β-Chloro-L-alanine_COA_26449_MedChemExpress
反相高效液相色谱法测定迷迭香中鼠尾草酚、鼠尾草酸和熊果酸的含量

反相高效液相色谱法测定迷迭香中鼠尾草酚、鼠尾草酸和熊果酸的含量作者:何默忠葛秀丹陈正收徐瑾朱珠来源:《上海医药》2009年第10期中图分类号:R282.71文献标识码:B文章编号:1006-1533(2009)10-0469-02迷迭香(Rosmarinus officinalis L.)系唇形科迷迭香属多年生草本植物,原产于欧洲及北非地中海沿岸,现在我国云南、湖南、四川、贵州等地也有种植。
迷迭香是一种多用途的经济作物,从中可提取抗氧化剂、迷迭香精油和医药中间体。
抗氧化剂是从迷迭香植物中提取得到的粉末状物质,主要成分是具有抗氧化功能的酚、酸、黄酮类成分,结构明确的活性成分有鼠尾草酸、鼠尾草酚、迷迭香酚等;迷迭香提取物具有高效、无毒的抗氧化效果,可广泛应用于食品、功能食品、香料、调味品和日用化工等行业中。
笔者以反相高效液相色谱法(HPLC)测定迷迭香中鼠尾草酚、鼠尾草酸和熊果酸的含量,重现性好,灵敏度高,操作简便,适用于迷迭香的质量控制。
1仪器与试药Agilent 1100高效液相色谱仪(美国);AE 240电子天平(METrLER TOLEDO,上海);RE 52-86A旋转蒸发仪(上海亚荣)。
8批样品经中南大学药学院鉴定,均为唇形科迷迭香属植物迷迭香。
甲醇为HPLC级,水为重蒸水,其余试剂均为分析纯。
鼠尾草酚和鼠尾草酸对照品购自SIC-MA公司;熊果酸购自中国药品生物制品检定所。
2实验方法与结果2.1色谱条件色谱柱:Kromasil 100-5C18(4.6 mm×250 mm,5μm);流动相:甲醇-0.2%磷酸溶液(82:18);流速为1 mL/min;紫外检测器测定鼠尾草酚和鼠尾草酸检测波长:284 mm,测定熊果酸检测波长:210 nm。
进样量10μL。
2.2对照品溶液的制备精密称取鼠尾草酚、鼠尾草酸对照品适量,加乙醇溶解,定容至25 mL量瓶中,其浓度分别为0.1和0.3 mg/mL,作为对照品混合贮备液。
海洋萜类

海洋萜类主要来源
海藻 海绵
腔肠动物 软体动物
海洋萜类化合物的结构特征
海洋生物萜类的基本结构与陆地天然萜类类似, 其分子中具有异戊二烯(C5H8) 的基本单位。
因为海洋高压、低营养、低温、无光照、高盐 等独特的环境,海洋生物萜类与陆地萜类在结 构上又有着明显的不同。
海洋萜类化合物的结构特征
在陆地生物体中,主要合成单萜;在海洋生物 体内主要生成分子量较高的萜类,特别是二萜、 二倍半萜。
海洋萜类 Marine Terpenoids
海洋萜类
海洋孕育了丰富的生物资源,特殊的环境使海 洋生物产生了多种独特的活性物质,这些都成 为人类保健和药品的天然宝库。其中萜类化合 物是最常见的海洋天然有机化合物。
到目前为止,从海洋生物中分离得到的萜类已 达千余种。海洋萜类主要来自海藻、海绵、腔 肠动物和软体动物。
海洋二萜类
从生长在希腊海域的海草 Cymodocea nodosa 中分离得到 4 个新的代谢物 其中 化合物D是从海洋生物中发现的第 1 个含溴原子的西松烷 (briarane) 型二萜化 合物
A
B
C
D
在对它们进行多药耐药性 (multidrug-resistant, MDR) 以及抗菌活性试验中发现化合物 A、B、C的活性均强于现有抗金葡球菌ATCC-25923 药物标准, 特别是化合物 C的活性 最强, 。由于它们结构比较简单, 或可成为通过结构修饰或化学合成开发抗菌药物的前体 化合物
海洋倍半萜
从生长在西太平洋巴布亚新几内亚海域海绵Diacarnus levii 中分 离得到的具有细胞毒性的降倍半萜类化合物diacarnoxides A、B
A:R=CH3 B :R=H Diacarnoxide B 是第 1 个与广泛应用于研究缺氧活化细胞毒素 替拉扎明 (tirapazamine) 结构完全不同的、新类型的缺氧细胞 毒素, 研究还发现其结构中的过氧基团会使活性增加
参与抗生素生物合成的FADH_2依赖型卤化酶研究进展

参与抗生素生物合成的FADH_2依赖型卤化酶研究进展李航;朱丽;陈代杰【摘要】从发现第一个天然卤化物到现在已有100多年了.在已发现的约4500个天然卤化物中有很多在医药等领域应用广泛,其中包括许多重要的抗生素.直到19世纪90年代中期,人们一直认为生物卤化反应主要由卤过氧化物酶负责催化.近期在许多关于抗生素生物合成基冈簇的研究中发现FADH_2依赖型卤化酶催化的卤化反应殖是许多微生物及其它生物中的主要卤化机制.本文综述了参与抗生素生物合成的FADH_2依赖型卤化酶的发现,催化机制及各种来源的该类卤化酶研究进展,并介绍了该类酶在组合生物合成及寻找天然卤化物等方面的应用.【期刊名称】《中国抗生素杂志》【年(卷),期】2010(035)001【总页数】6页(P1-6)【关键词】FADH_2依赖型卤化酶;抗生素;卤化物【作者】李航;朱丽;陈代杰【作者单位】上海医药工业研究院,上海,200040;上海来益生物药物研发中心,上海,201203;上海医药工业研究院,上海,200040【正文语种】中文【中图分类】TQ465从发现第一个天然卤化代谢物到现在已有100多年了,很长一段时间内该类化合物被认为是很特殊的一类化合物[1]。
直到1960年,被发现的天然卤化物才共有29个[2]。
随着该类化合物广泛的应用于医药,杀虫剂等领域,人们对该类化合物越来越感兴趣,目前为止共发现天然卤化物约4500个[3]。
该类化合物由许多不同的生物产生,且其中很多有较强的生物活性,例如:由土壤细菌产生的万古霉素[4],由哺乳动物甲状腺产生的甲状腺素[5],由藻青菌产生的抗肿瘤物质自念珠藻环肽A等[6]。
在这些卤化物中,陆地环境中发现的主要为氯化物,而海洋生物中发现的主要为溴化物。
天然氟化物则较稀少,仅在一些植物和细菌代谢物中被发现[7]。
随着越来越多的卤化物被发现,人们对相关的卤化酶及其作用机制的研究也越来越感兴趣。
从1966年在Caldariomyces fumago中发现第一个卤化酶[8]以来,目前为止共有4大类的卤化酶被发现,分别为:卤过氧化物酶(haloperoxidase)[9],非血红素Fe2+α-酮戊二酸及O2依赖型卤化酶(non-heme FeII αketoglutarate- and O2-dependent halogenase)[10],S-腺苷甲硫氨酸依赖型氯化酶(S-adenosyl-L-methionine dependent chlorinase)[11]和FADH2依赖型卤化酶(FADH2-dependent halogenase)。
大肠杆菌高产L丙氨酸

APPLIED GENETICS AND MOLECULAR BIOTECHNOLOGYProduction of L-alanine by metabolically engineered Escherichia coliXueli Zhang&Kaemwich Jantama&J.C.Moore&K.T.Shanmugam&L.O.IngramReceived:23May2007/Revised:13August2007/Accepted:16August2007/Published online:15September2007 #Springer-Verlag2007Abstract Escherichia coli W was genetically engineered to produce L-alanine as the primary fermentation product from sugars by replacing the native D-lactate dehydroge-nase of E.coli SZ194with alanine dehydrogenase from Geobacillus stearothermophilus.As a result,the heterolo-gous alanine dehydrogenase gene was integrated under the regulation of the native D-lactate dehydrogenase(ldhA) promoter.This homologous promoter is growth-regulated and provides high levels of expression during anaerobic fermentation.Strain XZ111accumulated alanine as the primary product during glucose fermentation.The methyl-glyoxal synthase gene(mgsA)was deleted to eliminate low levels of lactate and improve growth,and the catabolic alanine racemase gene(dadX)was deleted to minimize conversion of L-alanine to D-alanine.In these strains,re-duced nicotinamide adenine dinucleotide oxidation during alanine biosynthesis is obligately linked to adenosine triphosphate production and cell growth.This linkage provided a basis for metabolic evolution where selection for improvements in growth coselected for increased glycolytic flux and alanine production.The resulting strain, XZ132,produced1,279mmol alanine from120g l−1 glucose within48h during batch fermentation in the mineral salts medium.The alanine yield was95%on a weight basis(g g−1glucose)with a chiral purity greater than99.5%L-alanine.Keywords Alanine.Fermentation.E.coli.Evolution. GlycolysisIntroductionWorldwide production of L-alanine has been estimated at 500tons per year(Ikeda2003).In pharmaceutical and veterinary applications,L-alanine is used with other L-amino acids as a pre-and postoperative nutrition therapy(Hols et al.1999).Alanine is also used as a food additive because of its sweet taste(Lee et al.2004).The use of L-alanine is limited in part by the current high cost.L-Alanine is pro-duced commercially by the enzymatic decarboxylation of L-aspartic acid using immobilized cells or cell suspensions of Pseudomonas dacunhae as a biocatalyst with a yield greater than90%(Shibatani et al.1979).The substrate for this enzymatic production process,L-aspartate,is usually pro-duced from fumarate by enzymatic catalysis with aspartate ammonia-lyase.Fumaric acid is produced primarily from petroleum,a nonrenewable feedstock.An efficient fermen-tative process with a renewable feedstock such as glucose offers the potential to reduce L-alanine cost and facilitate a broad expansion of the alanine market into other products.Alanine is a central intermediate(Fig.1)and an essential component of cellular proteins.Most microorganisms produce alanine only for biosynthesis using a glutamate–pyruvate transaminase(Hashimoto and Katsumata1998). Some organisms such as Arthrobacter oxydans(Hashimoto and Katsumata1993;Hashimoto and Katsumata1998; Hashimoto and Katsumata1999),Bacillus sphaericus (Ohashima and Soda1979),and Clostridium sp.P2Appl Microbiol Biotechnol(2007)77:355–366DOI10.1007/s00253-007-1170-yElectronic supplementary material The online version of this article (doi:10.1007/s00253-007-1170-y)contains supplementary material, which is available to authorized users.X.Zhang:J.C.Moore:K.T.Shanmugam:L.O.Ingram(*) Department of Microbiology and Cell Science,University of Florida,Box110700,Gainesville,FL32611,USAe-mail:ingram@K.JantamaDepartment of Chemical Engineering,University of Florida, Gainesville,FL32611,USA(Orlygsson et al.1995)produce alanine from pyruvate and ammonia using an reduced nicotinamide adenine dinucleo-tide (NADH)-linked alanine dehydrogenase (ALD).How-ever,fermentations are slow,and yields from the best natural producers are typically 60%or less because of coproduct formation (Hashimoto and Katsumata 1998;Table 1).Plasmid-borne genes encoding NADH-linked ALD have been tested as an approach to develop improved biocatalysts with varying degrees of success (Table 1).Engineered strains of Zymomonas mobilis CP4expressing the B.sphaericus alaD gene produced low levels of racemic alanine during the anaerobic fermentation of 5%glucose (Uhlenbusch et al.1991).A native chromosomal lactate dehydrogenase gene (ldhA )-deleted strain of Lactococcus lactis containing a mutation in alanine racemase was engineered in a similar fashion and produced 12.6g l −1L -alanine from 1.8%glucose (Hols et al.1999).An Escherichia coli aceF ldhA double mutant containing pTrc99A-alaD plasmid produced 32g l −1racemic alanine in 27h during a two-stage (aerobic and anaerobic)fermentation with a yield of 0.63g alanine g −1glucose (Lee et al.2004).With further gene deletions and process optimization,the racemic alanine titer wasincreasedFig.1Alanine pathway in recombinant E.coli .a Native and recom-binant fermentation pathways.The foreign gene,G.stearothermophilus alaD ,is shown in bold .G.stearothermophilus alaD coding region and transcriptional terminator were integrated into the native ldhA gene under transcriptional control of the ldhA promoter.Solid stars represent deletions of native genes in XZ132.Note that the native biosynthetic route for alanine production is omitted for simplicity.ackA Acetate kinase,adhE alcohol/aldehyde dehydrogenase,alaD alanine dehydro-genase (Geobacillus stearothermophilus XL-65-6),aldA aldehyde dehydrogenase A,aldB aldehyde dehydrogenase B,alr alanine race-mase 1,dadX alanine racemase 2,frd fumarate reductase,gloA glyoxalase I,gloB glyoxalase II,gloC glyoxalase III,ldhA D -lactate dehydrogenase,mdh malate dehydrogenase,mgsA methylglyoxal synthase,pflB pyruvate –formate lyase,ppc phosphoenolpyruvate carboxylase,pta phosphate acetyltransferase.b Coupling of ATP production and growth to NADH oxidation and L -alanine production.Glucose is metabolized to pyruvate,ATP,and NADH.Energy conserved in ATP is utilized for growth and homeostasis,regenerating ADP.NADH is oxidized by alanine formation allowing glycolysis and ATP production to continueT a b l e 1C o m p a r i s o n o f a l a n i n e -p r o d u c i n g s t r a i n sO r g a n i s m s M o d i f i e d p r o p e r t yM e d i a ,s u b s t r a t e a n d p r o c e s s c o n d i t i o n sT i m e (h )A l a n i n e (g l −1)Y i e l d (%)L -A l a n i n ep u r i t y (%)R e f e r e n c e E .c o l i X Z 132I n t e g r a t e d G .s t e a r o t h e r m o p h i l u s a l a D ;Δp f l ,Δa c k A ,Δa d h E ,Δl d h A ,Δm g s A ,Δd a d XM i n e r a l m e d i u m ,b a t c h ,g l u c o s e 120g l −148.0114.095>99T h i s s t u d yA r t h r o b a c t e r o x y d a n s H A P -1M i n e r a l m e d i u m ,t w o -s t a g e f e d -b a t c h ,g l u c o s e 150g l −1120825560.0H a s h i m o t o a n d K a t s u m a t a 1998A .o x y d a n s D A N 75A l a n i n e r a c e m a c e d e f i c i e n tM i n e r a l m e d i u m ,t w o -s t a g e f e d -b a t c h ,g l u c o s e 150g l −1,0.2g l −1D -a l a n i n e120775198H a s h i m o t o a n d K a t s u m a t a 1998E c o l i A L 1(p O B P 1)P l a s m i d w i t h A .o x y d a n s H A P -1a l a D M i n e r a l m e d i u m ,g l u c o s e 20g l −1,l i m i t e d o x y g e n40841N o t r e p o r t e dK a t s u m a t a a n d H a s h i m o t o 1996C o r y n e b a c t e r i u m g l u t a m i c u m A L 107(p O B P 107)P l a s m i d w i t h A .o x y d a n s H A P -1a l a DC o r n s t e e p l i q u o r ,g l u c o s e 200g l −1,4g l −1D L -a l a n i n e ,l i m i t e d o x y g e n 707136>99K a t s u m a t a a n d H a s h i m o t o 1996Z y m o m o n a s m o b i l i s C P 4(p Z Y 73)P l a s m i d w i t h B .s p h a e r i c u s I F O 3525a l a D M i n e r a l s a l t s m e d i u m ,s i m p l e b a t c h ,g l u c o s e 50g l −126816N o t r e p o r t e dU h l e n b u s c h e t a l .1991L a c t o c o c c u s l a c t i s N Z 3950(p N Z 2650)P l a s m i d w i t h B .s p h a e r i c u s I F O 3525a l a D Δl d h AR i c h m e d i u m (M 17),g l u c o s e 18g l −117137085–90H o l s e t a l .1999L .l a c t i s P H 3950(p N Z 2650)P l a s m i d w i t h B .s p h a e r i c u s I F O 3525a l a D Δl d h A ,Δa l rR i c h m e d i u m (M 17),g l u c o s e 18g l −1,0.2g l −1D -a l a n i n e 17N o t k n o w nN o t k n o w n >99H o l s e t a l .1999E .c o l i A L S 887(p T r c 99A -a l a D )P l a s m i d w i t h B .s p h a e r i c u s I F O 3525a l a D Δl d h A ,Δa c e FY e a s t e x t r a c t ,t w o -s t a g e b a t c h ,g l u c o s e 50g l −1,a e r o b i c a i r 1l m i n −1273263N o t r e p o r t e d L e e e t a l .2004E .c o l i A L S 929(p T r c 99A -a l a D )P l a s m i d w i t h B .s p h a e r i c u s I F O 3525a l a D Δp f l ,Δp p s ,Δp o x B ,Δl d h A ,Δa c e E FY e a s t e x t r a c t a n d c a s a m i n o a c i d s ,t w o -s t a g e b a t c h (a e r o b i c c e l l g r o w t h a n d a n a e r o b i c f e r m e n t a t i o n )223486N o t r e p o r t e d S m i t h e t a l .2006E .c o l i A L S 929(p T r c 99A -a l a D )P l a s m i d w i t h B .s p h a e r i c u s I F O 3525a l a D Δp f l ,Δp p s ,Δp o x B ,Δl d h A ,Δa c e E FY e a s t e x t r a c t a n d c a s a m i n o a c i d s ,t w o -s t a g e f e d -b a t c h (a e r o b i c c e l l g r o w t h a n d a n a e r o b i c f e r m e n t a t i o n )4888100N o t r e p o r t e d S m i t h e t a l .2006to88g l−1in a more complex process with yields ap-proaching the theoretical maximum(Smith et al.2006). However,this strain produced only racemic alanine,utilized multicopy plasmids requiring antibiotic selection,and required complex media with a complex multistage fermen-tation process(Smith et al.2006).In this study,we developed novel biocatalysts that pro-duce chirally pure L-alanine in batch fermentations without using plasmid-containing biocatalysts,antibiotics,or com-plex nutrients.The resulting strains are based on a deriva-tive of E.coli W(strain SZ194)that produces D-lactate (Zhou et al.2006b).The ldhA gene in SZ194was replaced with a single,chromosomally integrated copy of the ALD gene from the thermophile,Geobacillus stearothermophilus XL-65-6(formerly B.stearothermophilus;Lai and Ingram 1993).After additional deletions of alanine racemase (dadX)and methylglyoxal synthase(mgsA)and metabolic evolution,the resulting strain produced L-alanine at high titers(over1M)and yields in batch fermentations using the mineral salts medium.Materials and methodsStrains,plasmids,media,and growth conditionsThe strains and plasmids used in this study are listed in Table2.Strain SZ194was previously engineered from a derivative of E.coli W(ATCC9637)and served as a starting point for constructions(Zhou et al.2006b).G. stearothermophilus XL-65-6(Lai and Ingram1993)was used for cloning the ALD gene.During sequencing of chro-mosomal genes,we discovered a20-year-old error in culture labeling.Strain SZ194,the parent used to construct the alanine strains,is a derivative of E.coli W(ATCC9637). Other constructs for ethanol production and lactate produc-tion that have been reported previously as derivatives of E. coli B are now known to be derivates of E.coli W(ATCC 9637).Primers used in this study are listed in Table3.During strain construction,cultures were grown aerobi-cally at30,37,or39°C in Luria broth(10g l−1Difco tryptone,5g l−1Difco yeast extract,and5g l−1NaCl) containing2%(w/v)glucose or5%(w/v)arabinose. Ampicillin(50mg l−1),tetracycline(12.5mg l−1), kanamycin(50mg l−1),or chloramphenicol(40mg l−1) were added as needed.For initial tests of fermentative alanine production,strains were grown without antibiotics at37°C in NBS mineral salts medium(Causey et al.2004) supplemented with100mM ammonia sulfate,1mM betaine,and2%(w/v)glucose.Fermentation experiments (2–12%sugar)were carried out in NBS medium and AM1 medium(Martinez et al.2007).Broth was maintained at pH 7by the automatic addition of5M NH4OH.Genetic methodsStandard methods were used for genomic deoxyribonucleic acid(DNA)extraction(Qiagen,Valencia,CA),polymerase chain reaction(PCR)amplification(Stratagene,La Jolla CA,and Invitrogen,Carlsbad,CA),transformation,plas-mid extration(Qiagen),and restriction endonuclease diges-tion(New England Biolabs,Ipswich,MA).Methods for foreign gene(alaD)integration and for chromosomal gene (mgsA and dadX)deletion are described below.DNA sequencing was provided by the University of Florida Interdisciplinary Center for Biotechnology Research.The Biocyc and Metacyc databases(Karp et al.2005)were instrumental in the design and completion of these studies. Cloning the alanine dehydrogenase gene alaD from G. stearothermophilus XL-65-6and detection of the enzyme activityThe primers for amplifying alaD from G.stearothermophilus XL-65-6were designed based on the alaD sequence of G. stearothermophilus strain10.The forward primers(5′–3′GGAAAAA GGAGGAAAAAGTG ATGAAGATCGG CATT)included the ribosomal-binding region(bold)and the amino terminus(italicized).The reverse primer(5′–3′GAA GGAGTTGATCATTGTTTAACGAGAGAGG)was down-stream from the putative transcriptional terminator region (Table3).ALD was verified in clones using an activity stain (Kuroda et al.1990).E.coli TOP10F′harboring plasmids containing alaD was grown on Luria–Bertani(LB)plates at 37°C,then transferred to a Whatman7.0-cm filter paper. The filter was immersed in10mM potassium phosphate buffer(pH7.2)and incubated for20min at80°C for lysis of the cells and denaturation of the E.coli proteins.The dried filter paper was assayed in a reaction mixture containing50mM L-alanine,50mM Tris–HCl buffer (pH9.0),0.625mM NAD+,0.064mM phenazine metho-sulfate,and0.24mM nitro blue tetrazolium.The cells with ALD appeared as blue spots on the filter.Integration of alaD into E.coli SZ194The alaD gene was integrated into the chromosomal ldhA gene of SZ194.The fragment(Sma I–Kpn I,1.7kb)con-taining a tet gene flanked by two FRT sites was isolated from pLOI2065and cloned into pLOI4211between a unique Bam HI site(Klenow-treated)and Kpn I site to produce plasmid pLOI4213(6.0kb).In this plasmid,transcription of alaD and tet are oriented in the same direction.The Apa I(treated with T4DNA polymerase to produce a blunt end)–Kpn I fragment(2.2kb)containing alaD and tet was isolated from pLOI4213and cloned into pLOI2395Table2 E.coli strains and plasmids used in this studyRelevant characteristics Source or referenceStrainsSZ194plfB frd adhE ackA deletions Zhou et al.2006bXZ103-110SZ194,ldhA::FRT-tet-FRT::This studyG.stearothermophilus alaDXZ111XZ105,ldhA::G.stearothermophilus alaD This studyXZ112XZ111,metabolic evolution in NBS medium with2%glucose This studyXZ113XZ112,metabolic evolution in NBS medium with5%glucose This studyXZ115XZ113,metabolic evolution in NBS medium with8%glucose This studyXZ121XZ115,mgsA deletion This studyXZ123XZ121,metabolic evolution in NBS medium with8%glucose This studyXZ126XZ123,dadX deletion This studyXZ129XZ126,metabolic evolution in NBS medium with8%glucose This studyXZ130XZ129,metabolic evolution in AM1medium with8%glucose This studyXZ131XZ130,metabolic evolution in AM1medium with10%glucose This studyXZ132XZ131,metabolic evolution in AM1medium with12%glucose This studyPlasmidspCR2.1-TOPO bla kan;TOPO TA cloning vector InvitrogenDatsenko and Wanner2000pKD46Blaγβexo(Red recombinase),temperature conditionalpSC101repliconpFT-A Bla flp,temperature conditional pSC101replicon Posfai et al.1997pEL04cat-sacB targeting cassette Lee et al.2001;Thomason et al.2005 pLOI2224kan;R6K conditional integration vector Martinez-Morales et al.1999pLOI2065bla;FRT-tet-FRT cassette Zhou et al.2003bpLOI2395bla;ldhA franked by two Asc I site Zhou et al.2003apLOI3421 1.8kbp SmaI fragment containing aac Wood et al.2005pLOI4151bla cat;cat-sacB cassette This studyalaD integrationThis studypLOI4211bla kan alaD;alaD(PCR)from G.stearothermophilus XL-65-6cloned into pCR2.1-TOPO vectorpLOI4213bla kan;alaD-FRT-tet-FRT Kpn I-Sma I fragment(FRT-tet-FRT)This studyfrom pLOI2065cloned into Kpn I-BamH I(blunted)site of pLOI4211This studypLOI4214bla kan;ldhA’-alaD-FRT-tet-FRT-ldhA”Apa I(blunted)-Kpn I fragment(alaD-FRT-tet-FRT)from pLOI4213cloned into ldhA at Hinc II-Kpn Isites of pLOI2395This studypLOI4215kan;ldhA’-alaD-FRT-tet-FRT-ldhA”Asc I fragment(ldhA’-alaD-FRT-tet-FRT-‘ldhA)from pLOI4214cloned into Asc I sites of pLOI2224mgsA deletionThis studypLOI4228bla kan;yccT’-mgsA-helD’(PCR)from E.coli W clonedinto PCR2.1-TOPO vectorThis studypLOI4229cat-sacB cassette PCR amplified from pLOI4151(Eco RV digested)cloned into mgsA in pLOI4228This studypLOI4230PCR fragment amplified from pLOI4228(using mgsA-1/mgsA-2primers),kinase treated,and self-ligateddadX deletionThis studypLOI4216bla kan;dadA’-dadX-cvrA’(PCR)from E.coli W clonedinto PCR2.1-TOPO vectorpLOI4218cat-sacB cassette PCR amplified from pLOI4151(Eco RV digested)This studycloned into dadX in pLOI4216This studypLOI4220PCR fragment amplified from pLOI4216(using dadX-4/dadX-5primers),kinase treated,and self-ligated(Hinc II to Kpn I sites)to produce pLOI4214(6.5kb).In this plasmid,ldhA ,alaD ,and tet genes are transcribed in the same direction.The Asc I fragment (4.3kb)containing these three genes was isolated from pLOI4214and cloned into the R6K integration vector pLOI2224to produce pLOI4215(6.2kb).Plasmid pLOI4215contains resistance genes for both tetracycline and kanamycin (Fig.2).The Asc I fragment (4.3kb)containing ldhA ,alaD ,and tet genes was isolated from pLOI4215,further cut by Xmn I to eliminate any remaining uncut plasmid DNA,and electroporated into SZ194containing the Red recombinase plasmid pKD46(Datsenko and Wanner 2000).Integrants were selected for tetracycline resistance,confirmed by sensitivity to kanamycin and ampicillin and by PCR analysis using the primers of ldhA and its neighboring genes ydbH and hslJ (Table 3).Deletion of mgsA and dadX genesA modified method for deleting E.coli chromosomal genes was developed using two steps of homologous recom-bination (Thomason et al.2005).With this method,no antibiotic genes or scar sequences remain on the chromo-some after gene deletion.In the first recombination,part of the target gene was replaced by a DNA cassette containing a chloramphenicol resistance gene (cat )and levansucrase gene (sacB ).In the second recombination,the cat –sacBcassette was removed by selection for resistance to sucrose.Cells containing the sacB gene accumulate levan during incubation with sucrose and are killed.Surviving recombi-nants are highly enriched for loss of the cat –sacB cassette.A new cassette was constructed as a template to facilitate gene deletions.The cat –sacB region was amplified from pEL04(Lee et al.2001;Thomason et al.2005)by PCR using the JM catsacB up Nhe I and JM catsacB down Nhe I primers (Table 3),digested with Nhe I,and ligated into the corresponding site in pLOI3421to produced pLOI4151.The cat –sacB cassette was amplified by PCR using pLOI4151as a template with the cat -up2and sacB -down2primers (Eco RV site included in each primer),digested with Eco RV ,and used in subsequent ligations.The mgsA gene and neighboring 500-bp regions (yccT ′–mgsA –helD ′,1,435bp)were amplified using the mgsA -up and mgsA -down primers and cloned into the pCR 2.1-TOPO vector (Invitrogen)to produce plasmid pLOI4228.A 1,000-fold diluted plasmid preparation of this plasmid served as a template for inside-out amplification using the mgsA -1and mgsA -2primers (both within the mgsA gene and facing outward).The resulting 4,958-bp fragment containing the replicon was ligated to the Eco RV-digested cat –sacB cassette from pLOI4151to produce pLOI4229(Fig.3a).This 4,958-bp fragment was also used to construct a second plasmid,pLOI4230(Fig.3b),by phosphorylation and self-ligation.In pLOI4230,the central region of mgsA is deleted (yccT ′–mgsA ′–mgsA ″–helD ′).After digestion of pLOI4229and pLOI4230with Xmn I (within the vector),each served as a template for amplifica-tion using the mgsA -up and mgsA -down primers to produce linear DNA for integration step 1(yccT ′–mgsA ′–cat –sacB –mgsA ″–helD ′)and step II (yccT ′–mgsA ′–mgsA ″–helD ′),respectively.After electroporation of the step 1fragment into XZ115containing pKD46(Red recombinase)and 2h ofTable 3Primers used in this study Primers SequencealaD -forward GGAAAAAGGAGGAAAAAGTGATGAA GATCGGCATTalaD -reverse GAAGGAGTTGATCATTGTTTAACGA GAGAGGldhA -forward AGTACCTGCAACAGGTGAAC ldhA -reverse CAGGCGACGGAATACGTCAT ldhA -up (ydbH )CTGATAACGCAGTTGCTGGA ldhA -down (hslJ )TTCATTAAATCCGCCAGCTTJM catsacB up NheI TTAGCTAGCATGTGACGGAAGATC ACTTCGJM catsacB down NheI CCGCTAGCATCAAAGGGAAAACTGT CCATATcat -up2AGAGAGGATATCTGTGACGGAAGAT CACTTCGsacB -down2AGAGAGGATATCGAATTGATCCGGT GGATGACmgsA -up CAGCTCATCAACCAGGTCAA mgsA -down AAAAGCCGTCACGTTATTGG mgsA -1AGCGTTATCTCGCGGACCGT mgsA -2AAGTGCGAGTCGTCAGTTCC dadX -up AGGCTACTCGCTGACCATTC dadX -down GGTTGTCGGTGACCAGGTAG dadX -4TGGGCTATGAGTTGATGTGC dadX -5CTGTATCGGACGGGTCATCTFig.2Integration vector used for chromosomal insertion of G.stearothermophilus alaD into E.coli ldhA .Sequence encoding the N-terminal and C-terminal regions are designated ldhA ′and ldhA ″,respectivelyincubation at 30°C to allow expression and segregation,recombinants were selected for chloramphenicol (40mg l −1)and ampicillin (50mg l −1)resistance in Luria broth at 30°C (18h).Three clones were selected,grown in Luria broth containing ampicillin and 5%(w/v)arabinose (to induce expression of red recombinase),and prepared for electro-poration.After electroporation with the step 2fragment,cells were incubated at 30°C for 4h and then transferred into a 250-ml flask containing 100ml of modified LB (100mM 3-(N -morpholino)propanesulfonic acid [MOPS]buffer added and NaCl omitted)containing 10%sucrose.After overnight incubation (30°C),clones were selected on modified LB plates (no NaCl;100mM MOPS added)containing 6%sucrose (39°C,16h).Resulting clones were tested for loss of ampicillin and chloramphenicol resistance.Construction was confirmed by PCR using the mgsA-up/down primer set.A clone containing a deletion in the central region of mgsA was selected and designated XZ121.The dadX gene was deleted in a manner analogous to that used to delete the mgsA gene.Primers for dadX deletion are shown in Table 3,and the corresponding plasmids are shown in Table 2.FermentationNBS mineral salts medium (Causey et al.2004)with 1mM betaine (Zhou et al.2006a )was used in the initial fermentation (pH 7.0).Preinoculum was grown by inocu-lating three colonies into a 250ml flask (100ml NBS medium,2%glucose,and 100mM ammonium sulfate).After 16h (37°C,120rpm),this preinoculum was diluted into 500-ml fermentation fleakers containing 300ml NBS medium (2–8%glucose,100mM ammonium sulfate,and 1mM betaine)with 33mg cell dry weight (CDW)l −1.In early experiments,pH was maintained at 7.0by automat-ically adding 2M potassium hydroxide.In later experi-ments,5M ammonium hydroxide was used to maintain pH,and a low salt medium,AM1(Martinez et al.2007),was used to replace the NBS medium for fermentation (8–12%glucose).AM1medium contains much less salt and has been optimized for E.coli .Metabolic evolutionCells from pH-controlled fermentations were serially transferred at 24-h intervals to facilitate metabolic evolution through competitive,growth-based selection (Fig.1b).At the beginning,sequentially transferred cultures were inoc-ulated with an initial density of 33mg CDW l −1.As growth increased,the inoculum was changed to a 1:100dilution and subsequently to a 1:300dilution.Periodically,clones were isolated from these experiments,assigned new strain designations,and frozen for storage.AnalysesCell mass was estimated by measuring the optical density at anic acids and glucose concentrations were mea-sured by high-performance liquid chromatography (HPLC,Underwood et al.2002).Analysis of fermentation products by mass spectroscopy and amino acid analyzer were provided by the University of Florida Interdisciplinary Center for Bio-technology Research.Alanine was found to be the predominant product.The alanine concentration and isomeric purity were further measured by HPLC using the Chiralpak MA(+)chiral column (Chiral Technologies,West Chester,PA).ResultCloning of the alanine dehydrogenase geneALD is found in Bacillus (and Geobacillus )species where it plays a pivotal role in energy generation during sporulation (Ohashima and Soda 1979;Kuroda et al.1990).ALD from B.sphaericus IFO3525has beenwidelyFig.3Plasmids used to delete mgsA .Plasmid pLOI4229(a )was used to delete the mgsA gene and insert the cat-sacB cassette in the first recombina-tion step.Plasmid pLOI4230(b )was used to remove the cat-sacB cassette to create a deletion devoid of foreign sequence.Se-quence encoding the N-terminal and C-terminal regions are des-ignated mgsA ′and mgsA ″,respectivelyused with varying degrees of success to engineer alanine production in recombinant bacteria(Uhlenbusch et al. 1991;Hols et al.1999;Lee et al.2004;Smith et al.2006). Selection of the B.sphaericus IFO3525is presumed to be due in part to the high specific activity(Ohashima and Soda 1979).In contrast,we have selected a thermostable ALD from the thermophile,G.stearothermophilus XL-65-6, based on our prior experience in expressing genes from this organism in recombinant E.coli(Burchhardt and Ingram 1992;Lai and Ingram1993;Lai and Ingram1995).The ribosomal-binding region,coding region,and tran-scriptional terminator of alaD were amplified from G. stearothermophilus XL-65-6and sequenced(EF154460in GenBank).The deduced amino acid sequence was identical to that reported for Geobacillus kaustophilus HTA426and very similar to G.stearothermophilus strain10(99%iden-tity)and G.stearothermophilus strain IFO12550(94% identity).The nucleotide sequence(65%identity)and the deduced ALD amino acid sequence(74%identity)were quite different from the B.sphaericus IFO3525gene,the gene pre-viously used for alanine production in recombinant bacteria.Modification of E.coli W for homoalanine productionE.coli W strain SZ194(pflB frdBC adhE ackA)was previously constructed to produce only D-lactic acid.All major fermentation pathways except lactate have been blocked in this strain by gene deletions(Fig.1a).To convert this strain to the production of alanine,part of the native ldhA-coding region was replaced by a DNA fragment containing the ribosomal-binding region,coding region,and transcriptional terminator of alaD from G. stearothermophilus XL-65-6.The promoterless alaD was oriented in the same direction as ldhA to allow expression from the native ldhA promoter(Fig.2).After electroporation,approximately500colonies were recovered with tetracycline resistance and sensitivity to kana-mycin,consistent with a double-crossover event.These colo-nies were further examined by PCR using ldhA forward and reverse primer set(Table3).Only eight colonies of the500 tested were correct based on an analysis of PCR fragments. These eight colonies were further verified using primer sets for alaD,ldhA forward and alaD reverse,alaD forward and ldhA reverse,and ldhA outside primers(Table3)and de-signated XZ103,XZ104,XZ105,XZ106,XZ107,XZ108, XZ109,and XZ110,respectively.These eight strains were initially tested in15-ml screw-cap tubes containing NBS medium with2%glucose and100mM ammonium sulfate, which were filled to the brim.Strain XZ105appeared to grow faster than the other strains(37°C for48h)and was selected for further development.XZ105was transformed with pFT-A,which contains an inducible flippase(FLP)recombinase(Martinez-Morales et al.1999;Posfai et al.1997).The chromosomal FRT-flanked tet gene in XZ105was removed by inducing the FLP recombinase.After growing in39°C to eliminate the temperature-sensitive plasmid pFT-A,resulting strain was designated XZ111.Expression of G.stearothermophilus alaD in XZ111is transcriptionally regulated by the ldhA promoter,the same promoter that regulates the production of lactate dehydrogenase(dominant fermentation pathway) in native E.coli.pH-controlled batch fermentation for alanine production Alanine production by strain XZ111was tested in500-ml fermentation vessels containing300ml NBS medium, 20g l−1glucose,100mM ammonium sulfate,and1mM betaine.Broth pH was automatically controlled by adding 2N potassium hydroxide.After96h,181mM alanine was produced.The alanine yield from total glucose was 81%(g/g),and84%based on glucose that had been metabo-lized.The chiral purity of L-alanine was96.1%(Table4). Very low levels of other products(lactate,succinate,ace-tate,ethanol)were present,typically below1mM.This result demonstrated that the integrated G.stearothermophilus alaD gene as a single chromosomal copy under the control of the native ldhA promoter can provide sufficient levels of ALD to support E.coli growth from the production of alanine as the sole fermentation product.Metabolic evolution of strain XZ111Although XZ111could accumulate alanine as the primary product,incubation times were long,and volumetric productivity was limited.When using a high-glucose concentration(80g l−1),growth and alanine productivity were further reduced(Table4).In this strain,adenosine triphosphate(ATP)production and growth are tightly coupled to NADH oxidation and alanine production by ALD(Fig.1b).This coupling provided a basis for strain improvement by selecting for increased growth during serial cultivation,i.e.,metabolic evolution.Cells with increased growth because of spontaneous mutations will successively displace their parents while coselecting for increased alanine productivity.Serial transfers of XZ111were carried out at24-h intervals in NBS mineral salts medium with1mM betaine.Cultures were first transferred in the medium containing20g l−1 glucose,and the pH was controlled by automatically adding 2N potassium hydroxide.However,after ten transfers to strain XZ112,little improvement was observed(data not shown).Because ammonia is essential for alanine pro-duction,it was thought that ammonia may be limiting for fermentation.Two normals potassium hydroxide containing 1N ammonia carbonate and5N ammonia hydroxide alone。
红树林植物海榄雌化学成分研究

年版已经将麻黄与麻黄根分别收入。
麻黄的主要功用是发汗散寒、宣肺平喘、利水消肿,其中麻黄碱起到升压作用[1],而根的功用是治疗体虚自汗,其中麻黄根碱具有明显的降压作用[1],所以根中麻黄碱自然愈少愈好,这也为科学用药提供了依据。
312 野生和栽培麻黄茎部挥发油中成分的异同:通过GC 2M S ,确定了麻黄茎部两种挥发油中的45种物质,约占总挥发油的80%。
在已确定的成分中,野生和栽培麻黄茎部挥发油中,相同成分有:己醛、Β2水芹烯、42蒈烯、聚伞花素、32异丙烯基25,52二甲基2环戊烯、Χ2萜品烯、四甲基吡嗪、异丙烯基甲苯、芳樟醇、1,3,52三亚甲基2环庚烷、L 242萜品醇、Α2萜品醇、Β2紫罗(兰)酮。
其中两者挥发油的主要成分均为Α2萜品醇,野生和栽培中相对质量分数分别达到27162%和35122%,远远超过其他成分。
栽培品中四甲基吡嗪,32异丙烯基25,52二甲基2环戊烯两者的相对质量分数也较大,超过6%,约为野生品的2倍。
从表1中可见野生和栽培品的主要成分大致相似,但仍存在许多不同,这跟两者的生长环境以及两者接触的肥料很有关系。
313 麻黄茎部挥发油中未发现麻黄碱相关物质:在GC 2M S 的结果中,未发现麻黄碱相关物质。
可能原因有:提取时采用的是《中国药典》2005年版一部附录中挥发油测定法中的甲法,此法适用于相对密度小于110的挥发油的提取[6],可能对麻黄碱提取效果不好;麻黄碱的沸点为225℃,沸点相对较高,一般GC 2M S 检测时需要将其衍生化[5],本实验没有衍生,可能对其检测效果也不佳。
致谢:参与麻黄采集的暑假社会实践队其他队员:张杰、葛丽、王伟伟、高海燕、周世林、曹沁园。
References :[1] Zheng H C ,Cai S Q 1P ha r m aceu tica l B otany and P ha r m a 2cog nosy (药用植物学与生药学)[M ]1Beijing :Peop le ′s M edical Publish ing House ,20031[2] Shen H ,D i L Q ,H uang Y Z 1Effect of different extracti onm ethods on ephedrine extracti on yield [J ]1J N anj ing U n iv T rad it Ch in M ed (南京中医药大学学报),2004,20(3):17021721[3] D ing M Y ,M a Y H 1D eter m inati on of ephedrine alkalo ids inextract pow der of Ep hed ra sin ica by h igh perfo r m ance liquid ch rom atography [J ]1A na l L ab (分析实验室),2004,23(7):1231[4] J i L ,Xu Z L ,Pan G G,et a l 1GC 2M S A nalysis of cons 2tituents of essential o ils from stem s of Ep hed ra sin ica Stapf ,E 1in ter m ed ia Sch renk et C 1A 1M ey 1and E 1equ isetina Bge 1[J ]1Ch ina J Ch in M a ter M ed (中国中药杂志),1997,22(8):48924921[5] H e F ,L uo J B ,Chen F L ,et a l 1H um an phar m acok inetics ofephedrine and p seudoephedrine in M ahuang T ang by GC 2M S [J ]1T rad it Ch in D rug R es C lin P ha r m (中药新药与临床药理),2004,15(5):1231[6] Ch P (中国药典)[S ]1V o l 120051红树林植物海榄雌化学成分研究冯 妍1,2,李晓明1,王斌贵13①(11中国科学院海洋研究所实验海洋生物学重点实验室,山东青岛 266071;21中国科学院研究生院,北京 100039)摘 要:目的 研究红树林植物海榄雌A v icennia m arina 枝、叶的化学成分。
甘西鼠尾草中三个新的萜类化合物

甘 西 鼠尾 草 为 常 用 中 药 丹 参 ( .mii ri g. 的 同属植 物 ,其 主 要 成 分 为 脂 溶 性 二 萜 醌 S lo h aB e ) tr z
类 和水溶 性 酚酸类 化合物 ’ .已报 道 的药理 活性 主要有 醛糖 还原 酶 抑制 活性 、抑制 超 氧 自由綦及抗 氧化 、 保护 心肌 缺血 及心 肌 缺 血再 灌 注 损 伤 、 炎 及 抑 菌 。 抗 等.本 课 题 组 在 对甘 两 鼠尾草 活性 部位 的筛选 中 , 证实 了甘西 鼠尾 草对 血清病 性 肾小球 肾炎模 型 大 鼠具有 较 好 的治疗 作用 , 可显 著降低 大 鼠尿蛋 白含量 、减轻 肾小 球 肿胀 ,还 可降 低 正 常大 鼠的血 液 黏 度 ,并 具有 显 著且 温和 的利尿 作用 , 以调整 肾脏 疾病 中的 电解 质紊 乱 ,对 于 治 疗 和 缓 解 肾脏 疾 病 的发 展 有 积 极 的 意 可 义.为 了进 一 步考察 其活性 物质 ,本文对 其活性 部位 的化 学 成分 进 行 了深 入研 究 ,从甘 西 鼠尾草 根 和 根茎 的 5 % 乙醇 提取 物 中分离鉴定 了 2个新 的二萜 化 合物 和 1个新 的单 萜苷 化 合物, 0
1 实验 部 分
1 1 试 剂 与仪器 .
柱色谱 硅胶 与薄层 色谱 硅 胶 板 为烟 台江 友 硅胶 开 发 公 司产 品 ;Sp a e 0凝 胶 为 P am c e hd xI 2 H hr ai a
公 司产 品 ; D I O S C 相硅胶 为 Me k公 司产 品 ; I e C P 0 8反 r c MC l H 2 P凝 胶 和 HP2 g - 0大孔 吸附树 脂 为 ■菱
杨 阳 ,吴志军’ ,杨颖博 ,来 威 ,孙连娜 ,陈万生
戈那瑞林

化合物简介
基本信息
物化性质
基本信息
中文名称:戈那瑞林 中文别名:黄体激素释放激素 英文名称:gonadorelin 英文别名:Glp-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2;(pyroglutamic acid)-His-Trp-SerTyr-Gly-Leu-Arg-Pro-Gly-NH2;Gonadorelin;GONADERELIN;luteinizing hormone-releasing hormone isoform I; CAS号:-09-2 分子式:C55H75N17O13 分子量:1182. 结构式: 精确质量:1181. PSA:472.
适应症
1.用于诊断下丘脑-垂体-生殖腺功能障碍。 2.治疗闭经与促性腺激素分泌不足和多滤泡性卵巢引起的不孕症。 3.戈那瑞林或其同类物布舍瑞林、戈舍瑞林、亮丙瑞林、那法瑞林和曲普瑞林还可用于避孕、隐睾症、恶性 肿瘤(尤其前列腺癌)、延迟的和提前的青春期。 4.还可用于子宫内膜异位。 5.用于促排卵以治疗下丘脑性闭经所致不孕、原发性卵巢功能不足,特别是对氯米芬无效的患者。 6.还用于小儿隐睾症及雄激素过多、垂体肿瘤等。 。
氨基酸比值取本品约2mg,置10ml量瓶中,加6mol/L盐酸溶液使溶解并稀释至刻度,摇匀,量取1ml置2ml硬 质安瓶中,在-5℃减压封口,置110℃加热24小时,冷却,启封,将内容物移至蒸馏瓶中,在减压下蒸干,残留物 加0.02mol/L盐酸溶液1ml溶解,摇匀,作为供试品溶液;另取与供试品溶液浓度相应的各个氨基酸对照品混合溶 液,作为对照品溶液。精密量取上述两种溶液各20μl,分别注入氨基酸分析仪,记录色谱图,按外标法以峰面 积计算,即得。以L一组氨酸、L一谷氨酸、L一亮氨酸、L一脯氨酸、甘氨酸和L一精氨酸摩尔数之和的七分之一 值为1,供试品溶液中各个氨基酸相应的摩尔比应符合以下规定:L一丝氨酸为0.7—1.05,L一谷氨酸为0.9—l.
洛塞那肽结构

洛塞那肽结构洛塞那肽(Lorcaserin)是一种用于治疗肥胖症的药物,它属于选择性5-羟色胺2C受体激动剂(5-HT2C受体激动剂)。
洛塞那肽通过作用于大脑内的5-HT2C受体,抑制食欲,从而帮助患者减少摄入的食物量,达到减肥的效果。
洛塞那肽的化学结构是一种类似芳香环的化合物,它的化学名为(R)-8-Chloro-1-methyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride。
洛塞那肽的分子式为C11H14ClN·HCl,相对分子质量为232.15。
洛塞那肽在体内的作用机制是通过与5-HT2C受体结合并激活该受体。
5-HT2C受体是一种G蛋白偶联受体,其激活将导致下游信号通路的改变。
在中枢神经系统中,5-HT2C受体的激活可抑制食欲,从而减少摄入的食物量。
此外,洛塞那肽还可以通过激活5-HT2C受体调节神经递质的释放,进一步影响食欲中枢的调控。
洛塞那肽的临床应用主要是用于治疗体质指数(BMI)大于30 kg/m²的肥胖患者,或者BMI大于27 kg/m²的伴有高血压、高胆固醇或2型糖尿病等相关疾病的患者。
洛塞那肽的用法是口服,每次10 mg,每天2次。
在使用洛塞那肽的同时,患者还需要进行饮食调整和增加体力活动,以达到更好的减肥效果。
洛塞那肽的副作用主要包括头痛、恶心、便秘、乏力等,其中恶心是最常见的副作用之一。
此外,洛塞那肽还可能引起心脏瓣膜病的风险增加,因此在使用洛塞那肽时需要密切监测患者的心脏功能。
洛塞那肽的禁忌症包括对该药物过敏的患者、妊娠期妇女和哺乳期妇女。
此外,洛塞那肽还不能与一些药物同时使用,如抗抑郁药物、三环类抗抑郁药物等。
在使用洛塞那肽之前,患者应告知医生自己的过敏史、疾病史和正在使用的药物,以避免不良反应的发生。
洛塞那肽是一种用于治疗肥胖症的药物,通过作用于大脑内的5-HT2C受体,抑制食欲,从而帮助患者减少摄入的食物量。
巨噬细胞自噬在慢性阻塞性肺疾病中的作用

tJRe
sp
i
r,
Augus
t2020,
Vo
l.
40,
No.
16
促进细胞存活。过度 的 自 噬 也 会 导 致 细 胞 死 亡。 自 噬 分 为
使 ERK 通 路 激 活 所 致;ERK 抑 制 剂 和 BRF1 同 时 作 用 于
3 种类型:大 自 噬、 小 自 噬 和 分 子 伴 侣 介 导 的 自 噬。 自 噬
t
o
ryandCr
i
c
i
ca
lCar
e Medi
c
i
net
heSe
c
ond Ho
spi
t
a
lof He
b
e
iMedi
ca
l
1
Un
i
v
e
r
s
i
t
i
i
azhuang050000 Ch
i
na 2Depar
tmen
tof Br
ea
s
tSurge
ry t
heFi
r
s
t Ho
spi
t
a
lof
y Sh
j
Qi
nhuangdao Qi
lmona
r
i
s
e
a
s
e COPD
pa
yd
Au
t
ophagya
sa me
chan
i
sm t
o
ma
i
n
t
a
i
nc
e
l
lhome
o
s
盐酸伐昔洛韦的合成路线

Valacyclovir盐酸伐昔洛韦的合成路线制药工程一班刘金贵3010207301Valacyclovir一中文名:维德思,盐酸伐昔洛韦,盐酸万乃洛韦二化学名称为:L-缬氨酸-2-(6-氧代-2-氨基-1,6-二氢- 9H -嘌呤-9-基)甲氧基乙基酯盐酸盐三化学结构式(如下图) 分子式C13-H20-N6-O4 分子量324.342四药代动力学本品口服进入人体后迅速分解为L-缬氨酸和阿昔洛韦,前者在体内参与正常生理生化代谢,后者在被疱疹病毒感染的细胞中,血中阿昔洛韦达峰时间为0.88~1.75小时。
口服生物利用度为67±13%,是阿昔洛韦的3~5倍。
药物进入体内后广泛分布,可分布至多种组织中,其中胃、小肠、肾、肝、淋巴结和皮肤组织中浓度最高,脑组织中的浓度最低。
药物在体内全部转化为阿昔洛韦,代谢物主要从尿中排除,其中阿昔洛韦占46%~59%,8-羟基-9-鸟嘌呤占25%~30%,9-羟基甲氧基鸟嘌呤占11%~12%。
阿昔洛韦原形为单相消除,血消除半衰期(t1/2β)为2.86±0.39小时。
五药理毒理药理作用:伐昔洛韦是一特异性疱疹病毒抑制剂,为阿昔洛韦(嘌呤核苷类似物)的L-缬氨酸酯。
伐昔洛韦在体内可能是通过伐昔洛韦水解酶迅速几乎完全转化为阿昔洛韦和缬氨酸。
阿昔洛韦在体外具有抑制单纯疱疹病毒(HSV)Ⅰ型和Ⅱ型、水痘带状疱疹病毒(VZV)、巨细胞病毒(CMV)、Epstein-Barr病毒(EBV)和人类疱疹病毒6(HHV-6)的作用。
病毒的胸苷激酶使其磷酸化,成为单磷酸化合物,再由细胞激酶磷酸化变成二磷酸和三磷酸化合物。
三磷酸化合物是抗病毒的活性物质,可抑制病毒的DNA聚合酶,终止其DNA合成,显示抗病毒效力。
由于本品是ACV 的氨基酸酯,没有游离羟基提供给磷酸化,因而在未转化为ACV之前,并无抗病毒活性,这一点使其不象其它前体药物如地昔洛韦(desciclovir)等另外增加对细胞的毒性。
益生菌对阿尔茨海默病作用的研究进展

益生菌对阿尔茨海默病作用的研究进展发布时间:2021-12-14T06:08:15.523Z 来源:《中国结合医学杂志》2021年12期作者:宋鑫萍1,2,李盛钰2,金清1[导读] 阿尔茨海默病已成为威胁全球老年人生命健康的主要疾病之一,患者数量逐年攀升,其护理的经济成本高,给全球经济造成重大挑战。
近年来研究显示,益生菌在适量使用时作为有益于宿主健康的微生物,在防治阿尔茨海默病方面具有积极影响,其作用机制可能通过调节肠道菌群,影响神经免疫系统,调控神经活性物质以及代谢产物,通过肠-脑轴影响该病发生和发展。
宋鑫萍1,2,李盛钰2,金清11.延边大学农学院,吉林延吉 1330022.吉林省农业科学院农产品加工研究所,吉林长春 130033摘要:阿尔茨海默病已成为威胁全球老年人生命健康的主要疾病之一,患者数量逐年攀升,其护理的经济成本高,给全球经济造成重大挑战。
近年来研究显示,益生菌在适量使用时作为有益于宿主健康的微生物,在防治阿尔茨海默病方面具有积极影响,其作用机制可能通过调节肠道菌群,影响神经免疫系统,调控神经活性物质以及代谢产物,通过肠-脑轴影响该病发生和发展。
本文综述了近几年来国内外益生菌对阿尔茨海默病的作用进展,以及其预防和治疗阿尔茨海默病的潜在作用机制。
关键词:益生菌;阿尔茨海默病;肠道菌群;机制Recent Progress in Research on Probiotics Effect on Alzheimer’s DiseaseSONG Xinping1,2,LI Shengyu2,JI Qing1*(1.College of Agricultural, Yanbian University, Yanji 133002,China)(2.Institute of Agro-food Technology, Jilin Academy of Agricultural Sciences, Chanchun 130033, China)Abstract:Alzheimer’s disease has become one of the major diseases threatening the life and health of the global elderly. The number of patients is increasing year by year, and the economic cost of nursing is high, which poses a major challenge to the global economy. In recent years, studies have shown that probiotics, as microorganisms beneficial to the health of the host, have a positive impact on the prevention and treatment of Alzheimer’s disease. Its mechanism may be through regulating intestinal flora, affecting the nervous immune system, regulating the neuroactive substances and metabolites, and affecting the occurrence and development of the disease through thegut- brain axis. This paper reviews the progress of probiotics on Alzheimer’s disease at home and abroad in recent years, as well as its potential mechanism of prevention and treatment.Key words:probiotics; Alzheimer’s disease; gut microbiota; mechanism阿尔茨海默病(Alzheimer’s disease, AD),系中枢神经系统退行性疾病,属于老年期痴呆常见类型,临床特征主要包括:记忆力减退、认知功能障碍、行为改变、焦虑和抑郁等。
羟氯喹体内代谢物-概述说明以及解释

羟氯喹体内代谢物-概述说明以及解释1.引言在1.1 概述部分,我们将简要介绍羟氯喹及其代谢物的研究背景和重要性。
羟氯喹是一种广泛用于治疗疟疾和类风湿关节炎的药物,近年来也被广泛应用于新型冠状病毒感染的治疗中。
羟氯喹在体内通过代谢产生不同的代谢物,这些代谢物可能具有重要的药理作用,对药物疗效和副作用产生影响。
因此,对羟氯喹代谢物的研究具有重要意义,可以深化对羟氯喹药理作用的理解,为临床应用提供更有效的指导,同时也为新药研发和治疗策略的制定提供参考。
本文将系统回顾羟氯喹体内代谢物的研究进展,探讨其在药理学上的意义和未来研究方向。
1.2 文章结构文章结构部分是对整个文章内容的一个简要概括,包括文章的各个章节和各个部分的主要内容和重点。
在这里,可以简要介绍文章的主要结构和内容安排,让读者在阅读之前有一个整体的了解。
具体内容可以包括每个章节的标题和主要内容概述,如引言部分介绍了羟氯喹的基本信息和研究背景,正文部分包括了羟氯喹的药理作用和在体内的代谢过程,结论部分对文章进行了总结和展望未来研究方向等。
这样的文章结构部分可以让读者更好地理解文章的内在逻辑和组织,帮助读者更好地把握整个文章的内容和主题。
1.3 目的羟氯喹是一种常用的抗疟药物,近年来也被广泛用于治疗风湿性关节炎和类风湿性关节炎等自身免疫性疾病。
在羟氯喹的体内代谢过程中,会生成一系列代谢物,这些代谢物可能具有更广泛的生物活性和潜在的药理作用。
因此,本文旨在系统总结羟氯喹体内代谢物的生成途径、生物活性和潜在应用,并展望未来的研究方向,以期为进一步深入了解和开发羟氯喹的药理作用提供参考。
通过对羟氯喹代谢物的研究,有望为拓展其在药物治疗领域的应用提供新的思路和可能性。
2.正文2.1 羟氯喹的药理作用羟氯喹是一种抗疟药物,主要用于治疗疟疾和风湿性关节炎。
其作用机制主要包括两个方面:2.1.1 作用机制羟氯喹能够通过干扰寄生虫对人体红细胞的侵染和生长,从而达到治疗疟疾的效果。
天然产物(+)-Perophoramidine,(+)-Psychotrimine,(Iso)rhy

天然产物(+)-Perophoramidine,(+)-Psychotrimine,(Iso)rhynchophylline和Strychnofoline的合成研究天然产物(+)-Perophoramidine,(+)-Psychotrimine,(Iso)rhynchophylline和Strychnofoline的合成研究天然产物在药物合成领域一直受到研究人员的关注。
(+)-Perophoramidine,(+)-Psychotrimine,(Iso)rhynchophylline 和Strychnofoline是一类重要的生物碱,以其独特的生物活性引起了科学家们的兴趣。
这些天然产物具有多种生物学活性,如抗肿瘤、抗炎症、神经保护等。
因此,发展合成这些物质的方法对于深入研究其药理学作用以及发展新型药物具有重要意义。
首先,我们来讨论(+)-Perophoramidine的合成研究。
Perophoramidine是一种藤黄酮和生物碱的混合物,具有明显的抑制肿瘤细胞生长活性。
研究发现,该化合物的抗肿瘤活性主要来源于其中的生物碱成分。
目前针对Perophoramidine的合成,主要有合成生物碱骨架、连接两个生物碱单元、构建官能团等策略。
其中,合成生物碱骨架的方法是非常重要的一步,可以通过大环化反应、酮肟互变反应等多种方法实现。
此外,连接两个生物碱单元和构建官能团也需要合适的试剂和条件,通常采用的方法有亲核取代、羟基保护和去保护等。
通过这些方法,目前已成功合成了Perophoramidine及其衍生物,并进行了相关的生物活性研究。
接下来,我们来讨论(+)-Psychotrimine的合成研究。
Psychotrimine是一种重要的中枢神经系统药物,具有镇静、抗焦虑和抗抑郁等药理学作用。
研究发现,该化合物的生物活性主要通过对多种神经递质受体的亲和力产生影响。
目前,Psychotrimine的合成研究主要集中在开发高效的合成路线和改进反应条件。
Keap1

非小细胞肺癌(non-small cell lung cancer,NSCLC)发病率占据肺癌的75%~80%。
肿瘤细胞进展快且易扩散转移,临床常采用手术、放化疗等进行治疗,但5年生存率低于60%[1-2]。
氧化应激是由活性氧(ROS)生成量增加所致,ROS积累可诱导肺癌细胞凋亡,清除ROS 可阻止癌细胞凋亡,即肺癌细胞存活依赖于癌细胞自身抗氧化能力[3]。
Kelch样环氧氯丙烷相关蛋白-1 (kelch-like epichlorohydrin-associated protein-1,Keap1)/核因子E2相关因子2(nuclear factor E2related factor 2,Nrf2)信号通路在癌症中发挥重要调控作用,氧化应激可激活Keap1,促使Keap1-Nrf2复合物裂解,Nrf2转移至细胞核内,可激活下游靶基因表达,参与肺癌发生发展过程[4]。
Nrf2可维持氧化还原稳态,ROS侵袭细胞时,Nrf2可进入细胞核,结合抗氧化反应元件(ARE)转录编码各种抗氧化蛋白、代谢酶基因,抑制氧化应激反应[5-6]。
目前氧化应激、Keap1/Nrf2信号通路在NSCLC发生过程中的机制尚未明确。
基于此,本研究尝试分析Keap1/Nrf2信号通路与临床病理参数、氧化应激指标的相关性,探讨其在NSCLC氧化应激机制中的作用,为临床研制新药提供参考依据。
1资料与方法1.1一般资料选取2017年4月至2020年4月郑州市第三人民医院收治的100例NSCLC患者为研究对象。
纳入标准:符合NSCLC诊断标准[7];术前未接受放化疗、免疫治疗者;预计生存期≥6个月;符合手术适应证、禁忌证;Karnofsky功能状态评分≥70分;签署知情同意书。
排除标准:合并凝血功能障碍、肝肾功能障碍、其他恶性肿瘤者;伴有急/慢性感染者;伴有精神疾病者;既往腹部相关外科手术史者。
所有患者均行肺癌根治性切除术,术中收集癌组织、癌旁组织(距离癌组织5cm范围内正常组织),其中男性63例,女性37例;年龄46~67岁,平均(56.32±3.16)岁;体质量指数(BMI)17~30kg/m2,平均(23.16±2.03)kg/m2;病理类型:鳞癌58例、腺癌42例;病理分级[8]:Ⅰ~Ⅱ级51例、Ⅲ级49例;T分期[9]:T1~T253例、T3~T447例;N分期:N055例、N1~N245例。
CellTiter Glo Luminescent Cell Viability Assay Protocol

Promega Corporation ·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·1.Description (1)2.Product Components and Storage Conditions (4)3.Performing the CellTiter-Glo ®Assay (5)A.Reagent Preparation (5)B.Protocol for the Cell Viability Assay (6)C.Protocol for Generating an ATP Standard Curve (optional) (7)4.Appendix (7)A.Overview of the CellTiter-Glo ®Assay..............................................................7B.Additional Considerations..................................................................................8C.References............................................................................................................11D.Related Products. (12)1.DescriptionThe CellTiter-Glo ®Luminescent Cell Viability Assay (a–e)is a homogeneous method to determine the number of viable cells in culture based on quantitation of the ATP present, which signals the presence of metabolically active cells. The CellTiter-Glo ®Assay is designed for use with multiwell-plate formats, making it ideal for automated high-throughput screening (HTS) and cell proliferation and cytotoxicity assays. The homogeneous assay procedure (Figure 1) involves adding a single reagent (CellTiter-Glo ®Reagent) directly to cells cultured in serum-supplemented medium. Cell washing, removal of medium or multiple pipetting steps are not required.The homogeneous “add-mix-measure” format results in cell lysis and generation of a luminescent signal proportional to the amount of ATP present (Figure 2).The amount of ATP is directly proportional to the number of cells present in culture in agreement with previous reports (1). The CellTiter-Glo ®Assay relies on the properties of a proprietary thermostable luciferase (Ultra-Glo™ Recombinant Luciferase), which generates a stable “glow-type” luminescent signal and improves performance across a wide range of assay conditions. The luciferase reaction for this assay is shown in Figure 3. The half-life of the luminescent signal resulting from this reaction is greater than five hours (Figure 4). This extended half-life eliminates the need for reagent injectors and provides flexibility for continuous or batch-mode processing of multiple plates. The unique homogeneous format reduces pipetting errors that may be introduced during the multiple steps required by other ATP-measurement methods.CellTiter-Glo ®Luminescent Cell Viability AssayAll technical literature is available on the Internet at: /protocols/ Please visit the web site to verify that you are using the most current version of this Technical Bulletin. Please contact Promega Technical Services if you have questions on useofthissystem.E-mail:********************Figure 1. Flow diagram showing preparation and use of CellTiter-Glo ®Reagent.Promega Corporation ·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·3170M A 12_0ACellTiter-Glo CellTiter-Glo MixerLuminometer®System Advantages•Homogeneous:“Add-mix-measure” format reduces the number of plate-handling steps to fewer than that required for similar ATP assays.•Fast:Data can be recorded 10 minutes after adding reagent.•Sensitive:Measures cells at numbers below the detection limits of standard colorimetric and fluorometric assays.•Flexible:Can be used with various multiwell formats. Data can be recorded by luminometer or CCD camera or imaging device.•Robust:Luminescent signal is very stable, with a half-life >5 hours,depending on cell type and culture medium used.•Able to Multiplex:Can be used with reporter gene assays or other cell-based assays from Promega (2,3).Figure 3. The luciferase reaction.Mono-oxygenation of luciferin is catalyzed byluciferase in the presence of Mg 2+, ATP and molecular oxygen.Promega Corporation ·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·3171M A 12_0A L u m i n e s c e n c e (R L U )Cells per Well10,00060,00020,00030,00040,00050,0000R² = 0.9990.5 × 1061.0 × 1061.5 × 1062.0 × 1062.5 × 1063.0 × 1063.5 × 1064.0 × 106r² = 0.99020,00010,00030,00040,00050,000r² = 0.9900100200300400HO SN S N O S N S N OCOOH +ATP+O 2Ultra-Glo™ Recombinant Luciferase +AMP+PP i +CO 2+LightBeetle Luciferin OxyluciferinMg 2+0Figure 2. Cell number correlates with luminescent output.A direct relationship exists between luminescence measured with the CellTiter-Glo ®Assay and the number of cells in culture over three orders of magnitude. Serial twofold dilutions of HEK293cells were made in a 96-well plate in DMEM with 10% FBS, and assays wereperformed as described in Section 3.B. Luminescence was recorded 10minutes after reagent addition using a GloMax ®-Multi+ Detection System. Values represent the mean ± S.D. of four replicates for each cell number. The luminescent signal from 50HEK293 cells is greater than three times the background signal from serum-supplemented medium without cells. There is a linear relationship (r 2= 0.99)between the luminescent signal and the number of cells from 0to 50,000 cells per well.Figure 4. Extended luminescent half-life allows high-throughput batchprocessing.Signal stability is shown for three common cell lines. HepG2 and BHK-21cells were grown and assayed in MEM containing 10% FBS, while CHO-K1 cells were grown and assayed in DME/F-12 containing 10% FBS. CHO-K1, BHK-21 and HepG2 cells, at 25,000 cells per well, were added to a 96-well plate. After an equal volume of CellTiter-Glo ®Reagent was added, plates were shaken and luminescence monitored over time with the plates held at 22°C. The half-lives of the luminescent signals for the CHO-K1, BHK-21 and HepG2 cells were approximately 5.4, 5.2 and5.8hours, respectively.2.Product Components and Storage ConditionsProduct Size Cat.#CellTiter-Glo ®Luminescent Cell Viability Assay 10ml G7570Substrate is sufficient for 100 assays at 100µl/assay in 96-well plates or 400 assays at 25µl/assay in 384-well plates. Includes:• 1 × 10mlCellTiter-Glo ®Buffer • 1 vial CellTiter-Glo ®Substrate (lyophilized)Product Size Cat.#CellTiter-Glo ®Luminescent Cell Viability Assay 10 × 10ml G7571Each vial of substrate is sufficient for 100 assays at 100µl/assay in 96-well plates or 400 assays at 25µl/assay in 384-well plates (1,000 to 4,000 total assays). Includes:•10 × 10mlCellTiter-Glo ®Buffer •10 vials CellTiter-Glo ®Substrate (lyophilized)Promega Corporation ·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·R e l a t i v e L u m i n e s c e n c e (%)Time (minutes)CHO-K101020304050607080901003173M A 12_0AProduct Size Cat.# CellTiter-Glo®Luminescent Cell Viability Assay100ml G7572 Substrate is sufficient for 1,000 assays at 100µl/assay in 96-well plates or 4,000assays at 25µl/assay in 384-well plates. Includes:•1 × 100ml CellTiter-Glo®Buffer• 1 vial CellTiter-Glo®Substrate (lyophilized)Product Size Cat.# CellTiter-Glo®Luminescent Cell Viability Assay10 × 100ml G7573Each vial of substrate is sufficient for 1,000 assays at 100µl/assay in 96-well plates or4,000 assays at 25µl/assay in 384-well plates (10,000to 40,000 total assays). Includes:•10 × 100ml CellTiter-Glo®Buffer•10 vials CellTiter-Glo®Substrate (lyophilized)Storage Conditions:For long-term storage, store the lyophilized CellTiter-Glo®Substrate and CellTiter-Glo®Buffer at –20°C. For frequent use, the CellTiter-Glo®Buffer can be stored at 4°C or room temperature for 48hours without loss of activity. See product label for expiration date information. ReconstitutedCellTiter-Glo®Reagent (Buffer plus Substrate) can be stored at room temperaturefor up to 8hours with <10% loss of activity, at 4°C for 48hours with ~5% lossof activity, at 4°C for 4days with ~20% loss of activity or at –20°C for 21weekswith ~3% loss of activity. The reagent is stable for up to ten freeze-thaw cycles,with less than 10% loss of activity.3.Performing the CellTiter-Glo®AssayMaterials to Be Supplied by the User•opaque-walled multiwell plates adequate for cell culture•multichannel pipette or automated pipetting station for reagent delivery•device (plate shaker) for mixing multiwell plates•luminometer, CCD camera or imaging device capable of reading multiwell plates •optional:ATP for use in generating a standard curve (Section 3.C)3.A.Reagent Preparation1.Thaw the CellTiter-Glo®Buffer, and equilibrate to room temperature priorto use. For convenience the CellTiter-Glo®Buffer may be thawed andstored at room temperature for up to 48hours prior to use.2.Equilibrate the lyophilized CellTiter-Glo®Substrate to room temperatureprior to use.Promega Corporation·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·3.A.Reagent Preparation (continued)3.Transfer the appropriate volume (10ml for Cat.# G7570 and G7571, or 100mlfor Cat.# G7572 and G7573) of CellTiter-Glo ®Buffer into the amber bottlecontaining CellTiter-Glo ®Substrate to reconstitute the lyophilizedenzyme/substrate mixture. This forms the CellTiter-Glo ®Reagent.4.Mix by gently vortexing, swirling or inverting the contents to obtain ahomogeneous solution. The CellTiter-Glo ®Substrate should go intosolution easily in less than 1minute.3.B.Protocol for the Cell Viability AssayWe recommend that you perform a titration of your particular cells todetermine the optimal number and ensure that you are working within thelinear range of the CellTiter-Glo ®Assay. Figure 2 provides an example of sucha titration of HEK293 cells using 0 to 50,000 cells per well in a 96-well format.1.Prepare opaque-walled multiwell plates with mammalian cells in culturemedium, 100µl per well for 96-well plates or 25µl per well for 384-wellplates.Multiwell plates must be compatible with the luminometer used.2.Prepare control wells containing medium without cells to obtain a value forbackground luminescence.3.Add the test compound to experimental wells, and incubate according toculture protocol.4.Equilibrate the plate and its contents at room temperature forapproximately 30 minutes.5.Add a volume of CellTiter-Glo ®Reagent equal to the volume of cell culturemedium present in each well (e.g., add 100µl of reagent to 100µl of mediumcontaining cells for a 96-well plate, or add 25µl of reagent to 25µl ofmedium containing cells for a 384-well plate).6.Mix contents for 2 minutes on an orbital shaker to induce cell lysis.7.Allow the plate to incubate at room temperature for 10 minutes to stabilizeluminescent signal.Note:Uneven luminescent signal within standard plates can be caused bytemperature gradients, uneven seeding of cells or edge effects in multiwellplates.8.Record luminescence.Note:Instrument settings depend on the manufacturer. An integration timeof 0.25–1 second per well should serve as a guideline.Promega Corporation ·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·3.C.Protocol for Generating an ATP Standard Curve (optional)It is a good practice to generate a standard curve using the same plate onwhich samples are assayed. We recommend ATP disodium salt (Cat.# P1132,Sigma Cat.# A7699 or GE Healthcare Cat.# 27-1006). The ATP standard curveshould be generated immediately prior to adding the CellTiter-Glo®Reagentbecause endogenous ATPase enzymes found in sera may reduce ATP levels.1.Prepare 1µM ATP in culture medium (100µl of 1µM ATP solution contains10–10moles ATP).2.Prepare serial tenfold dilutions of ATP in culture medium (1µM to 10nM;100µl contains 10–10to 10–12moles of ATP).3.Prepare a multiwell plate with varying concentrations of ATP standard in100µl medium (25µl for a 384-well plate).4.Add a volume of CellTiter-Glo®Reagent equal to the volume of ATPstandard present in each well.5.Mix contents for 2 minutes on an orbital shaker.6.Allow the plate to incubate at room temperature for 10 minutes to stabilizethe luminescent signal.7.Record luminescence.4.Appendix4.A.Overview of the CellTiter-Glo®AssayThe assay system uses the properties of a proprietary thermostable luciferase toenable reaction conditions that generate a stable “glow-type” luminescentsignal while simultaneously inhibiting endogenous enzymes released duringcell lysis (e.g., ATPases). Release of ATPases will interfere with accurate ATPmeasurement. Historically, firefly luciferase purified from Photinus pyralis(LucPpy) has been used in reagents for ATP assays (1,4–7). However, it hasonly moderate stability in vitro and is sensitive to its chemical environment,including factors such as pH and detergents, limiting its usefulness fordeveloping a robust homogeneous ATP assay. Promega has successfullydeveloped a stable form of luciferase based on the gene from another firefly,Photuris pennsylvanica(LucPpe2), using an approach to select characteristics thatimprove performance in ATP assays. The unique characteristics of this mutant(LucPpe2m) enabled design of a homogeneous single-reagent-addition approachto perform ATP assays with cultured cells. Properties of the CellTiter-Glo®Reagent overcome the problems caused by factors, such as ATPases, thatinterfere with ATP measurement in cell extracts. The reagent is physicallyrobust and provides a sensitive and stable luminescent output.Promega Corporation·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·4.A.Overview of the CellTiter-Glo®Assay (continued)Sensitivity and Linearity:The ATP-based detection of cells is more sensitivethan other methods (8–10). In experiments performed by Promega scientists,the luminescent signal from 50HEK293 cells is greater than three standarddeviations above the background signal from serum-supplemented mediumwithout cells. There is a linear relationship (r2= 0.99) between the luminescentsignal and the number of cells from 0 to 50,000 cells per well in the 96-wellformat. The luminescence values in Figure 2 were recorded after 10minutes ofincubation at room temperature to stabilize the luminescent signal as describedin Section3.B. Incubation of the same 96-well plate used in the experimentshown in Figure 2 for 360minutes at room temperature had little effect on therelationship between luminescent signal and number of cells (r2= 0.99).Speed:The homogeneous procedure to measure ATP using the CellTiter-Glo®Assay is quicker than other ATP assay methods that require multiple steps toextract ATP and measure luminescence. The CellTiter-Glo®Assay also is fasterthan other commonly used methods to measure the number of viable cells(such as MTT, alamarBlue®or Calcein-AM) that require prolonged incubationsteps to enable the cells’ metabolic machinery to convert indicator moleculesinto a detectable signal.4.B.Additional ConsiderationsTemperature:The intensity and decay rate of the luminescent signal from theCellTiter-Glo®Assay depends on the luciferase reaction rate. Environmentalfactors that affect the luciferase reaction rate will change the intensity andstability of the luminescent signal. Temperature is one factor that affects therate of this enzymatic assay and thus the light output. For consistent results,equilibrate assay plates to a constant temperature before performing the assay.Transferring eukaryotic cells from 37°C to room temperature has little effect onATP content (5). We have demonstrated that removing cultured cells from a37°C incubator and allowing them to equilibrate to 22°C for 1–2 hours hadlittle effect on ATP content. For batch-mode processing of multiple assayplates, take precautions to ensure complete temperature equilibration. Platesremoved from a 37°C incubator and placed in tall stacks at room temperaturewill require longer equilibration than plates arranged in a single layer.Insufficient equilibration may result in a temperature gradient effect betweenwells in the center and at the edge of the plates. The temperature gradientpattern also may depend on the position of the plate in the stack.Promega Corporation·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·Chemicals:The chemical environment of the luciferase reaction affects theenzymatic rate and thus luminescence intensity. Differences in luminescenceintensity have been observed using different types of culture media and sera.The presence of phenol red in culture medium should have little impact onluminescence output. Assaying 0.1µM ATP in RPMI medium without phenolred resulted in ~5% increase in luminescence output (in relative light units[RLU]) compared to assays in RPMI containing the standard concentration ofphenol red, whereas assays in RPMI medium containing twice the normalconcentration of phenol red showed a ~2% decrease in luminescence.Solvents for the various test compounds may interfere with the luciferasereaction and thus the light output from the assay. Interference with theluciferase reaction can be detected by assaying a parallel set of control wellscontaining medium without cells. Dimethylsulfoxide (DMSO), commonly usedas a vehicle to solubilize organic chemicals, has been tested at finalconcentrations of up to 2% in the assay and only minimally affects light output.Plate Recommendations:We recommend using standard opaque-walledmultiwell plates suitable for luminescence measurements. Opaque-walledplates with clear bottoms to allow microscopic visualization of cells also maybe used; however, these plates will have diminished signal intensity andgreater cross talk between wells. Opaque white tape may be used to decreaseluminescence loss and cross talk.Cellular ATP Content:Different cell types have different amounts of ATP,and values reported for the ATP level in cells vary considerably (1,4,11–13).Factors that affect the ATP content of cells may affect the relationship betweencell number and luminescence. Anchorage-dependent cells that undergocontact inhibition at high densities may show a change in ATP content per cellat high densities, resulting in a nonlinear relationship between cell numberand luminescence. Factors that affect the cytoplasmic volume or physiology ofcells also will affect ATP content. For example, oxygen depletion is one factorknown to cause a rapid decrease in ATP (1).Promega Corporation·2800 Woods Hollow Road ·Madison, WI 53711-5399 USA Toll F ree in USA 800-356-9526·Phone 608-274-4330 ·F ax 608-277-2516 ·4.B.Additional Considerations (continued)Mixing:Optimal assay performance is achieved when the CellTiter-Glo®Reagent is mixed completely with the cultured cells. Suspension cell lines (e.g., Jurkat cells) generally require less mixing to achieve lysis and extract ATP than adherent cells (e.g., L929 cells). Tests were done to evaluate the effect ofshaking the plate after adding the CellTiter-Glo® Reagent. Suspension cellscultured in multiwell plates showed only minor differences in light outputwhether or not the plates were shaken after adding the CellTiter-Glo®Reagent.Adherent cells are more difficult to lyse and show a substantial differencebetween shaken and nonshaken plates.Several additional parameters related to reagent mixing include the force ofdelivery of CellTiter-Glo®Reagent, sample volume and dimensions of the well.All of these factors may affect assay performance. The degree of reagent mixing required may be affected by the method used to add the CellTiter-Glo®Reagent to the assay plates. Automated pipetting devices using a greater or lesser force of fluid delivery may affect the degree of subsequent mixing required.Complete reagent mixing in 96-well plates should be achieved using orbitalplate shaking devices built into many luminometers and the recommended2-minute shaking time. Special electromagnetic shaking devices that use aradius smaller than the well diameter may be required to efficiently mixcontents of 384-well plates. The depth of medium and geometry of themultiwell plates may have an effect on mixing efficiency. We recommend that you take these factors into consideration when performing the assay andempirically determine whether a mixing step is necessary for the individualapplication.LuminometersFor highly sensitive luminometric assays, the luminometer model and settings greatly affect the quality of data obtained. Luminometers from differentmanufacturers will vary in sensitivities and dynamic ranges. We recommend the GloMax®products because these instruments do not require gainadjustments to achieve optimal sensitivity and dynamic range. Additionally, GloMax®instruments are preloaded with Promega protocols for ease of use.If you are not using a GloMax®luminometer, consult the operating manual for your luminometer to determine the optimal settings. The limits should beverified on each instrument before analysis of experimental samples. The assay should be linear in some portion of the detection range of the instrument used.For an individual luminometer there may be different gain settings. Werecommend that you optimize the gain settings.4.C.References1.Crouch, S.P. et al.(1993) The use of ATP bioluminescence as a measure of cellproliferation and cytotoxicity. J. Immunol. Methods160, 81–8.2.Farfan, A.et al.(2004) Multiplexing homogeneous cell-based assays. Cell Notes10, 2–5.3.Riss, T., Moravec, R. and Niles, A. (2005) Selecting cell-based assays for drugdiscovery screening. Cell Notes13, 16–21.4.Kangas, L., Grönroos, M. and Nieminen, A.L. (1984) Bioluminescence of cellular ATP:A new method for evaluating cytotoxic agents in vitro. Med. Biol.62, 338–43.5.Lundin, A. et al.(1986) Estimation of biomass in growing cell lines by adenosinetriphosphate assay.Methods Enzymol. 133, 27–42.6.Sevin, B.U. et al.(1988) Application of an ATP-bioluminescence assay in human tumorchemosensitivity testing. Gynecol. Oncol.31, 191–204.7.Gerhardt, R.T.et al.(1991) Characterization of in vitro chemosensitivity ofperioperative human ovarian malignancies by adenosine triphosphatechemosensitivity assay. Am. J. Obstet. Gynecol. 165, 245–55.8.Petty, R.D. et al.(1995) Comparison of MTT and ATP-based assays for themeasurement of viable cell number. J. Biolumin. Chemilumin.10, 29–34.9.Cree, I.A. et al.(1995) Methotrexate chemosensitivity by ATP luminescence in humanleukemia cell lines and in breast cancer primary cultures: Comparison of the TCA-100assay with a clonogenic assay. AntiCancer Drugs6, 398–404.10.Maehara, Y. et al.(1987) The ATP assay is more sensitive than the succinatedehydrogenase inhibition test for predicting cell viability. Eur. J. Cancer Clin. Oncol.23, 273–6.11.Stanley, P.E. (1986) Extraction of adenosine triphosphate from microbial and somaticcells. Methods Enzymol.133, 14–22.12.Beckers, B. et al.(1986) Application of intracellular ATP determination in lymphocytesfor HLA-typing. J. Biolumin. Chemilumin.1, 47–51.13.Andreotti, P.E. et al.(1995) Chemosensitivity testing of human tumors using amicroplate adenosine triphosphate luminescence assay: Clinical correlation forcisplatin resistance of ovarian carcinoma. Cancer Res. 55, 5276–82.4.D.Related ProductsCell Proliferation ProductsProduct Size Cat.# ApoLive-Glo™ Multiplex Assay10ml G6410 ApoTox-Glo™ Triplex Assay10ml G6320 CellTiter-Fluor™ Cell Viability Assay (fluorescent)10ml G6080 CellTiter-Blue®Cell Viability Assay (resazurin)20ml G8080 CellTiter 96®AQ ueous One SolutionCell Proliferation Assay (MTS, colorimetric)200 assays G3582 CellTiter 96®AQ ueous Non-RadioactiveCell Proliferation Assay (MTS, colorimetric)1,000 assays G5421 CellTiter 96®AQ ueous MTS Reagent Powder1g G1111 CellTiter 96®Non-RadioactiveCell Proliferation Assay (MTT, colorimetric)1,000 assays G4000 Additional sizes available.Cytotoxicity AssaysProduct Size Cat.# CytoTox-Glo™ Cytotoxicity Assay (luminescent)*10ml G9290Mitochondrial ToxGlo™ Assay*10ml G8000 MultiTox-Glo Multiplex Cytotoxicity Assay(luminescent, fluorescent)*10ml G9270 MultiTox-Fluor Multiplex Cytotoxicity Assay(fluorescent)*10ml G9200 CytoTox-Fluor™ Cytotoxicity Assay (fluorescent)*10ml G9260 CytoTox-ONE™ Homogeneous MembraneIntegrity Assay (LDH, fluorometric)*200–800 assays G7890 CytoTox-ONE™ Homogeneous MembraneIntegrity Assay, HTP1,000–4,000 assays G7892 CytoTox 96® Non-Radioactive Cytotoxicity Assay1,000 assays G1780 (LDH, colorimetric)*GSH-Glo™ Glutathione Assay10ml V691150ml V6912 GSH/GSSG-Glo™ Assay10ml V661150ml V6612 *Additional sizes available.LuminometersProduct Size Cat.# GloMax®-Multi+ Detection System with Instinct™ Software:Base Instrument with Shaking 1 each E8032 GloMax®-Multi+ Detection System with Instinct™ Software:Base Instrument with Heating and Shaking 1 each E9032 GloMax®-Multi+ Luminescence Module 1 each E8041Apoptosis ProductsProduct Size Cat.# Caspase-Glo®2 Assay*10ml G0940 Caspase-Glo®6 Assay*10ml G0970 Caspase-Glo®3/7 Assay* 2.5ml G8090 Caspase-Glo®8 Assay* 2.5ml G8200 Caspase-Glo®9 Assay* 2.5ml G8210Apo-ONE®Homogeneous Caspase-3/7 Assay1ml G7792 DeadEnd™ Fluorometric TUNEL System60 reactions G3250 DeadEnd™ Colorimetric TUNEL System20 reactions G7360Anti-ACTIVE®Caspase-3 pAb50µl G7481Anti-PARP p85 Fragment pAb50µl G7341Anti-pS473Akt pAb40µl G7441 Caspase Inhibitor Z-VAD-FMK, 20mM50µl G7231125µl G7232*Additional sizes available.(a)U.S. Pat. Nos. 6,602,677 and 7,241,584, European Pat. No. 1131441, Japanese Pat. Nos. 4537573 and 4520084 and other patents pending(b)U.S. Pat. No. 7,741,067, Japanese Pat. No. 4485470 and other patents pending.(c)U.S. Pat. No. 7,700,310, European Pat. No. 1546374 and other patents pending.(d)U.S. Pat. Nos 7,083,911, 7,452,663 and 7,732,128, European Pat. No. 1383914 and Japanese Pat. Nos. 4125600 and 4275715.(e)The method of recombinant expression of Coleoptera luciferase is covered by U.S. Pat. Nos. 5,583,024, 5,674,713 and 5,700,673.© 2001–2012 Promega Corporation. All Rights Reserved.Anti-ACTIVE, Apo-ONE, Caspase-Glo, CellTiter 96, CellTiter-Blue, CellTiter-Glo, CytoTox 96 and GloMax are registered trademarks of Promega Corporation. ApoTox-Glo, ApoLive-Glo, CellTiter-Fluor, CytoTox-Fluor, CytoTox-Glo, CytoTox-ONE, DeadEnd, GSH-Glo, GSH/GSSG-Glo, Instinct, Mitochondrial ToxGlo and Ultra-Glo are trademarks of Promega Corporation. alamarBlue is a registered trademark of Trek Diagnostic Ssystems, Inc.Products may be covered by pending or issued patents or may have certain limitations. Please visit our Web site for more information.All prices and specifications are subject to change without prior notice.Product claims are subject to change. Please contact Promega Technical Services or access the Promega online catalog for the most up-to-date information on Promega products.。
鲎血水解液美拉德反应产物的挥发性成分研究
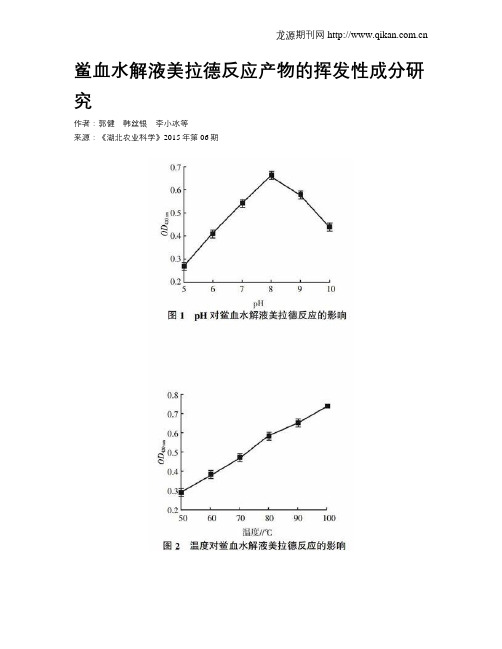
鲎血水解液美拉德反应产物的挥发性成分研究作者:郭健韩丝银李小冰等来源:《湖北农业科学》2015年第06期摘要:以制备鲎试剂产生的废料血浆为原料,经盐酸水解后,水解液进行美拉德反应,通过固相微萃取-气相色谱-质谱法(SPME-GC-MS)测定反应产物中的挥发性成分,分析了其呈香机理。
结果表明,鲎血水解液在pH为8.0,100 ℃加热2 h,其美拉德反应的褐变程度最强,确定了39种挥发性成分,主要包括烃类5.13%、酮类1.47%、酸类2.64%、醛类14.08%、酯类1.24%、酚类0.84%、醇类4.33%及含氮化合物0.26%。
该结果可为开发苯甲醛、食品香精及卷烟改良剂等方面提供参考。
关键词:鲎血;水解液;美拉德反应;挥发性成分中图分类号:TS202 文献标识码:A 文章编号:0439-8114(2015)06-1471-04DOI:10.14088/ki.issn0439-8114.2015.06.049Abstract: The waste blood of horseshoe crab, as the by-product of limulus reagent, was hydrolyzed by hydrochloric acid. The volatile components in Maillard reaction products of hydrolysate were detected with SPME-GC-MS. Its aroma mechanisms was studied. The results showed that the highest browning degree reached at pH 8 after incubating hydrolysate under 100 ℃for 2 h. 39 compositions including 5.13% hydrocarbons,1.47% ketones,2.64% acids,14.08% aldehydes,1.24% esters,0.84% phenols,4.33% alcohols and 0.26% nitrogenous compound were identified in this hydrolysate. It will provide a reference for developing benzaldehyde, food flavor and cigarette ameliorant.Key words:horseshoe crab′s blood; hydrolysate; Maillard reaction; volatile component鲎(Limulus)亦称马蹄蟹,属肢口纲剑尾目海生节肢动物,分为美洲鲎、东方鲎、巨鲎和圆尾鲎,常见于亚洲和北美东海岸,中国主要分布于广东、广西、福建沿海海域[1]。