Impact Systems• System Impact Assessment• Good Engineering Practice• Commissioning• Qualification Practices• Enhanced Design Review• Installation Qualification• Operational Qualification• Performance Qualification• Consistent Terminology• Documentation RequirementsSOME BRIEF EXPLANATION OF THESE IS AS FOLLOWS:It is the function of the facility, equipment or utility that determines what level of commissioning and qualification are needed.• ‘Direct Impact’ systems are expected to have an impact on product quality• ‘Indirect Impact’ systems are not expected to have an impact on product qualityBoth types of systems will require commissioning, however, the “Direct Impact” systems will be subject to supplementary qualification practices to meet the additional regulatory requirements of the FDA and other regulatory authorities.The determination of a system as either ‘Direct Impact’ or ‘Indirect Impact’ is critical. It is this differentiation between system types that determines the degree of effort and level of resources required for each system. System Impact Assessment provides the thought process and some key questions that must be asked in making the determination.During the production of this guide, regulatory authorities have expressed concern that designating a system “Indirect Impact” might be a means of doing less than full testing on a system that may actually require it. This is not the intention. The objective is that through a comprehensive impact assessment process, those systems presenting a risk to product quality are identified and given the attention appropriate to this level of risk, and by the right people (e.g. QA Departments).For this process to work it is essential that an explicit rationale is provided for the indirect/direct impact assessment and that the rationales are fully understood, documented and endorsed by QA Departments. This places a responsibility upon engineers to communicate clearly the nature of operation of engineering systems, and their potential impact on product quality.It will also be seen that throughout the Guide, the application of Good Engineering Practice is essential to the commissioning and qualification activities. Good Engineering Practice, commonly referred to as GEP, is proven and accepted, cost-effective, engineering methods and practices that ensure the effective satisfaction of stakeholder requirements. As such, GEP ensures that an engineering project meets the requirements of the user while being cost effective, compliant with regulations and well documented. Guidance and standards that have been defined by engineering institutes and other learned bodies support GEP. For direct impact systems, GEP is supplemented by enhanced documentation and qualification practices with the active participation of Quality Assurance personnel.The guide also attempts to clarify some misconceptions about how activities are defined, which activities are the subject of regulatory oversight and the sequence, if any, of these activities. For example, the guide discusses “Enhanced Design Review” and the components and criteria of this activity. The intent is to identify design aspects that are key to manufacturing facilities regulated by the FDA or other health authorities. How this enhanced design review is accomplished, either with a formal or informal process, is at the discretion of the individual company. The intent is not to establish new administrative requirements, especially for those activities not regulated by FDA or other regulatory authorities. The design review activities are part of GEP and are unregulated by FDA, i.e. these are good engineering practices, not regulatory requirements.Installation Qualification (IQ), Operational Qualification (OQ) and Performance Qualification (PQ) are activities that FDA may have an interest in, since these are the final activities before process validation can begin. IQ/OQ in many instances is done concurrently with commissioning and requires the enhanced documentation, QA involvement and additional tests and checks known as Qualification Practices.An overview of the Chapter structure is given in Figure 1-2.Figure 1-2: Chapter Structure1.4 GOALS OF THIS GUIDEThere are two primary goals of the Commissioning and Qualification Baseline® Guide. The first is to bring a common terminology and methodology to the commissioning and qualification process that can be used by manufacturers, facility designers, contractors and equipment suppliers. The second is to provide a system impact assessment process to bring structure and consistency to determining a direct and indirect impact system. An important secondary goal is to foster an interdisciplinary team approach to commissioning and qualification. Such an approach will help establish an effective basis for master planning and execution of facility projects. Specifically, the Guide is focused upon value added approaches that will eliminate duplication of effort and the costly practices of:• Repeating qualification steps during process validation• Qualifying systems that only require commissioning• Generating insufficient or excessive documentation• Excessively long project schedules• Delays which can result in product supply interruptions or delayed product launches2. GUIDE PHILOSOPHY AND KEY CONCEPTSThis Chapter describes the purpose and philosophy of the Commissioning and Qualification Baseline® Guide, and the differences between the commissioning and qualification processes in the context of this Guide. It is important to understand and apply the approaches outlined in this Baseline® Guide in a sound and well-reasoned manner, since every facility and project is different.The key terms used in the Guide are defined, including:• Direct Impact System• Indirect Impact System• No Impact System• Design for Impact• Good Engineering Practice• Enhanced Design Review• CommissioningAn overview of Qualification Practices is given, including Enhanced Design Review, Installation Qualification, Operational Qualification, and Performance Qualification. V-models are provided for both Direct Impact systems and Indirect Impact systems and the role of Quality Assurance is discussed.3. IMPACT ASSESSMENTImpact Assessment is the process of determining which systems and/or system components should be subject to Qualification Practices in addition to Good Engineering Practices (GEP). Impact Assessment assists in defining the Commissioning and Qualification scope of a project.This Chapter considers the Impact Assessment process. Terms specific to Impact Assessment are defined. A method is suggested for defining the steps of a system assessment process, including a discussion of the benefits, and a list of the criteria for determining system impact and component criticality.4. GOOD ENGINEERING PRACTICEThis Chapter provides an overview of the various project phases and sequence, from inception through commissioning, qualification, and operation. Concepts associated with “Good Engineering Practice” (GEP), the types of activities that occur, and documentation that is created through GEP are discussed. Overviews are provided of both effective project controls, and project team concepts and organization.The Requirements phase is considered in detail, including:• Project Purpose and Justification• User Requirements Brief• Requirements Specifications• Project Execution Plan• Maintenance and Technical Support Requirements• Compliance Requirements• DeliverablesStages in the design process are described with specific consideration of Piping and Instrumentation Diagrams, Specifications, and Construction drawings. Construction involves several elements, which are crucial to every project, including project site logistics and project quality control. This Chapter details typical requirements and elements of construction.The information given in the Chapter aims to demonstrate how GEP, as applied throughout the project lifecycle, provides a basis for effective qualification.5. COMMISSIONINGThis Chapter defines the term “commissioning” in the context of the Guide and describes the organization and content of the Commissioning Plan document. Commissioning is positioned within the context of the Qualification effort and guidance is provided in the management and execution of the commissioning activities. Typical commissioning deliverables and the associated commissioning team responsibilities are considered.Commissioning activities described include:• Inspection• Setting-to-Work• Regulation and Adjustment• Testing and Performance Testing• Training• Turnover• Commissioning Plan Close-Out6. QUALIFICATION PRACTICESDirect impact systems are subject to qualification practices that incorporate the enhanced review, control and testing against specifications and requirements necessary for compliance with current Good Manufacturing Practice. The purpose of this chapter is to introduce a high level overview of qualification practices that are required for direct impact systems. The Validation Master Plan and Qualification Rationale are described in detail. This Chapter contains detailed consideration of Enhanced Documentation.7. ENHANCED DESIGN REVIEWEnhanced Design Review (EDR) is the term adopted by this guide to describe the process by which engineering designs for pharmaceutical facilities, systems and equipment are evaluated. This process compliments Good Engineering Practice.This Chapter gives the regulatory perspective on EDR and relates EDR to the V-Model for Direct Impact systems. The EDR process is detailed. A structured design review method and a failure modes analysis method are suggested for evaluating designs.8. INSTALLATION QUALIFICATIONInstallation Qualification (IQ) is an activity that is regulated by the FDA, and is a part of final qualification activities before process validation begins.The primary objectives of this chapter are to:• Provide an overview of the Installation Qualification process• Describe the types of activities that occur and documentation that is needed for the Installation Qualification Process• Describe how Installation Qualification fits in with the overall qualification process• Describe how Commissioning integrates within the Installation Qualification process9. OPERATIONAL QUALIFICATIONOperational Qualification (OQ) is an activity that is regulated by the FDA, and is a part of final qualification activities before Performance Qualification or Process Validation begins.The primary objectives of this chapter are to:• Provide an overview of the Operational Qualification process• Describe the types of activities that occur and documentation that is needed for the Operational Qualification Process• Describe how Operational Qualification fits in with the overall qualification process• Describe how the commissioning process integrates within Operational Qualification10. PERFORMANCE QUALIFICATIONPerformance Qualification (PQ) is an activity that is regulated by the FDA, and is the final qualification activity before the remainder of Process Validation begins. For pharmaceutical grade utilities and certain support systems, PQ is the final qualification step.Once the system (or systems) have gone through IQ and OQ execution and have been approved/accepted the PQ can be performed.The primary objectives of this chapter are to:• Provide an overview of the Performance Qualification process• Describe the types of activities that occur and documentation that is needed for the Performance Qualification Process• Describe how Performance Qualification fits in with the overall qualification process• Describe how the commissioning process integrates within Performance Qualification11. RELATED PROGRAMSThis Chapter provides details of those programs that are undertaken to provide assistance and information in support of the qualification activities. Some of these programs can be applied to ‘Direct’, ‘Indirect’ and ‘No Impact’ systems and their components. Where these programs are undertaken in support of qualification activities, the appropriate qualification practices must be followed to ensure that the compliance of the over-all qualification effort is not compromised. Related programs considered include:• Safety• Standard Operating Procedures• Training• Preventative Maintenance and Calibration• Computer Systems Validation• Cleaning Validation• Analytical Method Validation• Process Validation• Revalidation12. GLOSSARYTerms and concepts used throughout the Commissioning and Qualification Baseline® Guide are defined and cross-referenced.13. ILLUSTRATIVE EXAMPLESThe illustrative examples given in this Chapter provide one interpretation of how the key concepts of this guidecan be applied in preparing for commissioning and qualification activities. Depending upon company policies or the intended use of the equipment listed, there may be additions or deletions to the listed activities. APPENDIXThe Appendix provides detail and references for Failures Modes Analysis.。
t8调考 2024年英语考试范文

t8调考2024年英语考试范文篇1Oh, my dear friends! Let's talk about the significance and influence of the T8 mock exam for English learning in 2024. It is truly a remarkable assessment that has a profound impact on our journey of mastering this language.For me, the T8 mock exam was like a mirror reflecting my weaknesses in grammar and vocabulary. I realized that I often made mistakes in complex sentence structures and had a limited vocabulary range. But, isn't this a precious opportunity to improve? With this awareness, I could conduct targeted review and practice, making my language skills more solid and refined.And look at my classmate Tom! His grades improved significantly through the T8 mock exam. This success not only enhanced his confidence but also inspired him to pursue higher goals. Isn't it amazing how a single exam can bring such a positive change?In conclusion, the T8 mock exam is of utmost importance. It helps us identify our shortcomings and encourages us to keep striving for better results. So, let's embrace it with enthusiasm and make the most of this valuable chance to excel in our English learning in 2024!篇2Oh my goodness! The T8 mock exam has just come to an end, and it's truly a golden opportunity for us to reflect and improve our English learning methods. How can we make the most of the results? Let me share some thoughts with you!Firstly, if we find that our listening skills are weak during the exam, we should definitely increase the time and frequency of our listening practice. For instance, we can listen to English podcasts or watch English movies and TV shows regularly. Isn't that a great way to enhance our listening comprehension?Secondly, looking at the answer strategies of top-performing classmates can be incredibly helpful. We can analyze how they approach different types of questions and adopt those effective techniques to improve our own problem-solving skills. Why not learn from the best?In conclusion, the T8 mock exam is not just a test, but a valuable guide for us to grow and progress in our English learning journey. Let's make the most of it and strive for better results! How exciting and challenging it is to keep improving our English!篇3Oh my goodness! The T8 mock exam has always been a significant indicator of the trends in English exams for the coming year. This year'sT8 exam is no exception! It has brought about some remarkable changes and new challenges.For instance, the emergence of new question types has caught many students off guard. The inclusion of more complex reading comprehension passages and the requirement of in-depth analysis in writing tasks have raised the bar significantly. Isn't it astonishing how these new elements are shaping the future of English exams?When we compare the changes in the T8 exams over the years, we can identify certain patterns. The focus on practical language usage and the ability to think critically has been steadily increasing. Why is this shift so important? It's because it reflects the real-world demands for language skills.So, what does all this mean for the 2024 English exams? Well, it's clear that students need to enhance their comprehensive language abilities, not just relying on rote memorization. They must be able to adapt quickly to new question types and think creatively. How exciting and challenging this journey will be!In conclusion, the T8 mock exam provides valuable insights into the evolving landscape of English exams, and we should take these cues seriously to prepare well for the exams in 2024.篇4As a teacher, the T8 mock exam results offer valuable insights foradjusting our teaching strategies. It's like a mirror reflecting the strengths and weaknesses of our students' learning. Oh, what an important tool it is!We notice that many students struggle with grammar comprehension. So, we could design specific remedial courses to address this common issue. How wonderful it would be if they could master those tricky grammar rules!For those students who perform well in some aspects but poorly in others, personalized guidance is essential. We should carefully analyze their test papers and find out the specific problems. For example, if a student is good at reading but weak in writing, we could provide more writing exercises and offer detailed feedback. Isn't it a practical approach?Also, we need to encourage students to actively participate in the learning process. We could organize group discussions or projects to enhance their communication and cooperation skills. Won't that make learning more interesting and effective?In conclusion, the T8 mock exam is not just a test, but a guide for us to improve our teaching and help students achieve better results. Let's work hard and make it happen!篇5The T8 mock exam in 2024 holds a significant position within the English education evaluation system. How could we underestimate its role? It provides a comprehensive assessment of students' English proficiency,covering various aspects such as grammar, vocabulary, reading comprehension, and writing skills. But how does it differ from other exams? Compared to some traditional tests that might focus mainly on theoretical knowledge, the T8 mock exam places more emphasis on practical application and comprehensive abilities. Isn't this a more scientific approach? It allows students to demonstrate their true language competence in real-life scenarios. However, there might be concerns about its weighting in the overall evaluation. Should it carry more weight than regular classroom tests? Or should it be balanced with other assessment methods? This is a question that requires careful consideration. After all, a fair and effective evaluation system should take into account multiple factors to accurately reflect students' English learning achievements. The T8 mock exam is undoubtedly an important part, but it's not the sole criterion. We need to view it in the broader context of English education to ensure that it serves its purpose of promoting students' all-round development in the language!。
制药英文缩写

Enhanced Design Review European directorate for the Quality of Medicines European agency for the evaluation of medicinal product
Factory Acceptance Test FOOD AND DRUG ADMINISTRATION Function Desgn Speciation Faliure Mode and Effects Analysis Fan Filter Unit Factory Monitoring System Functional Specifiaction
Master Batch Record
NEW DRUG APPLICATION
Quality Assurance Quality by Design
QC OFD OOS OOT OQ P PAT PCS
PICS/S
PID PLC PMD POU PQ PQR PW PS PPM PV PVP PVR Q QM QMS QA QC
危害分析和关键环节控制点 高效空气过滤器 人机界面 供热,通风,空调系统 人机界面或用户界面或使用者界面
人用药品注册技术要求国际协调会
临床研究申请 国家非专利名 安装确认 国际制药工程协会 影响评估 安装和运行确认 过程控制
实验室信息管理系统 单向流 检测限 定量限
上市许可 上市申请 主生产批记录
新药申请
药物主文件 设计说明 文件控制协调者 设计确认
欧洲委员会
欧洲药品与健康质量理事会
环境,健康和安全 欧洲药监局 欧洲药典
增加的设计审核 欧洲药品理事会
欧洲药品评价局
GEP 的基本概念

GEP 和GMP的联系和区别
GEP *规范准则 *设计指南 *标准技术规范 •国际标准 •工程设计摘要和具体说 明 •项目工程质量方案筹备 依据 •ISO •咨询顾问 •承包商 •专业团队 •设计 •制造和安装 •调试
GMP 活动 确认操作规范 影响评估 确认原理 QA的积极参与 严格的文档编制和严谨的文 件管理 扩大的最终用户的参与 审查及测试
• 举例:水系统 SAT 和 IQOQPQ • 包装线的SAT 和IQOQPQ
GEP 的基本概念
• GEP (Good Engineering Practice)良好的工程规范 在项目全过程中使用已确定的工程方案和标准达到合理的经济有效的结果。 GEP : 相关的设施 设备及公用设施,按照FDA相关的规定包括以下方面: 1。项目的设计和安装要充分考虑到现行GMP规范, 安全,健康,环保,功效学,操作 ,维修,认可的工业规范和法定要求 2。 专业的能胜任的项目管理,工程设计,采购,建筑,安装,调试移交 3。合理的文档记录,包括设计概念,设计规范图纸,调试记录,维修和操作手册,法定 检测证书等。
Enhanced design review (增强的设计审核)
• 一种对设计的存档审核,在项目的一个合适阶段,目的是为了保持和预期的一致性。 经一部设计审核不是必须遵守FDA生产设施规范, • EDR是一种商业风险选择,不是规定 • • • • 1。系统的影响 2。系统的复杂程度 3。系统和供应商的熟悉度或者新颖性 4。应用标准设备的新颖性
Qualification
• 对于直接影响系统,为了满足FDA和其他监管部门的要求, GEP的实施应该依据确认 操作规范得到加强和完善。 • 确认操作规范包括: • 1。系统影响评估 • 2。QA的参与 • 3。严格的文件编制,严谨的文档管理,和一个有条理的批准程序 • 4。QA变更 程序 • 5。最终用户的参与 • 6。培训 • 7。使用确认原理鉴别什么系统应该确认,确认到什么程度,为什么确认,谁来确认 • 8。确定不需要检查的项及理由
国际制药协会ISPE基础指南05卷

A GUIDE FOR NEW FACILITIES VOLUME 5: COMMISSIONING AND QUALIFICATIONEXECUTIVE SUMMARYJUNE 2000A DOCUMENT DEVELOPED IN PARTNERSHIP BY:23ISPE PHARMACEUTICAL ENGINEERING GUIDECOMMISSIONING AND QUALIFICATIONFOREWORDAs noted in the Baseline® Guides, Volume 1, the pharmaceutical industry has experienced a ratcheting effect in the cost of new facilities. This increase in cost has been driven in part by uncertainty about the requirements for regulatory compliance. Some significant areas of concern are validation, particularly related to automation systems, and the trend to validate back to source utilities, architectural and HVAC. The absence of a consistent and widely accepted interpretation of regulatory requirements has led to one-upmanship. This practice of building increasingly technically advanced facilities has led to increased cost, longer lead times and, in some cases, delays in bringing new products to market.In May 1994, engineering representatives from the pharmaceutical industry engaged in a discussion with the International Society for Pharmaceutical Engineering (ISPE) and the Food and Drug Administration (FDA). That first discussion allowed for the creation of 10 facility engineering guides, now known as the Baseline® Pharmaceutical Engineering Guides. These guides are intended to assist pharmaceutical manufacturers in the design, construction and commissioning of facilities that comply with the requirements of the FDA. Volume 1, covering Bulk Pharmaceutical Chemicals (BPC), was published in June of 1996. This Guide, for Commissioning and Qualification, is the fifth volume in the series.As with the BPC Guide, the Commissioning and Qualification Guide, has been sponsored by ISPE’s Pharmaceutical Advisory Council, made up of senior pharmaceutical engineering executives from owner companies, the FDA and ISPE senior management. Overall planning, direction and technical guidance in the preparation of the Commissioning and Qualification Guide was provided by a Steering Committee most of whom were involved in the BPC Guide. The Commissioning and Qualification Guide itself was produced by a task force of around 60 individuals who expended a great deal of their own time in its preparation and development.Editors’ Disclaimer:This guide is meant to assist pharmaceutical manufacturers in the design and construction of newfacilities that comply with the requirements of the Food and Drug Administration (FDA). TheInternational Society for Pharmaceutical Engineering (ISPE) cannot ensure, and does not warrant,that a facility built in accordance with this guide will be acceptable to FDA.4ISPE PHARMACEUTICAL ENGINEERING GUIDECOMMISIONING AND QUALIFICATIONACKNOWLEDGEMENTSThis guide was developed by an integrated US-European team under the co-leadership of Alan Philips of Pfizer and Christopher Wood of Glaxo Wellcome.The Core Team on the guide was comprised:Alan Philips PfizerChristopher Wood Glaxo WellcomeBob Myers KvaernerGeorgia Keresty, Ph.D. Bristol Myers SquibbThe Extended Review Team was comprised the Core Team plusJan Gustafsson Novo NordiskGraham Shewell SmithKline BeechamTodd Troutman KvaernerSimon Shelley Glaxo WellcomeGene Yuan Hoffman LaRocheThe Chapter Credits are as follows:Introduction Alan PhilipsGeorgia Keresty, Ph.D. PfizerBristol-Myers SquibbKey Concepts & Philosophy Chris WoodGeorgia Keresty, Ph.D. Glaxo Wellcome Bristol-Myers SquibbImpact Assessment Bob MyersSimon ShelleyTodd Troutman KvaernerGlaxo WellcomeKvaerner5Good Engineering Practice John FadoolGraham Shewell Glaxo Wellcome SmithKline BeechamCommissioning Mark E. Miller1Chris WoodGraham Shewell GenentechGlaxo Wellcome SmithKline BeechamQualification Practices Jan GustafssonGene YuanSue Bacso Novo Nordisk Hoffman LaRoche MerckEnhanced Design Review Graham ShewellChris Wood SmithKline Beecham Glaxo WellcomeInstallation Qualification Bob MyersBob AdamsonTodd Troutman Kvaerner Foster Wheeler KvaernerOperational Qualification Bob MyersTodd Troutman Kvaerner KvaernerPerformance Qualification Chris Dell Cioppia KvaernerRelated Programs Georgia Keresty, Ph.D.Todd Troutman Bristol-Myers Squibb KvaernerIllustrative Examples Bob Myers KvaernerThe guide co-team leaders would also like to acknowledge the contributions made by the following part-time members of the guide team:Flemming Steen Jensen (then of) Novo NordiskSteve Heidel MerckCecilia Luna Novartis1 With the support of John Hughes (TVS Inc,), Jon Sheh (Alza Inc.) and Gary Schoenhouse (Genentech)6Tony deClaire APDC ConsultingFDA Reviewers:We would like to thank the following FDA review team for their input to this guide:Robert Sharpnack InvestigatorEric S. Weilage NDA/ANDA Pre-approval Inspection ManagerRobert Coleman National Drug Expert, DEIOBrian Nadel Compliance Officer, CDERWe also appreciate FDA support from the following representatives:Sharon Smith-Holston Deputy Commissioner for External AffairsSusan Setterberg Regional Director, Mid-Atlantic RegionJoe Phillips Deputy Regional Manager, Mid-Atlantic RegionIn addition, we would like to acknowledge the support and contribution of the ISPE Technical Documents Steering Committee, in particular the following members:Paul D’Eramo Committee Chairman, Johnson & JohnsonMel Crichton Eli LillyBruce Davis Astra ZenecaPaul Lorenzo (Retired)71. INTRODUCTION1.1 BACKGROUNDThe design, construction, commissioning and qualification of manufacturing facilities regulated by FDA or other regulatory authorities pose significant challenges to manufacturers, engineering professionals and equipment suppliers. These facilities are required to meet cGMP regulations while remaining in compliance with all other governing codes, laws, and regulations.The cost and time required to bring such facilities on line has been increasing, in many cases due to inconsistent interpretation of regulatory requirements. The ISPE and engineering representatives from a broad base of healthcare companies (e.g. pharmaceutical, device, biotechnology, etc.) have entered into a partnership with the Food and Drug Administration (FDA) to enhance understanding of Baseline cGMP requirements for facilities. This Guide is intended to define key terms and offer a consistent interpretation, while still allowing a flexible and innovative approach to facility design, construction, commissioning and qualification. A fundamental goal of the Guide is to provide value added guidance to industry that will facilitate timely and cost effective commissioning and qualification of facilities.This guide is one in a series of Baseline® Guides being planned and produced by ISPE. The majority of these are specific to one functional area (e.g. Oral Solid Dosage Forms). However, this guide provides advice and guidance that may be applied to all types of facilities, utilities and equipment found in the healthcare industry.This Guide was prepared by the ISPE, and has incorporated comments from:Industry representatives from all areas and disciplinesFDA Field Investigators and personnel from The Center for Drug Evaluation and ResearchIt is recognized that industry standards evolve and this document reflects the understanding of these standards, as of publication date.1.2 SCOPE OF THIS GUIDEThis is a Guide to be used by industry for the design, construction, commissioning and qualification of new or newly renovated manufacturing facilities that are regulated by FDA or other health authorities. It is neither a standard nor a GMP. It is not intended to replace governing laws, codes, standards or regulations that apply to facilities of this type. These are mentioned only for completeness and where their impact affects facility, equipment and utility design relative to cGMP’s. The use of this document for new or existing facilities, equipment or utilities is at the discretion of the owner or operator.This Guide focuses on the engineering approaches and practices involved in providing cost effective manufacturing facilities in a timely manner that meet their intended purposes. Specifically, the Guide addresses the process of designing, constructing, commissioning and qualifying the facilities, utilities and equipment regulated by FDA or other health authorities.This Guide is not intended to address any aspect of process/product validation. This is a subject that has been well defined by FDA and other authorities and for which substantial guidance documentation exists.It must be recognized, however, that Commissioning and Qualification activities are the foundation upon which Process Validation is built. Furthermore, these activities play a crucial role in delivering operationally effective, safe and efficient facilities, utilities and equipment. Therefore, it is important to ensure that a comprehensive approach is 8undertaken during the commissioning and qualification process. A well conceived and executed commissioning and qualification plan can greatly facilitate a timely and cost effective validation effort.Where non-engineering issues are covered (e.g. support systems, documentation, decision processes), the guidance is provided to show engineers the importance of such topics and the impact they have on the commissioning and qualification process. Consequently, non-engineering topics are not covered comprehensively. Specialist advice from QA Departments should be sought where additional information is required.The Guide is intended primarily for facilities, equipment and utilities meeting regulatory requirements to supply the United States (US) market and is aligned with US standards and references. The Guide may also be helpful to manufacturers needing to meet European requirements.91.3 KEY FEATURES AND CHAPTERS OF THIS GUIDEThe following key concepts are defined and used as a basis for guidance:• Direct Impact Systems• Indirect Impact Systems• System Impact Assessment• Good Engineering Practice• Commissioning• Qualification Practices• Enhanced Design Review• Installation Qualification• Operational Qualification• Performance Qualification• Consistent Terminology• Documentation RequirementsSOME BRIEF EXPLANATION OF THESE IS AS FOLLOWS:It is the function of the facility, equipment or utility that determines what level of commissioning and qualification are needed.• ‘Direct Impact’ systems are expected to have an impact on product quality• ‘Indirect Impact’ systems are not expected to have an impact on product qualityBoth types of systems will require commissioning, however, the “Direct Impact” systems will be subject to supplementary qualification practices to meet the additional regulatory requirements of the FDA and other regulatory authorities.The determination of a system as either ‘Direct Impact’ or ‘Indirect Impact’ is critical. It is this differentiation between system types that determines the degree of effort and level of resources required for each system. System Impact Assessment provides the thought process and some key questions that must be asked in making the determination.During the production of this guide, regulatory authorities have expressed concern that designating a system “Indirect Impact” might be a means of doing less than full testing on a system that may actually require it. This is not the intention. The objective is that through a comprehensive impact assessment process, those systems presenting a risk to product quality are identified and given the attention appropriate to this level of risk, and by the right people (e.g. QA Departments).For this process to work it is essential that an explicit rationale is provided for the indirect/direct impact assessment and that the rationales are fully understood, documented and endorsed by QA Departments. This places a responsibility upon engineers to communicate clearly the nature of operation of engineering systems, and their potential impact on product quality.It will also be seen that throughout the Guide, the application of Good Engineering Practice is essential to the commissioning and qualification activities. Good Engineering Practice, commonly referred to as GEP, is proven and accepted, cost-effective, engineering methods and practices that ensure the effective satisfaction of stakeholder requirements. As such, GEP ensures that an engineering project meets the requirements of the user while being cost effective, compliant with regulations and well documented. Guidance and standards that have been defined by engineering institutes and other learned bodies support GEP. For direct impact systems, GEP is supplemented by enhanced documentation and qualification practices with the active participation of Quality Assurance personnel.The guide also attempts to clarify some misconceptions about how activities are defined, which activities are the subject of regulatory oversight and the sequence, if any, of these activities. For example, the guide discusses “Enhanced Design Review” and the components and criteria of this activity. The intent is to identify design aspects that are key to manufacturing facilities regulated by the FDA or other health authorities. How this enhanced design review is accomplished, either with a formal or informal process, is at the discretion of the individual company. The intent is not to establish new administrative requirements, especially for those activities not regulated by FDA or other regulatory authorities. The design review activities are part of GEP and are unregulated by FDA, i.e. these are good engineering practices, not regulatory requirements.Installation Qualification (IQ), Operational Qualification (OQ) and Performance Qualification (PQ) are activities that FDA may have an interest in, since these are the final activities before process validation can begin. IQ/OQ in many instances is done concurrently with commissioning and requires the enhanced documentation, QA involvement and additional tests and checks known as Qualification Practices.An overview of the Chapter structure is given in Figure 1-2.Figure 1-2: Chapter Structure1.4 GOALS OF THIS GUIDEThere are two primary goals of the Commissioning and Qualification Baseline® Guide. The first is to bring a common terminology and methodology to the commissioning and qualification process that can be used by manufacturers, facility designers, contractors and equipment suppliers. The second is to provide a system impact assessment process to bring structure and consistency to determining a direct and indirect impact system. An important secondary goal is to foster an interdisciplinary team approach to commissioning and qualification. Such an approach will help establish an effective basis for master planning and execution of facility projects. Specifically, the Guide is focused upon value added approaches that will eliminate duplication of effort and the costly practices of:• Repeating qualification steps during process validation• Qualifying systems that only require commissioning• Generating insufficient or excessive documentation• Excessively long project schedules• Delays which can result in product supply interruptions or delayed product launches2. GUIDE PHILOSOPHY AND KEY CONCEPTSThis Chapter describes the purpose and philosophy of the Commissioning and Qualification Baseline® Guide, and the differences between the commissioning and qualification processes in the context of this Guide. It is important to understand and apply the approaches outlined in this Baseline® Guide in a sound and well-reasoned manner, since every facility and project is different.The key terms used in the Guide are defined, including:• Direct Impact System• Indirect Impact System• No Impact System• Design for Impact• Good Engineering Practice• Enhanced Design Review• CommissioningAn overview of Qualification Practices is given, including Enhanced Design Review, Installation Qualification, Operational Qualification, and Performance Qualification. V-models are provided for both Direct Impact systems and Indirect Impact systems and the role of Quality Assurance is discussed.3. IMPACT ASSESSMENTImpact Assessment is the process of determining which systems and/or system components should be subject to Qualification Practices in addition to Good Engineering Practices (GEP). Impact Assessment assists in defining the Commissioning and Qualification scope of a project.This Chapter considers the Impact Assessment process. Terms specific to Impact Assessment are defined. A method is suggested for defining the steps of a system assessment process, including a discussion of the benefits, and a list of the criteria for determining system impact and component criticality.4. GOOD ENGINEERING PRACTICEThis Chapter provides an overview of the various project phases and sequence, from inception through commissioning, qualification, and operation. Concepts associated with “Good Engineering Practice” (GEP), the types of activities that occur, and documentation that is created through GEP are discussed. Overviews are provided of both effective project controls, and project team concepts and organization.The Requirements phase is considered in detail, including:• Project Purpose and Justification• User Requirements Brief• Requirements Specifications• Project Execution Plan• Maintenance and Technical Support Requirements• Compliance Requirements• DeliverablesStages in the design process are described with specific consideration of Piping and Instrumentation Diagrams, Specifications, and Construction drawings. Construction involves several elements, which are crucial to every project, including project site logistics and project quality control. This Chapter details typical requirements and elements of construction.The information given in the Chapter aims to demonstrate how GEP, as applied throughout the project lifecycle, provides a basis for effective qualification.5. COMMISSIONINGThis Chapter defines the term “commissioning” in the context of the Guide and describes the organization and content of the Commissioning Plan document. Commissioning is positioned within the context of the Qualification effort and guidance is provided in the management and execution of the commissioning activities. Typical commissioning deliverables and the associated commissioning team responsibilities are considered.Commissioning activities described include:• Inspection• Setting-to-Work• Regulation and Adjustment• Testing and Performance Testing• Training• Turnover• Commissioning Plan Close-Out6. QUALIFICATION PRACTICESDirect impact systems are subject to qualification practices that incorporate the enhanced review, control and testing against specifications and requirements necessary for compliance with current Good Manufacturing Practice. The purpose of this chapter is to introduce a high level overview of qualification practices that are required for direct impact systems. The Validation Master Plan and Qualification Rationale are described in detail. This Chapter contains detailed consideration of Enhanced Documentation.7. ENHANCED DESIGN REVIEWEnhanced Design Review (EDR) is the term adopted by this guide to describe the process by which engineering designs for pharmaceutical facilities, systems and equipment are evaluated. This process compliments Good Engineering Practice.This Chapter gives the regulatory perspective on EDR and relates EDR to the V-Model for Direct Impact systems. The EDR process is detailed. A structured design review method and a failure modes analysis method are suggested for evaluating designs.8. INSTALLATION QUALIFICATIONInstallation Qualification (IQ) is an activity that is regulated by the FDA, and is a part of final qualification activities before process validation begins.The primary objectives of this chapter are to:• Provide an overview of the Installation Qualification process• Describe the types of activities that occur and documentation that is needed for the Installation Qualification Process• Describe how Installation Qualification fits in with the overall qualification process• Describe how Commissioning integrates within the Installation Qualification process9. OPERATIONAL QUALIFICATIONOperational Qualification (OQ) is an activity that is regulated by the FDA, and is a part of final qualification activities before Performance Qualification or Process Validation begins.The primary objectives of this chapter are to:• Provide an overview of the Operational Qualification process• Describe the types of activities that occur and documentation that is needed for the Operational Qualification Process• Describe how Operational Qualification fits in with the overall qualification process• Describe how the commissioning process integrates within Operational Qualification10. PERFORMANCE QUALIFICATIONPerformance Qualification (PQ) is an activity that is regulated by the FDA, and is the final qualification activity before the remainder of Process Validation begins. For pharmaceutical grade utilities and certain support systems, PQ is the final qualification step.Once the system (or systems) have gone through IQ and OQ execution and have been approved/accepted the PQ can be performed.The primary objectives of this chapter are to:• Provide an overview of the Performance Qualification process• Describe the types of activities that occur and documentation that is needed for the Performance Qualification Process• Describe how Performance Qualification fits in with the overall qualification process• Describe how the commissioning process integrates within Performance Qualification11. RELATED PROGRAMSThis Chapter provides details of those programs that are undertaken to provide assistance and information in support of the qualification activities. Some of these programs can be applied to ‘Direct’, ‘Indirect’ and ‘No Impact’ systems and their components. Where these programs are undertaken in support of qualification activities, the appropriate qualification practices must be followed to ensure that the compliance of the over-all qualification effort is not compromised. Related programs considered include:• Safety• Standard Operating Procedures• Training• Preventative Maintenance and Calibration• Computer Systems Validation• Cleaning Validation• Analytical Method Validation• Process Validation• Revalidation12. GLOSSARYTerms and concepts used throughout the Commissioning and Qualification Baseline® Guide are defined and cross-referenced.13. ILLUSTRATIVE EXAMPLESThe illustrative examples given in this Chapter provide one interpretation of how the key concepts of this guidecan be applied in preparing for commissioning and qualification activities. Depending upon company policies or the intended use of the equipment listed, there may be additions or deletions to the listed activities. APPENDIXThe Appendix provides detail and references for Failures Modes Analysis.。
国际制药协会ISPE基础指南06卷

A GUIDE FOR NEW FACILITIESVOLUME 6: BIOPHARMACEUTICALS EXECUTIVE SUMMARY OFDRAFT FOR REVIEWJANUARY 2003Copyright:This document is owned by ISPE. No reproduction of the whole or any part of this document is to be made without the written authority of ISPE.CONTENTS1INTRODUCTION1.1BACKGROUND1.2SCOPE OF THE GUIDE1.3KEY CONCEPTS OF THE GUIDE1.4USING THE GUIDE2THE REGULATORY BASIS FOR FACILITY REQUIREMENTS2.1EXECUTIVE SUMMARY2.2SCOPE2.3DEFINITIONS2.4REGULATORY CONSIDERATIONS2.5GENERAL CONCEPTS2.6BIBLIOGRAPHY2.7SIGNIFICANT REGULATORY DOCUMENTS3MANUFACTURING OPERATIONS AND ACTIVITIES3.1EXECUTIVE SUMMARY3.2OPEN VERSUS CLOSED SYSTEMS3.3PYROGEN-CONTROLLED PROCESSING3.4CONSIDERATIONS FOR MULTI-PRODUCT OPERATIONS3.5VIRAL CLEARANCE3.6STAGE OF PRODUCT DEVELOPMENT3.7OPERATIONAL UPSET3.8OPERABILITY AND MAINTAINABILITY3.9CLEANING AND HOUSEKEEPING CONSIDERATIONS4CHAPTER 4: PROCESS AND EQUIPMENT4.1EXECUTIVE SUMMARY4.2TYPICAL BIOPHARMACEUTICAL PROCESSES4.3CRITICAL PROCESS PARAMETERS4.4GENERAL CONSIDERATIONS FOR EQUIPMENT DESIGN4.5SPECIFIC EQUIPMENT DESIGN CONSIDERATIONS4.6SUMMARY5PROCESS SUPPORT AND UTILITIES5.1EXECUTIVE SUMMARY5.2REGULATORY ISSUES5.3SYSTEM IMPACT DESCRIPTIONS5.4SYSTEM LAYOUT AND ROUTING5.5SPECIFIC SERVICE CONSIDERATIONS6FACILITY6.1EXECUTIVE SUMMARY6.2PROCESS CONSIDERATIONS6.3OPERATIONAL CONSIDERATIONS6.4FACILITY LAYOUT CONSIDERATIONS6.5OPERATIONAL SUPPORT6.6AREA ENVIRONMENT6.7ARCHITECTURE AND FINISHES6.8DISCRETIONARY CONSIDERATIONS7PROCESS CONTROLS7.1EXECUTIVE SUMMARY7.2BIOPHARMACEUTICAL AUTOMATION ISSUES7.3LEVEL OF AUTOMATION7.4BIOPHARMACEUTICAL UNIT OPERATIONS7.5CONTROL SYSTEMS MAINTENANCE7.6VALIDATION OF BIOPHARMACEUTICAL AUTOMATION SYSTEMS8COMMISSIONING AND QUALIFICATION8.1EXECUTIVE SUMMARY8.2IMPACT ASSESSMENT8.3QUALIFICATION9GLOSSARY10APPENDIX - EUROPEAN ASPECTS10.1INTRODUCTION (GENERAL)10.2WATER QUALITY (REF: CHAPTER 4 AND 5)10.3BIO-CONTAINMENT AND ENVIRONMENTAL PROTECTION (SEE CHAPTER 5) 10.4ENVIRONMENTAL IMPACT ISSUES10.5CHROMATOGRAPHY SKID SHARING10.6QUALIFICATION AND VALIDATION (REF: CHAPTER 8)1 INTRODUCTION1.1 BACKGROUNDThe design, construction, commissioning, and qualification of biopharmaceutical facilities will challenge manufacturers, engineering professionals and equipment suppliers. These facilities must not only meet cGMP regulations but must comply with local codes, laws, and regulations.The current situation is one of confusion and sometimes little science:• •• ••• • Solutions are applied out of context (one product’s approaches inappropriately applied to a different type of product)Product and process are not considered in decisions. A common reason is “Company X did it, so we should, too.”Capital concerns:Capital funds may be limited, so wise use of funds is importantThe need to get quick facility approval at all costs has led to overspending to remove potentialsnags during inspectionsMoney is not going toward product protection as much as to “fluff”:Money that could have been used for protecting the product is diverted to features with noproduct impact:– Mirror finishes, “stainless steel” facilities– Confusion regarding required process water quality, often over-specified without economic or scientific justification– Classified spaces (cleanrooms) where they are not needed, as for closed processes1.2 SCOPE OF THE GUIDEThis guide may be used by industry for the design, construction, commissioning, and qualification of new biopharmaceutical facilities. It is neither a standard nor a GMP, nor is it a detailed design guide. It is not intended to replace governing laws or regulations that apply to facilities of this type. The application of this document for new or existing facilities is at the discretion of the facility owner or operator. Approaches to meeting GMP provided in this guide need not be retroactively applied to currently licensed facilities.This guide applies to large molecule biotech products, cell-cultured or fermented:It does not apply to blood, vaccines, etc. However, most concepts in this guide may be applied to these productsIt applies to biopharmaceutical Active Pharmaceutical Ingredient (API) products licensed by both CBER (Center for Biologics Evaluation and Research) and CDER (Center for Drug Evaluationand Research)U.S. GMPs:•• •• Not much is specifically stated in the U.S. GMPs but best practices are covered here. It is ultimately the owner’s responsibility to justify decisions and approaches to regulators.Other GMPs are covered in the Appendix.National Institute of Health (NIH) and other safety issues are mentioned in the guide where they affect GMPs or design.The audience for this guide is professionals involved in the design, construction, validation and operation of licensed biopharmaceutical manufacturing facilities:The mission of the Baseline® Guides is to help operating companies to satisfy the GMPs andproduce product in a manner that allows the manufacturer to stay in businessThis guide is not a GMP, but instead it focuses on use of resources to meet GMP. This Guide is but one approach to satisfying the intent of the GMPs. Other methods of protecting the productmay exist now or evolve in the future. If an issue is not covered in this guide, or if alternativesappear feasible, the reader is advised to discuss them with the appropriate regulatory agencies before significant financial commitments are made.It is intended that this Guide will be used by regulators and quality control to understand thetechnical issues regarding the facility or process. This Guide does not attempt to cover the basics of the engineering sciences, nor does it attempt to cover biopharmaceutical GMPs that do notaddress the facility or the manufacturing process technology.1.3 KEY CONCEPTS OF THE GUIDE1.3.1 Does the process equal product?There is a continuum of process and facility approaches based on the product and processes used to make the product. The best engineering solution makes optimal use of people, materials, and capital while protecting the product. There is not one “right” or “perfect” way to design and operate the facility. However, the design of a facility has a profound impact on process design and on how the facility is operatedDue to limitations in analytical methodologies and only superficial understandings of the relationships between process variables and final product quality, biopharmaceutical processes have historically been viewed as “black boxes”. Thus, there also has been a prevailing view that the “process equals the product”. This view has led to reluctance to alter biopharmaceutical processes, a reluctance that has been reinforced by conservative regulatory approaches. How could manufacturers assure the identity of the final product in the case of process variations? How could manufacturers assure the final product with changes in scale or changes in the facilities of manufacture?As the industry has developed a better understanding of biopharmaceutical processes and as analytical methods have improved, we have developed a better understanding of the “cause and effect” relationship between process variables and products. This evolution has caused a change in focus to those issues that are critical to the consistent manufacture of high quality products. Products and processes have been proven to be transportable between facilities and can be operated on different scales.1.3.2 Process design is tied to facility designThis Guide covers the variables that most directly affect the process and facility:•• • • •• • Open versus closed processing:– Closed processing places more emphasis on protecting the product INSIDE the process.– Open processing places more emphasis on the facility and its people.What works best for one product, facility, or process scale may not work best for another product, facility, or process scaleFeatures that work well in a single product facility may be inadequate for a multiple product facility Chapter 3 discusses these issues as well as viral clearance and clinical materials manufactureProcess controls:Automation is not a GMP requirement, but if automation is used then there are GMP implications.Chapter 7 provides more insight regarding automation.For subjects generic to all pharmaceutical facilities, the reader is directed to other sources for more in-depth information.Qualification basics are covered in ISPE Baseline® Guide for Commissioning and Qualification– Commission everything in accordance with Good Engineering Practice, but– Qualify only direct impact systems and critical components of those systems.– Design Qualification or Enhanced Design Review will help comply with ICH Q7A– Qualification considerations specific to Biopharmaceutical systems are covered in Chapter 8, with reference to topic-specific qualification activities in Chapters 3 through 7 Water and steam systems are covered in the ISPE Water and Steam Systems Baseline® Guide The Guide user is encouraged to work with the regulators to iron out “unique” issues before they become significant issues.1.3.3 Controlled ProcessingThe product must be protected by controlling the process and often its surroundings. This requires knowledge of the product and process and protection utilizing segregation and flow patterns. Chapter 3 discusses controlled processing in more detail.1.3.3.1 Know the Product (and its process)Intimate knowledge of the product, its critical parameters, the processes involved, and processing parameters is essential. Evaluation of potential contamination routes is needed. Data that demonstrate control of the process and to justify processing decisions will be key to a successful facility.1.3.3.2 The process can not add contaminationThe process’s contamination profile must be known and the process controlled to specifications.• Process Water should reflect the product purity profile.• Chapter 3 discusses recovery from upsets and prevention of contamination in manufacturing operations.1.3.3.3 Contamination control strategyAs discussed in Chapter 3, bulk biopharmaceutical manufacturing is low “bioburden” production. Aseptic-like processing steps or “sterile” processing operations utilizing sterilized process equipment are usually operated closed.Chapter 3 also discusses housekeeping, cleaning, and fumigation. Chapter 4 discusses equipment cleanability, and closure.1.3.4 Segregation and FlowSegregation protects the product from contamination in its surroundings (i.e., from the facility and other products). Segregation may be accomplished via procedures, timing, or by physical means. Flow patterns in the facility influence segregation, especially if more than one product is manufactured there. Chapter 6 provides more detail to help decision-making regarding segregation and flow.1.3.4.1 Primary and secondary segregation:As discussed in Chapters 2, 3, and 6, protection of product may be accomplished through primary and secondary segregation.Primary Segregation = used to mitigate a known risk of product contamination, usually supported by a strong GMP driver and process data. It is the foundation of the basic organization and operation of the facility and identifies process steps at risk.Secondary Segregation = used when there is little demonstrated risk to product, but segregation is desirable to minimize the risk of human error and mix-ups. There is no direct impact on product quality, but it helps define the facility and its operation, although more from a management standpoint than intrinsic process protection. Secondary segregation is open to interpretation as to applications and methodology.1.3.4.2 Flow and traffic patterns in the facilityImplementation of the segregation strategies results in “flow”• • • Flow patterns should address scale, volume and duration of expected traffic.Flow patterns should also address upset conditions (such as maintenance and change out of large equipment) and future construction.A mature materials handling philosophy must be in place before establishing flow patternsPhilosophies of primary and secondary segregation affect flow patterns: Raw materials flowProduct flow, including intermediates and hold pointsPersonnel flowGlass and equipment flow (through cleaning protocols)Waste flowFlow patterns may force issues with the cleanliness of the facility:• Materials of construction and architectural details• Building layout and potential contamination routes (via air, people, equipment)• Issues with cleaning of equipment and piping:– CIP/SIP– Wash facilities1.3.5 Open versus Closed Processing1.3.5.1 If a unit operation is demonstrated closed, it may operate in controlled non-classified(CNC) space.• • • •• • • Closed = segregation by physical means (hardware) to protect the product and process from contamination by the surrounding environment (outside the process)“Closure” and its measures must be defined by the Owner, and demonstrated to prevent contamination of the product.Various operating systems have varying degrees of closure, some may be absolute, while others also provide segregation, but to a lesser degree. The use of a “hard” definition may limit the understanding of “closed.”The surrounding room environment is not part of the equation for a closed process, but it should be “controlled”.1.3.5.2 If a unit operation is open, the product must be protected by other means.Open = not “closed”:Product = process + facilitySurrounding environment is a factor in the process.Either a classified space or a controlled non-classified environment will likely be needed, but the need for area monitoring is driven by the open process.The choice between closed processing in controlled non-classified (CNC) space and open processing in classified space is often driven by scale of the process, cost of operations, and value of product at risk. Chapter 4 provides information to help in selecting process equipment to meet open or closed requirements. Chapter 6 discusses the effects of process closure on the facility.1.3.6 Scale Affects DecisionsChapters 3, 4 and 5 deal with process design and support utility design issues connected with process scale, and Chapter 6 covers facility layout options.One size does not fit all. As scale of the process increases, there is a shift toward:• Vertical layouts with gravity flow of materialsMore closed operationsMore Primary segregationEquipment fixed in place (often dedicated)More automationControlled non-classified space instead of classified space (due to closed processing)Small process scales tend to include:• Horizontal process flow with pumps• Open operations• Segregation by time (campaigning)• Manual operations (mixing, etc.)• Less automationMore portable equipment, often shared with other productsMore need for classified spaces1.3.7 Single product vs. multiple products manufactureAs discussed in Chapter 3, when more than one product is manufactured in a facility, ensuring the products’ safety and quality becomes more difficult but no less important. Multi-product manufacturing facilities may segregate products by campaigning (one product at a time) or may process multiple products concurrently.• Campaigning depends heavily on validated cleaning and changeover procedures. (Chapter 3) Concurrent manufacturing must avoid cross-contamination through physical segregation and operating procedures. (Chapters 3 and 6)1.4 USING THE GUIDE1.4.1 Organization of the GuideAn overview of the Guide’s structure is shown in Figure 1-1.Appendix 6 Chap 14Process technologiesAppendix 7 Chap 15FacilityFigure1.1 Proposed structure of the final ISPE Baseline® Guide for BioPharmaceuticals1.4.2 Application of the GuideAs shown in Figure 1-1 it is necessary to begin by understanding the GMP requirements (Chapter 2) and then addressing the product and operational requirements (Chapter 3). From there, once operational concepts have been established, User Requirements defined, and perhaps even a Functional Design created, the discipline designers may begin detail design.Users of this Guide are advised to refer to other ISPE Baseline® Guides for more detailed or complementary information. For example, water and steam systems are thoroughly discussed in the ISPE Baseline® Guide for Water and Steam systems, and the design of classified pharmaceutical manufacturing space is discussed at length in the ISPE Baseline® Guide for Sterile Manufacturing Facilities.Users of this Guide are also encouraged to understand GMP and specific product requirements thoroughly before attempting facility design. Where there is conflict or a lack of understanding, manufacturers and engineers are encouraged to discuss concepts with the appropriate regulatory agency. Such early discussion opens dialogue and helps to settle potentially thorny issues.1-112 THE REGULATORY BASIS FOR FACILITY REQUIREMENTSDuring the design of new facilities every manufacturer faces numerous issues that may significantly affect the facility cost. These include process definition, process equipment requirements, the definition of a suitable manufacturing environmental quality to support manufacturing, water requirements, and facility layout. While some of the issues faced may affect the quality of the active pharmaceutical ingredient (API or bulk drug substance), others may have no impact.The primary element to be considered in a biotech facility is the ability of the facility and the process to protect, i.e., prevent contamination of, the API. Product protection issues may be addressed in a voluntary Product Protection Control Strategy.The evolution of facilities for manufacturing biopharmaceutical products has led to many extremes in size, complexity, and capital/resources. Processing approaches and designs suitable for a small-scale process are often inadequate or inappropriate for a large-scale facility. The multi-product facility will differ in certain key areas from either of these dedicated facilities.Specifically, each company should determine the appropriate requirements to provide adequate protection for its product(s) and, thereby, the requirements for the completed facility. No single solution or design fits all drug substances or products, since the decisions made and incorporated in the facility will depend upon:• Nature of the process and product (i.e., contamination-sensitive processes to less sensitive processes, open versus closed processing, etc.)• Scale and complexity of the process• Number and types of the products in the facilityThis Chapter addresses some of the significant process-related concepts and facility attributes with regulatory implications to be considered when designing a facility. Key points developed include: • There is not one universal “GMP” standard or approach to biopharmaceutical facility and process design. The nature of the product and its processes greatly influences thesedecisions. Systems and their components will have varying effect on each product.• Biotech manufacturing operations are not usually intended to produce a sterile drug substance, but rather one of low bioburden. Although the voluntary adoption of asepticmanufacturing techniques and facility standards have occurred in the industry, suchstandards are not required. The production process and facility should include theappropriate controls to prevent, limit, and detect API contamination.• Processes may be closed or open. Closed processing presents less risk to product and presents fewer demands on the facility design. Local controls may be used with openprocesses to provide protection of the product.• Multiple products segregated by appropriate procedural or physical means may be produced within a single facility.• Water used in manufacture should be appropriate to the process; WFI may not be scientifically necessary throughout the entire process for most products.3 MANUFACTURING OPERATIONS AND ACTIVITIESThis Chapter involves the operational aspects of a biopharmaceutical facility, as opposed to the physical design of the facility itself, and addresses key regulatory issues and concepts defined in Chapter 2. The Chapter addresses the impact of facility and equipment design decisions on manufacturing operations. Conversely, the Chapter also describes how operability and maintainability considerations should influence the design of a biopharmaceutical facility. Concerns and issues of production management, process operators and other plant support personnel are included. Important concepts addressed in this Chapter are as follows:• • •• • Operational and Procedural Controls can play an important role in protecting the product, and must be factored into the “open versus closed” design decision. Application of these types of controls with a well trained manufacturing staff can often be a better solution than over-engineering a system.“Bioburden-Controlled Processing” and “Pyrogen (Endotoxin)-Controlled Processing” are key operational concepts that have a significant impact on process and facility design, and both are distinctly different from sterile processing. Some features of traditional sterile design and operation may be employed but are typically not required to establish the appropriate level of control.Viral Clearance (Reduction and Inactivation) – Biopharmaceutical processes commonly use raw materials from biological sources, starting with the cell line and often extending to supplements added during the cell culture and purification stages. Cell lines used in the biotechnology industry are extensively characterized for identity and purity, and are tested for the presence of infectious agents. Nevertheless, it is still a regulatory requirement for manufacturers using mammalian cell culture-based processes to demonstrate adequate viral clearance during the manufacturing process. In addition, increasing concern over the transmission of prions from animal-sourced raw materials has required manufacturers to take additional measures to minimize the risk of such contamination. The decision on how/where to accomplish viral clearance can have an impact on the equipment design, and may affect the design and layout of the facility.Segregation is critical in any biopharmaceutical operation to ensure product protection. Traditional applications include:– Between organisms, products, or technologies– Between processing steps (e.g., upstream and downstream operations)– Between raw materials or products at various stages of quality control or process step– Between components or equipment at different stages of cleanlinessSegregation can be accomplished by procedure, by spatial separation (physical), by time (temporal), by environmental control, or by process design (system closure).In a Multi-Product Operation, products can be either campaigned or processed concurrently.For campaigned products, the focus is on cleaning validation, changeover procedures between products, and line clearance procedures. For concurrent product manufacture, the focus is onsegregation, procedural controls, and avoidance of cross-contamination. In all cases, the overall guiding principle is to ensure the quality and safety of the product.Manufacturing at Different Stages of Product Development is important for many biopharmaceutical companies, particularly those facing their first major capital investment in manufacturing facilities. While the regulations are clear in stating that GMP compliance is required for all stages of clinical development, it is also recognized that in most cases the manufacturing process is not completely defined during early-stage clinical work. It is important that process issues having significant impact on the facility design be locked down as early as possible. The emphasis of process/facility design and validation during early-stage clinical manufacturing should be placed on areas that have the greatest impact on product quality and consistency.4 PROCESS AND EQUIPMENTThe Chapter on Process and Equipment is primarily concerned with design aspects of biopharmaceutical processes and equipment, as opposed to the related operational aspects addressed in Chapter 3. More specifically, this Chapter deals with the design of biopharmaceutical process equipment, and associated piping and instrumentation, which contact a product or its components at a stage in the process where such contact could influence the quality, safety, purity, strength or identity of the ultimate product. The primary audience for this Chapter is process and equipment engineers.In general, biopharmaceutical processes are similar in that nearly all have fermentation/cell culture production steps, harvest steps, purification steps, formulation steps, and final bulk filling steps. Although manufacturing processes may differ, certain Critical Process Parameters are consistent from product to product, and certain key considerations for each processing step apply to all processes.Within each process step there are process considerations driven by the overall philosophy of the organization operating the process. The design approach that is chosen based on these considerations (GMP and business drivers) will result in a set of criteria to be used for both equipment selection and overall facility design. There is no single answer to the majority of the process considerations mentioned. However, the combinations of the choices and solutions will define reasonable, compliant process designs.Various types of equipment share similar design considerations and requirements. Specifically, cleanability/drainability, surface finish, materials of construction, shear generation, closure level, containment level, and pressure/temperature requirements must be considered for virtually any piece of equipment or device used in biological manufacturing. Improper consideration can lead to processing systems that are either not operable (placing product at risk) or are operationally inefficient (lower process yields).Key topics addressed in this Chapter are:• Simplified process flow diagrams of several typical biopharmaceutical processes.• Critical process parameters that are consistent from product to product. Key processing (critical) parameters for various processing steps are identified for typical unit operations.Critical process parameters, such as temperature, pH, conductivity, bioburden, endotoxin,product concentration, by-product levels, purity, and stability are generally similar fromprocess to process. However, the acceptance criteria, implications and applicable designoptions from process to process may vary significantly.• General considerations for equipment design – design considerations common to most biopharmaceutical unit operations.• General equipment considerations, such as materials of construction, cleanability, avoiding cross contamination, open vs. closed, process monitoring, safety, containment, andmaintenance, can be applied to most process equipment, and design considerations areoutlined. Similarly, there are design considerations applying specifically to general particularareas such as cell culture and purification. These are outlined as well in the form ofchecklists for the process and equipment engineer.。
review的名词

ReviewIntroductionA review, in its noun form, refers to a careful examination, assessment, or appraisal of something. It involves critically analyzing and evaluating a particular subject or object, such as a book, movie, product, or performance, to provide an informed opinion. Reviews play a crucial role in guiding consumers’ decisions, influencing trends, and shaping public perceptions. In this article, we will delve into the significance of reviews, their types, and their impact on variousaspects of our lives.Different Types of Reviews1. Product ReviewsProduct reviews are among the most common and impactful types of reviews. They are typically written by consumers who have used or purchased a specific product. These reviews assess the product’s quality, performance, features, and overall value for money. Product reviews help potential buyers make informed decisions, as they provide valuable insights into the pros and cons of the item.2. Movie and Book ReviewsMovie and book reviews provide critical analysis and evaluation of films and literary works. These reviews discuss various elements such as the plot, characters, writing style, direction, cinematography, or any other relevant aspect. The review writer offers their perspective on the strengths and weaknesses of the movie or book, helping readers orviewers decide whether to engage with the content.3. Restaurant and Travel ReviewsRestaurant and travel reviews are essential for individuals seeking new dining experiences or planning vacations. Travelers and food enthusiasts share their evaluations of restaurants, hotels, tourist attractions, and travel destinations. These reviews cover aspects like quality of service, ambiance, cleanliness, menu options, location, and value for money. Travel and restaurant reviews assist consumers in choosing suitable establishments and ensuring memorable experiences.4. Academic and Scientific Paper ReviewsIn the academic and scientific community, peer reviews play a vital role in determining the quality and credibility of research papers. Expertsin the field evaluate these papers based on their methodology, significance, originality, and adherence to ethical standards. Peer reviews ensure the accuracy and reliability of published research, enabling the advancement of knowledge and fostering scientific integrity.The Impact of ReviewsReviews hold significant influence over several aspects of our lives, including:1. Consumer Decision-MakingBefore making a purchase, many consumers rely on reviews to gather information about products or services. Positive reviews often lead to increased sales, while negative reviews may deter potential buyers. Reviews provide social proof and help consumers gain confidence in their choices, especially in the increasingly digital age. They help establish trust between consumers and businesses.2. Business Reputation and SuccessFor businesses, reviews can have a profound impact on their reputation and success. Positive reviews attract new customers, build credibility, and boost sales. On the other hand, a cluster of negative reviews can harm a company’s image and result in financial losses. Maintaining agood online reputation through proactive customer engagement and addressing concerns raised in reviews has become crucial for businesses of all sizes.3. Influencing Trends and PopularityPopular culture and trends are often shaped and influenced by reviews. Positive reviews can propel a book, movie, or product to new heights of popularity, resulting in increased demand and widespread recognition. Likewise, negative reviews can hinder the success of a creative project or discourage the adoption of a particular product. The collective opinion reflected in reviews can significantly impact societal tastes and preferences.4. Personal Growth and DevelopmentReviews also offer opportunities for personal growth and improvement. Whether it be professional feedback from a supervisor or constructive criticism from customers, reviews can highlight areas of strengths and weaknesses. Acknowledging feedback and implementing necessary changes can lead to enhanced performance, increased customer satisfaction, and personal development.How to Write an Effective ReviewWriting an effective review requires certain key elements to provide valuable insights and guidance. Here are some tips to consider:1.Be informative: Include relevant and specific details about thesubject of your review. Describe its features, functionality,performance, or any other pertinent information that would helpreaders form an accurate understanding.2.Provide balanced criticism: While it is essential to highlightboth positive and negative aspects, ensure your criticism is fair and constructive. Strive to offer suggestions for improvementinstead of solely focusing on the negative aspects.3.Consider the target audience: Tailor your review to meet theneeds and interests of the intended readers. For example, a reviewaimed at parents may emphasize different aspects than onetargeting technology enthusiasts.e clear and concise language: Make your review easy to read andcomprehend. Avoid excessive technical jargon or ambiguousstatements that might confuse readers.5.Provide supporting evidence: Back up your statements andevaluations with examples or specific details. This will lendcredibility to your review and help readers understand the basisfor your opinions.6.Be respectful and unbiased: Maintain a respectful tone throughoutthe review, even if you are critical. Avoid personal attacks orinflammatory language. Also, strive to be unbiased and transparent, especially if there are any potential conflicts of interest.ConclusionReviews are an essential part of our decision-making processes, allowing us to make informed choices in various aspects of our lives. Whether it be assessing products, movies, books, restaurants, or academic papers, reviews enable us to navigate the vast array of options available to us. They shape consumer behaviors, influence trends, impact businesses, and provide opportunities for personal growth. By understanding thedifferent types of reviews, their impact, and how to write an effective review, we can make the most of this valuable tool in today’s information age.。
epc 设计控制措施

epc 设计控制措施英文回答:Engineering and Production Change (EPC) Design Control Measures.EPC design control measures are an essential part of ensuring product quality and patient safety in the medical device industry. By implementing effective EPC design controls, manufacturers can minimize the risk of errors and defects, and ensure that their products meet the highest standards of performance and safety.Key EPC Design Control Measures.The following are some of the key EPC design control measures that manufacturers should consider:Design review and verification: A systematic and independent review of the design to ensure that it meetsthe requirements of the product specification and that it has been properly implemented.Design validation: A process to demonstrate that the design meets the user needs and intended use of the product.Design transfer: A process to transfer the design from the design team to the manufacturing team, ensuring thatall design information is accurately and completely transferred.Design change control: A process to control and manage changes to the design, ensuring that all changes are madein a controlled and documented manner.Design history file: A record of all design activities, including changes, that provides a complete history of the design.Benefits of Effective EPC Design Controls.Effective EPC design controls can provide the followingbenefits:Reduced risk of errors and defects.Improved product quality and patient safety.Greater efficiency and productivity.Enhanced regulatory compliance.Increased customer satisfaction.Implementing EPC Design Controls.Manufacturers can implement EPC design controls by following these steps:Establish a design control plan that outlines the procedures and responsibilities for design control.Train employees on the design control plan and ensure that they understand their roles and responsibilities.Develop and implement design review and verification procedures.Develop and implement design validation procedures.Develop and implement design transfer procedures.Develop and implement design change control procedures.Maintain a design history file.Conclusion.EPC design control measures are an essential part of ensuring product quality and patient safety in the medical device industry. By implementing effective EPC design controls, manufacturers can minimize the risk of errors and defects, ensure that their products meet the highest standards of performance and safety, and ultimatelyincrease customer satisfaction.中文回答:工程和生产变更 (EPC) 设计控制措施。
制定学习计划的重要性英语

制定学习计划的重要性英语IntroductionIn the fast-paced world we live in, where almost everything is scheduled and planned, it is essential for students to have a study plan in place to ensure they can achieve their academic goals. A study plan is a well-thought-out schedule that outlines the learning objectives, study materials, and timelines for accomplishing the set goals. It is a useful tool that helps students stay organized, focused, and motivated to excel in their studies. The importance of setting a study plan cannot be overstated, and in this essay, we will discuss the reasons why it is vital for students to have a study plan, the benefits that can be derived from it, and how to effectively create a study plan.Reasons for Setting a Study Plan1. Time Management: One of the primary reasons for setting a study plan is to effectively manage time. With the vast amount of academic material that students need to cover, it is crucial to allocate specific time for studying each subject. A study plan allows students to allocate their time according to the priority and difficulty level of the subjects, thereby ensuring that they do not cram at the last minute and avoid unnecessary stress.2. Goal Setting: Setting a study plan helps students to set clear and achievable academic goals. By breaking down their study tasks into smaller, manageable steps, students can track their progress and stay motivated to achieve their targets. This sense of accomplishment encourages students to stay focused and committed to their studies.3. Organization: A study plan helps students to stay organized by providing a structured approach to their learning. It helps them to stay on track with their syllabus, assignments, exams, and other academic commitments. By organizing their study materials and priorities, students can avoid feeling overwhelmed and scattered, leading to better concentration and performance.4. Prioritization: With the help of a study plan, students can prioritize their study tasks based on their importance and urgency. It allows them to focus on the most crucial subjects or topics first, thus ensuring that they allocate sufficient time and effort to the areas that require the most attention.5. Resource Management: Setting a study plan enables students to manage their study resources, including textbooks, notes, online resources, and study aids. By allocating specific resources to each study task, students can make the most of the available materials and enhance their understanding of the subject matter.6. Stress Management: A well-structured study plan can help students manage stress and anxiety related to their academic workload. By breaking down their study tasks into manageable chunks, students can avoid feeling overwhelmed and reduce the pressure of completing everything at once.Benefits of Setting a Study Plan1. Enhanced Productivity: A study plan helps students to increase their productivity by providing a clear roadmap for their studies. It allows them to focus on specific tasks at hand, thus minimizing distractions and procrastination.2. Improved Time Management: With a study plan in place, students can effectively manage their time and allocate it to different subjects and tasks. This results in better time utilization and increased efficiency in completing their study goals.3. Better Learning Outcomes: Setting a study plan enables students to study systematically and consistently, leading to better retention of information and understanding of the subjects. This, in turn, leads to improved academic performance and results.4. Reduced Stress and Anxiety: By having a study plan, students can avoid last-minute cramming and feel more in control of their studies. This leads to a reduction in stress and anxiety, thus promoting a more positive and productive study environment.5. Increased Motivation: A study plan provides a sense of direction and purpose, which motivates students to stay committed to their studies. As they achieve their study milestones, students feel a sense of accomplishment, which further fuels their motivation and drive to succeed.6. Enhanced Self-Discipline: Setting a study plan instills self-discipline in students by requiring them to adhere to the scheduled study times and priorities. This habit of self-discipline translates into better study habits and overall personal development.7. Improved Performance: Ultimately, a study plan leads to improved academic performance and results. By staying organized, focused, and committed to their studies, students are better prepared to excel in their examinations and achieve their learning objectives.How to Create an Effective Study Plan1. Set Clear and Realistic Goals: The first step in creating a study plan is to set clear and achievable goals. This could be in the form of studying a certain number of chapters, completing assignments, or preparing for exams. It is essential to be realistic and specific about the goals to ensure that they are attainable.2. Prioritize Subjects and Tasks: Identify the most important and urgent subjects or tasks that require immediate attention. Prioritize them in your study plan to ensure that you allocate sufficient time and effort to each one.3. Allocate Time for Each Task: Create a schedule that outlines the specific time allocated for studying each subject or task. Be mindful of your energy levels and concentration spans, and plan your study times accordingly.4. Break Down Study Tasks: Break down your study tasks into smaller, manageable steps. This could involve dividing the chapters, topics, or assignments into smaller components, making it easier to tackle and track your progress.5. Consider Study Environment: Take into account the study environment that works best for you. Whether it's a quiet room, a library, or a coffee shop, ensure that your study environment is conducive to focus and concentration.6. Include Breaks and Rewards: Incorporate short breaks and rewards into your study plan to avoid burnout and maintain motivation. Taking regular breaks and rewarding yourself after accomplishing study milestones can help keep you refreshed and engaged.7. Review and Adjust Regularly: Regularly review your study plan and make adjustments as necessary. It's essential to be flexible and adapt to changes in your study priorities and time allocations.ConclusionIn conclusion, setting a study plan is crucial for students to effectively manage their time, stay organized, and achieve their academic goals. By prioritizing subjects, allocating time, and breaking down study tasks, students can enhance their productivity, motivation, and learning outcomes. An effective study plan leads to better time management, stress reduction, improved performance, and overall personal development. Therefore, it is imperative for students to recognize the importance of setting a study plan and utilize this valuable tool to excel in their academic endeavors.。
Side-by-SideComparison

The mission of the NIH is to support science in pursuit of knowledge about the biology and behavior of living systems and to apply that knowledge to extend healthy life and reduce the burdens of illness and disability. As part of this mission, applications submitted to the NIH for grants or cooperative agreements to support biomedical and behavioral research are evaluated for scientific and technical merit through the NIH peer review system.
GEP 的基本概念

项目工程师 供应商
A
A
A
W
W&E
W
W
W
W&E
W
E
W
W&E
W
W
W
W&E
W
W&E
W
W
W
W
调试报告
• 针对URS 提出的要求 • 测试文件的格式与确认文件基本一致, • 由项目经理和工程经理批准, 但没有QA的批准 • 如果确认文件需要引用调试报告的内容,该部分的测试报告必须得到QA的批准
• 举例:水系统 SAT 和 IQOQPQ • 包装线的SAT 和IQOQPQ
• Commission 包括以下的工作: • 1。物理完成和检查 • 2。设置运行 • 3。调试和校准 • 4。测试和性能检测 • 5。计划和筹备,管理以上操作
Qualification
• 对于直接影响系统,为了满足FDA和其他监管部门的要求, GEP的实施应该依据确认 操作规范得到加强和完善。
• 确认操作规范包括: • 1。系统影响评估 • 2。QA的参与 • 3。严格的文件编制,严谨的文档管理,和一个有条理的批准程序 • 4。QA变更 程序 • 5。最终用户的参与 • 6。培训 • 7。使用确认原理鉴别什么系统应该确认,确认到什么程度,为什么确认,谁来确认 • 8。确定不需要检查的项及理由
GEP 的基本概念
• GEP (Good Engineering Practice)良好的工程规范 在项目全过程中使用已确定的工程方案和标准达到合பைடு நூலகம்的经济有效的结果。
GEP : 相关的设施 设备及公用设施,按照FDA相关的规定包括以下方面: 1。项目的设计和安装要充分考虑到现行GMP规范, 安全,健康,环保,功效学,操作
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
•Design complies with requirements of any relevant rules, guides, standards etc •GMP
– – – – – Size and layout personnel and material flows changing regimes avoidance of x-contamination surface finishes GLP chemical testing microbiological testing in-process testing areas retention sample stores Document retention areas
Informs
•QA/QC
– – – – – –
•Warehousing practice
etc.
Review vs. Structured Review
•Structured review documents that design has quality using design review criteria •provides positive statements of compliance rather than just a list of design problems •the engineering review process can be made explicit •is a vehicle for knowledge capture and checklist growth •use approach either as self-check or part of Peer Review process
DQ ASSESSMENT SHEET Project : Development : DQ
Note s : 1. 2. lis te d. 3. 4. This is a ge ne ric form inte nde d for c omple tion a s pa rt of a pe e r group re vie w . The re vie w e r is a dvis e d to a s s e s s the de s ign ge ne ra lly prior to a s s e s s me nt a ga ins t e a c h c rite ria The re vie w e r is re que s te d to provide a s ta te me nt of the a s pe c ts of the de s ign ins pe c te d unde r e a c h c rite ria . If the de s ign is una c c e pta ble a ga ins t a ny c rite ria this mus t be jus tifie d a nd a re c omme nda tion give n.
EDR IQ OQ
EDR IQ OQ How, Acceptance Criteria and PQ results for systems working together What, Why, Who for systems that work together
PQ Rationale
PQOverview NhomakorabeaSys tem Name :
1
Des ign Meets Performance Criteria
Comment :
Satis factory : Recommendation :
2
Des ign for Reliability
Comment :
Satis factory: Recommendation
EDR Concepts
• Structured review of the design of facilities, utilities and equipment. • Smart way to prepare for IQ and OQ activities. • Method of examination of the design
â
â â
Impact of the system System complexity Degree of Novelty or Familiarity
Regulatory context
Enhanced Design Review NOT a regulatory requirement, BUT
• • • • • • • EDR definition and concepts Regulatory context V model position Rationale - Link with QP EDR structure Tools Workshop
EDR Structure
• Document search
The European Commission working party on Control of Medicines and Inspections Annex 15
Draft 4 states:
Regulatory context
“Qualification and Validation should establish and provide documentary evidence that: • the premises, the supporting utilities, the equipment and the processes have been designed in accordance with the requirements of GMP. This constitutes DESIGN QUALIFICATION or DQ.”
3
Des ign For Commis s ioning & Tes ting
Comment :
Satis factory : Recommendation :
FMEA and FMECA
Function Failure Modes Several failure modes Failure Causes Several causes for each mode? Failure Detection Method Are each of these causes detectable ? Effect of Failure Consequence of failure? Failure Probability How probable is this failure cause? Detection Probability How sure can we be of detecting the failure cause? Severity Factor What level of risk does this present?
GEP EDR
• • • • • • • • • • • • • •
Design meets performance criteria Design for Reliability Design for Commissioning Design for Construction & Installation Design for Efficiency Design for Maintenance Design for Safety Design for Low Environmental Impact Degree of Innovation Use of Standard Solutions Comparison with Previous Designs Design Value Engineered Design for Expansion Design Minimises Risk to Product
Operational Qualification
Detail Design i.e. How to make EDR & Impact Assessment
IQ Test Plan (inc. PDI)
Installation Qualification
Implementation
Overview
â â â â â â
URS Technical, Procurement specifications Design drawings „Engineering‟ change control Design Development/Scope changes Review meetings minutes
• Index and archive
EDR Structure
• Re-evaluate Impact Assessment
â
â
Component Assessment Adjust Boundary of systems
• Review to verify that Design process links construction to the User Requirement Specification .
EDR Structure
•?
â
â
â
Have we gotten what we asked for ? Is the design robust ? Can we demonstrate traceability ?
Chapter Overview
• • • • • • • EDR definition and concepts Regulatory context V model position Rationale - Link with QP EDR structure Tools Workshop