T载体插入位点附近序列+测序引物
一代测序常见问题及解决策略知识分享

一代测序常见问题及解决策略知识分享测序常见问题及解决策略一、PCR常见问题1.假阴性,不出现扩增条带PCR出现假阴性结果,可从以下几个方面来寻找原因:1)模板:①模板中有杂蛋白;②模板中有Taq酶抑制剂;③在提取制备模板时丢失过多;④模板核酸变性不彻底。
2)酶:酶失活或反应时忘了加酶。
3)Mg2+浓度:Mg2+浓度过高可降低PCR扩增的特异性,浓度过低则影响PCR扩增产量甚至使PCR扩增失败而不出扩增条带。
4)反应条件:变性对PCR扩增来说相当重要,如变性温度低,变性时间短,极有可能出现假阴性;退火温度过低,可致非特异性扩增而降低特异性扩增效率退火温度过高影响引物与模板的结合而降低PCR扩增效率。
5)靶序列变异:靶序列发生突变或缺失,影响引物与模板特异性结合,或因靶序列某段缺失使引物与模板失去互补序列,其PCR扩增是不会成功的。
2.假阳性假阳性:出现的PCR扩增条带与目的靶序列条带一致,有时其条带更整齐,亮度更高。
常见原因有:1)引物设计不合适:选择的扩增序列与非目的扩增序列有同源性,因而在进行PCR扩增时,扩增出的PCR产物为非目的性的序列。
靶序列太短或引物太短,容易出现假阳性。
需重新设计引物。
2)靶序列或扩增产物的交叉污染:这种污染有两种原因:一是整个基因组或大片段的交叉污染,导致假阳性。
这种假阳性可用以下方法解决:操作时应小心轻柔,防止将靶序列吸入加样枪内或溅出离心管外。
二是空气中的小片段核酸污染,这些小片段比靶序列短,但有一定的同源性。
可互相拼接,与引物互补后,可扩增出PCR产物,而导致假阳性的产生,可用巢式PCR方法来减轻或消除。
3.出现非特异性扩增带PCR扩增后出现的条带与预计的大小不一致,或大或小,或者同时出现特异性扩增带与非特异性扩增带。
非特异性条带的出现,其原因:一是引物与靶序列不完全互补、或引物聚合形成二聚体。
二是Mg2+离子浓度过高、退火温度过低,及PCR循环次数过多有关。
T-载体连接方案

T-载体连接全过程第一步:PCR扩增目的基因及纯化1. PCR反应体系:10×扩增缓冲液 5.0ul10mMdNTP 1.0ul引物1 1.0ul引物2 1.0 ul模板DNA 1.0ulTaq DNA聚合 1.0ul加双蒸水至50ul(相同的引物体系可配置PCR MIX体系)2. PCR反应条件:PCR反应条件为温度、时间和循环次数。
95℃ 5min95℃ 45s40~60℃ 50s 循环 35次72℃ 55s72℃ 10min3. PCR期间配置琼脂糖凝胶:PCR结束以后跑胶验证,并照相,标记保存。
4. PCR产物回收(1)单一PCR产物用氯仿法●把PCR产物转移至1.5ml的离心管,加500ul无菌水,500ul氯仿异戊醇(24:1),混匀后10000rpm离心5-10分钟●转移上清(约400ul)至灭菌的新1.5 mL的Eppendorf管,加入2倍体积(780 µL体积即可)的无水乙醇与40ul3MNaAc。
置冰上或-80℃冰箱30分钟以上。
●10000 rpm离心10 min。
弃上清,沉淀用适量的70%的乙醇洗一次,于纸上扣干。
小心不要将沉淀洗掉。
●置于恒温器上干燥(55℃)至无色透明或无乙醇气味,大至要5~10min。
●加入40 µL 无菌水溶解沉淀备用。
(2)多条带PCR产物用切胶回收试剂盒。
●PCR产物进行琼脂糖电泳(大容量上样系统)●切胶并回收目的条带至1.5ml的离心管,称重。
●参照试剂盒说明书进行操作(附录3)。
●用50ul无菌水洗脱离心柱2次备用。
第二步:T-载体连接和转化准备工作:制备感受态细胞,方法见附录一,LB培养基,Amp母液,氯化钙的配制,方法见附录二;1.在微量离心管(PCR管)中配制以下体系:注:T-载体试剂盒开封前要向pMD18-T Vector 管中加入25 ul 无菌水,同时再管上面做标记。
pMD18-T Vector 1.0 ul外源DNA 1.5 ulSolutionI 2.5 ul2.16℃或4℃低温连接1h-2h(最好过夜);3. 连接产物全量加入50 ul 的DH5α感受态细胞中,轻轻摇匀后冰上放置30min;DH5α感受态细胞加5 ul 0.1M的无菌氯化钙做阴性对照;4. 热击:42℃水浴中热击90秒(注:精确计算时间,过长将对细胞造成伤害),热击后立即置于冰上冷2-5分钟(禁止摇动)。
pcr产物连接t载体

pcr产物连接t载体PCR(聚合酶链式反应)是一种常用的分子生物学技术,可在体外迅速扩增特定DNA片段。
常见的应用之一是将PCR产物连接到T载体上,以便进一步研究和分析。
本文将介绍PCR产物连接T载体的步骤和注意事项。
一、材料准备1. PCR产物:经PCR扩增得到的目标DNA片段。
2. T载体:一种载体,其末端具有“T”碱基(胸腺嘧啶)的一对互补序列,可与PCR产物进行连接。
3. T4 DNA连接酶:用于催化连接反应的酶。
二、PCR产物连接T载体步骤1. 酶切T载体:将T载体进行线性化,以便与PCR产物进行连接。
按照酶切酶厂商提供的酶切条件,在适当的缓冲液中进行酶切反应。
2. 纯化线性化的T载体:通过凝胶电泳等方法纯化线性化的T载体,以去除酶切剂和副产物。
3. PCR产物末端处理:将PCR产物进行末端处理,以便与T载体连接。
这一步骤通常使用多聚核苷酸添入酶(如T4 DNA聚合酶)对PCR产物末端进行A尾化处理。
4. T载体与PCR产物连接:将线性化的T载体与末端处理后的PCR产物进行连接,通常在适当的缓冲液中,加入T4 DNA连接酶,进行连接反应。
5. 进一步处理:将连接反应混合液经过适当的热处理或其他方法,以便降低非特异性连接和副产物的生成。
6. 转化大肠杆菌:将连接后的混合物转化到大肠杆菌中,通过培养和筛选,获得含有PCR产物的重组质粒。
三、注意事项1. 杂交温度:连接反应中的杂交温度需根据T载体末端的“T”碱基特性来确定。
一般而言,约为55-65°C。
2. 酶切反应时间:酶切T载体的时间需根据酶切剂的推荐时间来确定,过长或过短的酶切时间都会影响连接效率。
3. 转化菌株的选择:应选择适合的大肠杆菌菌株进行转化,以获得较高的转化效率和选择性。
4. 正负对照:在进行PCR产物连接T载体时,可以设置正负对照实验,以确保连接结果的准确性。
总结:PCR产物连接T载体是一种常用的分子生物学技术,它为后续的DNA克隆和遗传工程研究提供了重要的工具和平台。
引物设计原则及酶切位点选择和设计

引物设计原则及酶切位点选择和设计[整理]:最初的时候,由于害怕设计酶切位点最后且不开,所以经常采用最通用的方法,用T载体克隆解决问题,但后来发现她也有问题,就是浓度提不上去,你需要体大量的载体来酶切,所以感到还是直接扩增好一点。
但这就需要你仔细设计引物。
连入质粒中的重要目的就是进行酶切和连接,当然首先就是在想要合成或者是进行PCR扩增出靶基因的时候在核酸的两端接入酶切位点,酶切位点是与你的质粒的特点相关的,可以在质粒的图谱说明书上找取相应的位点,进行设计。
(一)设计引物前应做的准备工作:准备载体图谱,大致准备把片断插在那个部分准备一本所买公司的酶的商品目录,便于查酶的各种数据及两种酶是否可以配用(二)设计引物所要考虑的问题两个位点应是载体上的,,所连接片断上没有这两个位点,且距离不能太近,往往导致两个酶都切不好。
因此,紧挨在一起,只能切一个,除非恰好是与上面两个酶在一起的酶切位点。
我看promega的说明书上说,最好隔四个。
还有一种情况是:不能有碱基的交叉,比如AGATCTTAAG,这样的位点比较难切。
两个酶切点最好不要是同尾酶(切下来的残基不要互补),否则效果相当于单酶切。
最好使用酶切效率高的。
最好使用双酶切有共同buffer的酶。
最好使用较常用的酶(如hind3, bamhl, ecorl等),最好使用自己实验室有的酶,这样可以省钱。
Tm的计算,关于Tm的问题,很多的战友都有疑惑。
其实园子里有很多的解释了。
Tm叫溶解温度(melting temperature, Tm),即是DNA双链溶解所需的温度。
大家可以理解,这个温度是由互补的DNA区域决定的,而不互补的区域对DNA的溶解是没有作用的。
因此,对于引物的Tm,只有和模板互补的区域对Tm才有贡献。
计算Tm时,只计算互补的区域(除非你的酶切位点也与模板互补)。
不少战友设计的引物都Tm过低,是因为他们误把保护碱基和酶切位点都计算到Tm里了,最后的结果是导致了PCR反应的诸多困难。
pGEM-T中文说明书

20 个反应
包括:
1.2µg
pGEM®-T Easy 载体(50ng/µl)
12µl
插入 DNA 对照(4ng/µl)
100 U 200µl
II. 载体图谱……………………………………………………………………………….. 3 III. 产品组分…………………………………………………………………………….. 6 IV. 采用 pGEM®-T 和 pGEM®-T Easy 载体和 2×快速连接缓冲液进行连接………. 7
A. 操作步骤………………………………………………………………………… 7 V. 采用 pGEM®-T 和 pGEM®-T Easy 载体的连接反应产物的转化………………... 7 VI. 注意事项…………………………….………………………………………………. 8
产品
包装
pGEM®-T 载体系统Ⅰ
20 个反应
包括:
1.2µg
pGEM®-T 载体(50ng/µl)
12µl
插入 DNA 对照(4ng/µl)
100 U 200µl
1
T4 DNA 连接酶 T4 DNA 连接酶的 2×快速连接缓冲液 操作手册
目录号 A3600
产品
包装
pGEM®-T 载体系统Ⅱ
20 个反应
注意:本中文操作手册仅供实验参考,在实际使用中请详细对照原英文技术手册 TM042。如遇到问题请与 Promega 公司北京
办事处联系,TEL: 010-68498287;E-mail: techserv@
技术手册号码:CTM042
第 5 页 共 23页
III.产品组成
1 10-128 139-158
141 176-197
总结了测序中经常遇到的问题及原因分析,希望对大家有帮助!

总结了测序中经常遇到的问题及原因分析,希望对大家有帮助!Q:测序结果中为什么找不到引物序列?A:找不到用来测序的引物,这是正常的,因为测序的方法是荧光标记测序法,仪器通过检测 ddNTP 上的荧光来读取所测序列,而引物本身是不被标记的,所以仪器无法检测到;找不到所测片段的扩增引物,这种情况是因为您所采用的酶切位点离所用的测序引物距离太近,一般荧光燃料会干扰几十个碱基的读取,这部分碱基会损失掉,损失掉的序列很可能就包含引物的序列,所以引物的序列无法找到;测出的引物序列是原引物序列的反向互补序列,这是因为 TA 克隆的插入没有方向性,如果插入片段是相反的,这时就要反向互补查找引物;测出的结果为空载体,这是因为由于某种原因导致质粒上没有插入外源片段,这时所测的为载体序列,所以就找不到引物了;存在单引物扩增,有一条引物的特异性不好,有多个结合位点,导致只有一条引物参与扩增。
Q:为什么测序结果中引物的序列有个别碱基不同了?A: 这是因为引物区同样存在错配的可能,出现这种可能性有两种: 合成错误和引物区有错配我们可以挑选同批次 2个以上的克隆进行测序验证,如果结果完全相同,应该是合成错误;如果在引物的相同位置错误的碱基不一样那就应该是引物区有错配。
Q:哪些引物不适合作为测序引物?A:①兼并引物:简并引物要在测序模板上有多个结合位点,直接影响测序结果;②随机引物:如 RAPD 引物,随机引物一般都比较短,所用退火温度低,在测序反应的条件下,不能很好地与模板结合;③过长的引物:一般要求测序引物不大于 24bp,过长的引物在测序反应的较低的条件下容易在测序模板上有多个结合位点,导致测序结果背景增高。
另外,较长的引物纯度也将难以保证。
通常用于测序的引物纯度要在 90%以上,引物纯度低时,测序反应的背景将明显增大,直接影响到测序结果;④有特殊标记的引物:该情况主要指荧光标记的引物。
我们测序反应的四种碱基都是荧光标记的,这样,荧光标记的引物将产生干扰。
M5HiPerpTOPO-TACloningKit使用说明书

M5 HiPer pTOPO-TACloning Kit使用说明书产品名称单位货号M5 Hiper pTOPO-TA Cloning Kit 20T MF019-01M5 Hiper pTOPO-TA Cloning Kit 4×20T MF019-04【储存条件】长期保存,请置于-20˚C,有效期12个月。
使用后请及时放入-20˚C保存以保证酶的活性。
【产品简介】本制品和传统的T4连接酶原理不同,它利用了Topoisomerase可以在瞬间(几秒钟-几分钟)、高效(接近100%)连接DNA片段的原理采用本公司独创的工艺制成。
【产品特点】1. 可以在瞬间(几秒钟-几分钟)完成连接。
2. 无需冰浴和热休克,室温5分钟内完成转化;无需1小时复苏,只需37˚C 10分钟复苏便可以涂板。
从连接到涂板只需15-20分钟。
3. 无自连、零背景,无需繁琐蓝白斑筛选,见到长出的克隆便是有插入的(接近100%)。
4. 可以连接长达10kb片段。
5. 测序可以采用M13F/M13R通用引物测序(见后面图谱)【产品组份】20T 4×20TM5 HiPer pTOPO-TA Vector(30ng/µl) 20µl 80µl1000bp Control(30ng/µl)5µl 5µl10x Enhancer 20µl 80µl【操作步骤】1. 连接反应的准备:<注意:PCR引物不能磷酸化>。
使用能在PCR产物末端加一个突出的3’-A的酶系列扩增(如Taq、T aqHiFi、Tth、AmpliTaq、KlenTaq DNA Polymerase)。
PCR产物(仅有目的条带、无非特异条带和引物二聚体)可直接进行连接反应,无需纯化;如果有非特异扩增,建议胶回收纯化(货号:MF029)。
如果是以质粒为模板的PCR产物,也必须进行切胶纯化,因为模板质粒也可能长出菌落(但不是想构建的目的载体)。
引物找周边序列及引物扩增目的序列
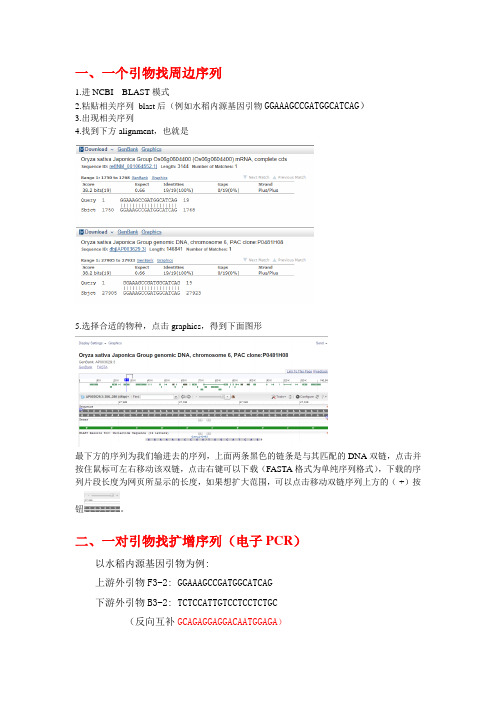
一、一个引物找周边序列1.进NCBI BLAST模式2.粘贴相关序列blast后(例如水稻内源基因引物GGAAAGCCGATGGCATCAG)3.出现相关序列4.找到下方alignment,也就是5.选择合适的物种,点击graphics,得到下面图形最下方的序列为我们输进去的序列,上面两条黑色的链条是与其匹配的DNA双链,点击并按住鼠标可左右移动该双链,点击右键可以下载(FASTA格式为单纯序列格式),下载的序列片段长度为网页所显示的长度,如果想扩大范围,可以点击移动双链序列上方的(-+)按钮。
二、一对引物找扩增序列(电子PCR)以水稻内源基因引物为例:上游外引物F3-2: GGAAAGCCGATGGCATCAG下游外引物B3-2: TCTCCATTGTCCTCCTCTGC(反向互补GCAGAGGAGGACAATGGAGA)进NCBI BLAST页面,找到下面的特殊blast中的primer-blast点开primer-blast页面,A.输入自己的引物B.选择数据库database(如Mrna,或genomes)C.输入物种(范围可大可小,如plant植物)D.点,就可以出结果。
E.点击合适物种链接如,得全基因图;F. 也可以继续点击graphics,得下图(点击-+按键调节显示序列,点击并按住可以拖动双链至合适位置(D上显示1750-1928为目的序列):G.在双链上点击右键,选save,选fasta,然后保存到电脑,此.fa文件可以用IE打开,得到下列目的序列>gi|115468835|ref|NM_001064552.1|:1741-1966 Oryza sativa Japonica Group Os06g0604400 (Os06g0604400) mRNA, complete cdsCATTTGCAT GGAAAGCCGATGGCATCAG ACCAGAAGACATTGAGGCGTTGCATCTGATTCCCAGAGAGA TTTCTCTGAAGATTGTGAACAAGATTGAAGCTGGTGAGCGTTTTGCAGTCTATGTTGTGCTGCCAATGT GGCCTGAAGGACCTCCTGCTAGTGGATCAGTGCAGGCAATACTGGATTG GCAGAGGAGGACAATGGAGA TGATGTACTATGATATTGC3.水稻内源基因LAMP扩增片段及引物水稻内源基因引物2:上游外引物F3-2: GGAAAGCCGATGGCATCAG下游外引物B3-2: TCTCCATTGTCCTCCTCTGC上游内引物FIP-2:GCAAAACGCTCACCAGCTTCAA(反向互补TTGAAGCTGGTGAGCGTTTTGC) -GACATTGAGGCGTTGCATCT下游内引物BIP-2:ATGTTGTGCTGCCAATGTGGC-CCAGTATTGCCTGCACTGAT(反向互补ATCAGTGCAGGCAATACTGG)内源基因内源基因扩增序列如下:CATTTGCAT GGAAAGCCGATGGCATCAG ACCAGAA GACATTGAGGCGTTGCATCT GATTCCCAGAGAGA TTTCTCTGAAGATTGTGAACAAGA TTGAAGCTGGTGAGCGTTTTGC AGTCT ATGTTGTGCTGCCAATGT GGC CTGAAGGACCTCCTGCTAGTGG ATCAGTGCAGGCAATACTGG ATTG GCAGAGGAGGACAATGGAGA TGATGTACTATGATATTGC。
测序常见问题及其分析
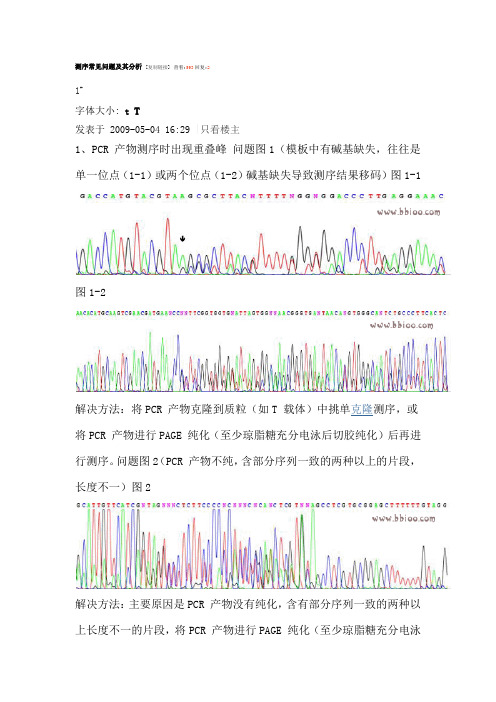
测序常见问题及其分析[复制链接]查看:593回复:21#字体大小: t T发表于 2009-05-04 16:29 |只看楼主1、PCR 产物测序时出现重叠峰问题图1(模板中有碱基缺失,往往是单一位点(1-1)或两个位点(1-2)碱基缺失导致测序结果移码)图1-1图1-2解决方法:将PCR 产物克隆到质粒(如T 载体)中挑单克隆测序,或将PCR 产物进行PAGE 纯化(至少琼脂糖充分电泳后切胶纯化)后再进行测序。
问题图2(PCR 产物不纯,含部分序列一致的两种以上的片段,长度不一)图2解决方法:主要原因是PCR 产物没有纯化,含有部分序列一致的两种以上长度不一的片段,将PCR 产物进行PAGE 纯化(至少琼脂糖充分电泳后切胶纯化)后再进行测序,便可解决。
问题图3(测序引物有碱基缺失)测序引物有碱基缺失(一般是引物的5'端缺失),和模板的碱基缺失即图1 有些类似,所不同的是模板碱基缺失一般是在一段正常测序序列后才出现移码,而引物碱基缺失的话,则从测序一开始就出现移码,表面在图形上便是一开始就是严重的峰形重叠。
解决方法:重新合成引物,或将引物进行PAGE 纯化2、克隆测序时出现峰形重叠原因:所挑选的重组子不是单克隆,所提供的测序用质粒中含有两种以上插入片段不同的质粒;或是是送测序的菌液污染解决方法:重新挑单克隆的菌落(划线分离单菌落),提质粒或送菌液再次测序。
3、样品有杂合/突变位点模板中有杂合型突变,也就说模板本身在这个位点出现突变;或者是从基因组中扩增出来的杂合位点。
如果模板有杂合(突变或缺失),那么测序图形中其他的位點一般都是單一的峰形,然后突然在某一個位點出現重叠峰(如图中箭頭所示)。
解决方法:建议将DNA 片段克隆到载体再测序。
4、polyA/T 和C/G cluster 导致的套峰和测序信号衰减图4-1图4-3图4-4RACE 测序时经常遇到图4-1 和图4-4 的情形,解决方法:从另一端测序;但如果这样的序列出现在中间,呵呵,目前还没有很好的解决方法,要看测序公司的本事了。
生工pUCm-T载体

T-载体PCR产物克隆试剂盒产品编号:SK2213/SK2214包装规格:20次/100次试剂盒组成组分SK2213,20次SK2214,100次T Vector 1 µg 5 µg10×Ligation Buffer 50 µl 200 µl50% PEG 4000 50 µl 200 µlT4 DNA Ligase, 5 U/µl 100 U 500 USterilized ddH2O 1 ml 1 ml操作手册1份1份保存方法及注意事项该产品低温运输,-20°C保存,有效期见包装。
产品介绍pUCm-T Vector是一种高效克隆PCR产物(TA Cloning)的专用载体。
本载体由pUC系列载体改建而成,在pUC载体的多克隆位点处插入特殊的限制性内切酶识别位点,酶切后能在载体的3’端形成悬挂的“T”末端,大大提高了3’末端A突出PCR产物的连接、克隆效率。
本载体在插入位点两端独特设计了两个Pst I 识别位点,可以用Pst I 单酶切对插入片段进行检测,大大方便了客户的酶切检测步骤。
经本载体克隆的PCR片段,可以用M13通用引物和T7启动子引物进行测序。
实验准备1. PCR产物3’末端带有突出的A碱基:Taq、Tth、AmpliTaq、KlenTaq DNA聚合酶扩增的PCR产物3’末端均带有突出的A碱基。
建议扩增的PCR产物通过琼脂糖凝胶电泳进行回收,再进行连接转化实验,可以使用生工产品柱式DNA胶回收试剂盒(SK8131/8132)。
如果PCR扩增产物特异性很高,可直接使用生工产品柱式PCR产物纯化试剂盒(SK8145/8146)纯化PCR产物。
2. PCR产物3’末端没有突出的A碱基:Pfu、Pwo、Tli或DeepVent等DNA聚合酶具有3’5’外切酶活性,其扩增的PCR产物末端可能不带有3’ A,要对这种平末端PCR产物进行克隆,应先对其进行3’末端加A,再进行连接转化实验。
一代测序常见问题及解决策略

测序常见问题及解决策略一、PCR常见问题1.假阴性,不出现扩增条带PCR出现假阴性结果,可从以下几个方面来寻找原因:1)模板:①模板中有杂蛋白;②模板中有Taq酶抑制剂;③在提取制备模板时丢失过多;④模板核酸变性不彻底。
2)酶:酶失活或反应时忘了加酶。
3)Mg2+浓度:Mg2+浓度过高可降低PCR扩增的特异性,浓度过低则影响PCR 扩增产量甚至使PCR扩增失败而不出扩增条带。
4)反应条件:变性对PCR扩增来说相当重要,如变性温度低,变性时间短,极有可能出现假阴性;退火温度过低,可致非特异性扩增而降低特异性扩增效率退火温度过高影响引物与模板的结合而降低PCR扩增效率。
5)靶序列变异:靶序列发生突变或缺失,影响引物与模板特异性结合,或因靶序列某段缺失使引物与模板失去互补序列,其PCR扩增是不会成功的。
2.假阳性假阳性:出现的PCR扩增条带与目的靶序列条带一致,有时其条带更整齐,亮度更高。
常见原因有:1)引物设计不合适:选择的扩增序列与非目的扩增序列有同源性,因而在进行PCR扩增时,扩增出的PCR产物为非目的性的序列。
靶序列太短或引物太短,容易出现假阳性。
需重新设计引物。
2)靶序列或扩增产物的交叉污染:这种污染有两种原因:一是整个基因组或大片段的交叉污染,导致假阳性。
这种假阳性可用以下方法解决:操作时应小心轻柔,防止将靶序列吸入加样枪内或溅出离心管外。
二是空气中的小片段核酸污染,这些小片段比靶序列短,但有一定的同源性。
可互相拼接,与引物互补后,可扩增出PCR产物,而导致假阳性的产生,可用巢式PCR方法来减轻或消除。
3.出现非特异性扩增带PCR扩增后出现的条带与预计的大小不一致,或大或小,或者同时出现特异性扩增带与非特异性扩增带。
非特异性条带的出现,其原因:一是引物与靶序列不完全互补、或引物聚合形成二聚体。
二是Mg2+离子浓度过高、退火温度过低,及PCR循环次数过多有关。
三是酶的质和量,往往一些来源的酶易出现非特异条带而另一来源的酶则不出现,酶量过多有时也会出现非特异性扩增。
测序通用引物序列

测序通用引物序列常见载体的测序引物:Primer of Vector:Vector:Primer(F);Primer(R)pACT T7 T3pACT2 GAL4 AD pACT2-RpAS2-1 GAL4 BD pAS2-1.RpB42AD pB42ADF pB42ADRpBACPAK8 BAC1 BAC2pBK-CMV T7 T3PBS(SK/KS)/M13- M13F/T7 T3/M13RpBV220 PBV220F PBV220RpCAMBIA 1301(1300) P1 P2pCAMBIA 2300 M13R(-48) M13F(-47)PCANTB5E S1/M13R S6pCAT3-enhancer RVP3 此载体无反向引物pcDNA3.0 CMV-F/T7 SP6/BGHpcDNA3.1 T7 BGHpcDNA4 T7/CMV-F BGHpcDNA6 T7/CMV-F(J21025在T7前面)BGHpcDNAII T7 SP6pCE2.1 M13F M13RpCEP4 pCEP-F EBV-RpCF-T M13F M13RpCI T7(17Base)此载体无反向引物pCI-neo T7(17Base)T3pCMS-EGFP T7 T3pCMV-3Tag-4A T3 /PFLAG-CMV-F T7pCMV5 pCMV5F pCMV5RpCMV5-Flag pCMV5F pCMV5RpCMV-MYC/HA pCMV-F pCMV-RpCMV-Sport M13F/T7 SP6/M13RpCMV-Tag(KAN+) T3 T7pCR2.1-TOPO M13F/T7 M13RpCR3.1 T7 BGHpCS2 SP6 T7pDonar M13F M13RpDONR221 M13F M13RpDrive T7 SP6pDRIveR(KAN+) T7 SP6pDsRED1-C1 pDsRED-ex-C1-F pEGFP-N-3’(距离很近,一般不用) pDsRED2-C1(KAN+) pDsRED-ex-C1-F pEGFP-N-3’pDSRED-N1(KAN+) pEGFP-N-5’ PDSRED-N-RpECFP-C pEGFP-C-5’ pECFP-C-3′pECFP-N1 pEGFP-N-5’ pEGFP-N-3’pEF/myc/ER pEF-F pCDNA3.1RpEGFP-C(KAN+) pEGFP-C-5’ pEGFP-C-3′pEGFP-N(KAN+) pEGFP-N-5’ pEGFP-N-3’pENTR/D-TOPO(KAN+) M13F M13RpET-*(His)(KAN+) T7 T7 TerpET22(a,b,c)(AMP+) T7 T7 TerpET28,30,24,49(KAN+) T7 T7 TerpET32(a,b,c)(AMP+) T7/S.tag T7 TerpET50(amp+) T7/s.tag T7TERpET44(AMP+) T7/s.tag T7 TerpEYFP-N1 pEGFP-N-5’ pEGFP-N-3’pEYFP-C1 pEGFP-C-5’ pEGFP-C-3′pFLAG-CMV pFLAG-CMV -F pFLAG-CMV -R pGAD424 GAL4 AD 3′ADpGADGH GAL4 AD 3′ADpGADT7-Rec GAL4 AD/T7 3′ADpGAPZa-A a-FACTOR 3′AOXpGBKT7(Kana+) T7 3′BDpGEM-T(-Easy) M13F/T7 SP6/M13RpGEX-(*)T pGEX-4T-5’ pGEX-4T-3’pGL2 GLP1 GLP2pGL3 RVP3 GLP2(RVP4)(没有特别说明用GLP2) PinpointTM Vector Pinpoint Primer SP6pIRES PIRES-F/T7 T3/PIRES-R (J40720)pIRES2-EGFP pIRES2-EGFP.P5’ pIRES2-EGFP.P3’pLXSN pLXSN-F pLXSN-RpMAL-c2E MalE primer M13F(-47)pMAL-C2x P5’ P3’pMAL-p2X MalE primer M13F(-47)PMD18-T M13R(-48) M13F(-47)PMD19-T M13F(-47) M13R(-48)pPIC9K(AMP+、ZEO+)5’AOX/a-factor 3’AOX pPICZa 5′AOX/PPICZa-F 3′AOXpPROEXHTA M13R(-48) 无反向引物pQE30or40 pQE30-F pQE-R:pRSET T7 T7TERpSilencer3.1-H1 hygro M13F(-47) 3.0REV pSILENCEV2.0-U6 T7 2.0REVpSK01-T M13F M13RpSK-CMV(KAN+) T7 T3pSP72 T7 SP6pSport1 SP6/M13F(-47) T7/M13RpSUPER T7 T3pTA2 M13F/T7(优先使用M13F)T3/M13RpT-Adv M13F /T7 M13RpTARGET TM pTarget.F(在T7前面)/T7 pTarget.R pTO-T7 T7 T7TERpTRC99a-c pTRC99C-F pTRC99C-RpTriPLEx2 5′pTriPLEx2 T7pTWIN-1 T7 T7TER(客户确认再用)pUC18(19)/118(119) M13F(-47) M13R(-48)pUCm-T M13F/T7 M13RpVAX1 T7 BGHpWSK29 T7/M13F/M13F(-47) T3/M13R/M13R(-48)pXT7 T7 SP6pYES2 T7 pYes2.RpLEXA pLEXA-F pLEXA-RpLNCX pLNCX-F pLNCX-RpRECEIVER CMV-F 此载体无反向引物pSHUTTLE-CMV PSHUTTLE-CMV-F PSHUTTLE-CMV-R pSP64/65 SP6 此载体无反向引物pSTBlue-1 T7 SP6pT7Blue(R) T7 M13F(-47)引物名称序列(5′-3′):M13R:CAG GAA ACA GCT ATG ACCM13F:TGT AAA ACG ACG GCC AGTM13F(-47):CGC CAG GGT TTT CCC AGT CAC GACM13R(-48):AGC GGA TAA CAA TTT CAC ACA GGA M13(-96):CCC TCA TAG TTA GCG TAA CGSP6:ATT TAG GTG ACA CTA TAGT7:TAA TAC GAC TCA CTA TAG GGT7 terminator:TGC TAG TTA TTG CTC AGC GGT3:ATT AAC CCT CAC TAA AGG GApGEX-4T-5′:GGG CTG GCA AGC CAC GTT TGG TG pGEX-4T-3′:CCG GGA GCT GCA TGT GTC AGA GG GLp1:TGT ATC TTA TGG TAC TGT AAC TGGLp2:CTT TAT GTT TTT GGC GTC TTC CARVp3:CTA GCA AAA TAG GCT GTC CCRVp4:GAC GAT AGT CAT GCC CCG CGpcDNA3.1R:TAG AAG GCA CAG TCG AGGPinPoint primer:CGT GAC GCG GTG CAG GGC G pCMV-F:TCT AAA AGC TGC GGA ATT GTpCMV-R:TCCAAACTCATCAATGTATCpTRC99C-F: TTG CGC CGA CAT CAT AACpTRC99C-R: CTGCGTTCTGATTTAATCTGpCEP-F: AGA GCT CGT TTA GTG AAC CGEBV-R : GTG GTT TGT CCA AAC TCA TCpIRES2-EGFP.P5’: GTA GGC GTG TAC GGT GGG AG pIRES2-EGFP.P3’: AAC GCA CAC CGG CCT TAT TC3′AD: AGA TGG TGC ACG ATG CAC AGCMV -F:CGC AAA TGG GCG GTA GGC GTGS1:CAA CGT GAA AAA ATT ATT ATT CGCS6:GTA AAT GAA TTT TCT GTA GTA GG5`AOX1:GAC TGG TTC CAA TTG ACA AGC3`AOX1:GCA AAT GGC ATT CTG ACA TCCα-Factor:TAC TAT TGC CAG CAT TGC TGCGAL4 AD:TAC CAC TAC AAT GGA TGpACT2-R:GTGCACGATGCACAGTTGAApB42ADF:CCA GCC TCT TGC TGA GTG GAG ATGpB42ADR:AAG CCG ACA ACC TTG ATT GGA GpEGFP-N-5’:TGG GAG GTC TAT ATA AGC AGA G pEGFP-N-3’:CGT CGC CGT CCA GCT CGA CCA G pEGFP-C-5’:CAT GGT CCT GCT GGA GTT CGT G pEGFP-C-3′ :TAT GGC TGA TTA TGA TCA GTPBV220F:AAG AAG GGC AGC ATT CAA AGPBV220R:CTG CGT TCT GAT TTA ATC TGU6:ATG GAC TAT CAT ATG CTT ACC GTA2.0rev primer:AGG CGA TTA AGT TGG GTA3.0rev:GAG TTA GCT CAC TCA TTA GGCS.tag:GAA CGC CAG CAC ATG GAC5′TriplEx2:CTC CGA GAT CTG GAC GAG CRVP4:GAC GAT AGT CAT GCC CCG CGRVP3:CTA GCA AAA TAG GCT GTC CCpQE30-R:GTT CTG AGG TCA TTA CTG GpQE30-F:TGA GCG GAT AAC AAT TTC ACpEF-F:TCA AGC CTC AGA CAG TGG TTCpDSRED-N1-R:TGA AGC GCA TGA ACT CCT TG pDsRED-express-C1-F:TCC CAC AAC GAG GAC TAC AC pCMV5R:ATT ATA GAG GAC ACC TAG TCpCMV5F:TTC CAA AAT GTC GTA ATA ACP5′:TGC GTA CTG CGG TGA TCA ACP3′:CTG CAA GGC GAT TAA GTT GGBAC2:ACG CAC AGA ATC TAG CGC TTBAC1:AAC CAT CTC GCA AAT AAA TA3′BD:TAA GAG TCA CTT TAA AAT TTG TAT AC pTarget.F:CCA GGA TTT TCC CAG TCA CpTarget.R:GGC TTT ACA CTT TAT GCT TCT7(17base):ACA TCC ACT TTG CCT TTC TCpYes2.R:TCG GTT AGA GCG GAT GTGGAL4 BD:TCA TCG GAA GAG AGT AGpAS2-1.R:AAG AGT TAC TCA AGA ACA AGA ApFlag-CMV-F: GGT AGG CGT GTA CGG TGGpFlag-CMV-R: GCA CTG GAG TGG CAA CTTpIRES-F: CTT ACT GAC ATC CAC TTT GCpIRES.R: CAC TGC ATT CTA GTT GTG GTP1:CCA GGC TTT ACA CTT TAT GCP2:GCG ATT AAG TTG GGT AAC GCpLEXA-F:CGT CAG CAG AGC TTC ACC ATpLEXA-R:TAA AAC CTA AGA GTC ACT TTpLNCX-F:AGC TCG TTT AGT GAA CCG TCA GAT CG pLNCX-R:ACC TAC AGG TGG GGT CTT TCA TTC CC pLXSN-F:CTT GAA CCT CCT CGT TCG ACpLXSN-R:GTT GCT GAC TAA TTG AGA TGpSHUTTLE-CMV-F:GGT CTA TAT AAG CAG AGG TG pSHUTTLE-CMV-R:GTG GTA TGG CTG ATT ATG ATC AG MalE Primer:GGT CGT CAG ACT GTC GAT GAA GCC。
引物步移法测序

引物步移法测序•复制网址•新浪微博•豆瓣社区•腾讯微博•开心网•人人网只看楼主听说长片段(>500)测序一般采用所谓“引物步移”的方法,根据测序结果设计引物继续walking,直至测通全长。
那么根据测序结果设计一个引物,另一端用什么引物?是用随机引物吗?如果用随机引物的话,这组随机引物怎么设计?各位高手有没有这方面的资料可以看看,先谢谢了0票票数good good study day day up flameofstar edited on 2006-08-20 13:57 举报•• 【专题讨论】将支气管镜操作进行到底。
热烈欢迎上海长海医院呼吸内科黄海东主治医师做客丁香园。
xrsgjt 2006-11-13 14:14 消息引用收藏分享测序引物只要一条就可以了啊。
为什么需要两条呢?呵呵。
只要在Re:请问什么是步移法步移法一般认为是由于反向PCR可用于研究与已知DNA片段相连的未知染色体序列,因此又被称为染色体步移法。
此法所选择的引物虽与已知DNA序列互补,但两引物3’端是反向的。
染色体步移技术主要应用于克隆启动子、步查获得新物种中基因的非保守区域、鉴定T-DNA或转座子的插入位点、染色体测序工作中的空隙填补,从而获得完整的基因组序列等方面。
screen.width-333)this.width=screen.width-333" width=640 height=404 title="Click to view full 染色体步移法1.JPG (700 X 442)" border=0align=absmiddle>克隆步移法作者:agui674提交日期:2007-1-22 16:22:00在基因组测序中,存在着两种不同的战略,即全基因组随机测序战略和以物理图为基础的以大片断克隆为单位的定向测序战略。
两者的最大区别在于是否依赖基因组作图。
全基因组随机测序战略主要采用鸟枪法(shotgun)。
T载体的应用及研究进展

T载体的应用及研究进展田生礼1 梁秀怡1 梁志成2【摘要】摘要T载体可直接用于克隆PCR产物,因此应用于大规模的基因克隆中,具有快速、高效、操作简单等优点。
对T载体进行了综述,介绍了T载体在生物学众多领域的广泛应用,总结了当今T载体的技术创新和改良,特别介绍了目前最新的新型多功能T载体——定向克隆表达型T载体,并且探讨了T载体未来的发展趋势。
【期刊名称】科学技术与工程【年(卷),期】2014(014)030【总页数】8【关键词】关键词 T载体 TA克隆定向克隆表达型T载体生物科学以基因的扩增和克隆为中心的分子生物学实验操作在生命科学发展进程中始终起着重要的作用。
近年来,随着基因组测序工作的完成,产生大量的基因组信息,将日益丰富的信息通过分析与归类成为有用的知识,首先需要面临大量的基因克隆工作[1,2]。
聚合酶链式反应(PCR)是常用的DNA体外扩增技术,T 载体作为一种新型的载体,可直接用于克隆PCR产物且操作简单,因此应用于大规模的基因克隆与表达的研究[3—6]。
T载体对PCR产物克隆的原理是由于PCR反应过程中Taq DNA聚合酶具有非模板依赖性的末端转移酶活性,因此PCR产物3′端被添加单个腺嘌呤残基(dA)形成突出的末端,可与T载体的3′末端悬挂的胸腺嘧啶残基(dT)形成互补配对,快速进行PCR产物的克隆,该克隆方法被称为TA克隆[7—9],该方法通过黏性末端将载体与片段连接一起,连接效率比平末端连接效率高。
但目前市面上供应的T载体多为进口试剂盒,其价格昂贵,质量批次不稳定,制约了T载体的进一步广泛使用[10]。
在此,笔者简要综述了T载体的常规的制备方法,以及多个针对常规制备方法的弊端和实际需求对T载体进行一些技术创新和改良的尝试。
1 T载体的种类和应用1.1 T载体的分类随着生物学研究和基因工程的进步,已发展出多种T载体,根据其功能可分为克隆型T载体和表达型T载体。
克隆型T载体允许外源的DNA插入,储存,并可在宿主中扩增[11,12],主要是DNA水平上的操作,如Takara公司的pMDTM系列克隆载体。
t载体与目标基因连接[说明]
![t载体与目标基因连接[说明]](https://img.taocdn.com/s3/m/3ba03115b42acfc789eb172ded630b1c59ee9b2c.png)
一. 重组质粒的构建T质粒载体重组的DNA分子是在DNA连接酶的作用下,有Mg2 、ATP存在的连接缓冲系统中,将分别经酶切的载体分子与外源DNA分子进行连接。
DNA连接酶有两种:T4噬菌体DNA连接酶和大肠杆菌DNA连接酶。
两种DNA连接酶都有将两个带有相同粘性末端的DNA分子连在一起的功能,而且T4噬菌体DNA连接酶还有一种大肠杆菌DNA连接酶没有的特性,即能使两个平末端的双链DNA分子连接起来。
但这种连接的效率比粘性末端的连接率低,一般可通过提高T4噬菌体DNA 连接酶浓度或增加DNA浓度来提高平末端的连接效率。
T4噬菌体DNA 连接酶催化DNA 连接反应分为3 步:首先,T4 DNA 连接酶与辅因子ATP形成酶-ATP复合物;然后,酶-ATP复合物再结合到具有5’磷酸基和3’羟基切口的DNA上,使DNA腺苷化;最后产生一个新的磷酸二酯键,把切口封起来。
连接反应通常将两个不同大小的片断相连。
很多DNA聚合酶在进行PCR扩增时会在PCR产物双链DNA每条链的3’端加上一个突出的碱基A。
pUCm-T载体是一种已经线性化的载体,载体每条链的3’端带有一个突出的T。
这样,pUCm-T载体的两端就可以和PCR产物的两端进行正确的AT配对,在连接酶的催化下,就可以把PCR产物连接到pUCm-T载体中,形成含有目的片断的重组载体。
连接反应的温度在37℃时有利于连接酶的活性。
但是在这个温度下粘末端的氢键结合是不稳定的。
因此采取折中的温度,即12-16℃,连接12-16h(过夜),这样既可最大限度地发挥连接酶的活性,又兼顾到短暂配对结构的稳定。
二. 感受态制备原理细菌在0°C CaCl2低渗溶液中胀成球形,丢失部分膜蛋白,成为容易吸收外源DNA的感受态。
三. β-半乳糖甘酶显色反应选择法LacZ基因是大肠杆菌乳糖操纵子中的一个基因,可以编码β—半乳糖核苷酶。
β—半乳糖核苷酶是由4个亚基组成的四聚体,可催化乳糖的水解.用X-Gal为底物进行染色时,呈蓝色。
蛋白透析时产生的絮状沉淀真的令不少人痛心不已,有时明

蛋白透析时产生的絮状沉淀真的令不少人痛心不已,有时明明看了又看,都没发现哪里不对,那么,这其中可能是哪里出现问题,还有这些问题从哪些方面着手解决最合适呢?请看下文分析:蛋白透析产生沉淀可能有以下原因:1.由高浓度的蛋白变性试剂(如8m尿素),到低浓度,再到不含变性剂的buffer,如果条件变化剧烈,使得蛋白不正确折叠而沉淀,这种情况比较常见。
如果选择透析的方法,一定要多加几个中间浓度梯度,而且注意低温(4度),这有利于蛋白正确折叠。
2.透析袋本身有一定的吸附,可以通过磁力搅拌来降低吸附。
3.透析液PH靠近等电点,像catzp说的,换buffer。
4.透析袋不干净?楼主不会范这种错误吧。
或者没认真处理,有杂质、金属离子建议:1 可以采用梯度浓度透析,先采用与你纯化后蛋白盐浓度相近的透析液透析,4-8小时后换更小浓度的盐溶液透析,最后采用生理盐水或PBS(pH 7.4)透析。
例如:用6M盐酸胍沉淀的目的蛋白,第一步透析可采用4M盐酸胍透析,4-8小时后采用2M 盐酸胍透析,再之后可以使用PBS或生理盐水。
2 透析液的pH值不要和目的蛋白的等电点接近,这样目的蛋白可能会更容易沉淀。
3 保持蛋白浓度在mg/ml量级上面。
4 用20mM的醋酸透析也能降低沉淀5 加一些保护剂,如蔗糖,甘油,巯基乙醇等。
6 减少脱盐时间。
可考虑用超滤或者G系列的凝胶脱盐,这样时间较快,减少蛋白质的变性。
7 增加蛋白质浓度也可在一定程度上减少透析过程中蛋白质的沉淀,不过效果不是特别好的。
8 最后,不排除是蛋白不稳定的可能性,可在提纯开始时加点甘油,浓度控制在10-25%左右。
测序过程常见问题分析与解答1、为什么找不到PCR引物?答:以下几种情况,将无法找到做PCR时的引物序列(1)用PCR引物作为测序引物进行测序时,所测序列是从引物3’末端后第一个碱基开始的,所以自然就找不到您的引物序列了。
有两种方法可以得到您的引物序列。
对于较短的PCR产物(<800bp),可以用另一端的引物进行测序,从另一端测序可以一直测到序列的末端,就可以在序列的末端得到您的引物的反向互补序列。
pMD19-T载体说明书

TaKaRa Code:D102ApMD®19-T Vector宝生物工程(大连)有限公司目录内 容 Page●制品说明 1●制品内容 1●保 存 1●纯 度 1●用 途 1●pMD®19-T Vector的结构 1 ●实验操作 2■Control DNA片段的克隆实验2■一般DNA片段的克隆实验2●相关说明 3 ●使用注意 4 ●Q&A4●制品说明pMD ®19-T Vector是一种高效克隆PCR产物(TA Cloning)的专用载体。
本载体由pUC19载体改建而成,在pUC19载体的多克隆位点处的Xba I和Sal I识别位点之间插入了Eco R V识别位点,用Eco R V进行酶切反应后,再在两侧的3'端添加“T”而成。
因大部分耐热性DNA聚合酶进行PCR反应时都有在PCR 产物的3'末端添加一个“A”的特性,所以使用本制品可以大大提高PCR产物的连接、克隆效率。
由于本载体以pUC19载体为基础构建而成,所以它具有同pUC19载体相同的功能。
此外,本制品中的高效连接液Solution I 可以在短时间内(约30分钟)完成连接反应,其连接液可以直接用于细菌转化,大大方便了实验操作。
本制品中的Control Insert(500 bp)还可以用于Control 反应。
本载体与pMD ®18-T Vector 相比,本制品的 β-半乳糖苷酶的表达活性更高,菌落显示蓝色的时间缩短,菌落显示的蓝色更深。
因此,克隆后更容易进行克隆体的蓝白筛选。
●制品内容pMD ®19-T Vector(50 ng/μl) 20 μl×1支 Control Insert(50 ng/μl)10 μl×1支 Solution I*75 μl×2支* 使用时请于冰中融解。
●保存: -20℃●纯度■ Control Insert 经克隆后的白色菌落中,有90%以上含有Insert DNA 片段。
常见载体引物表
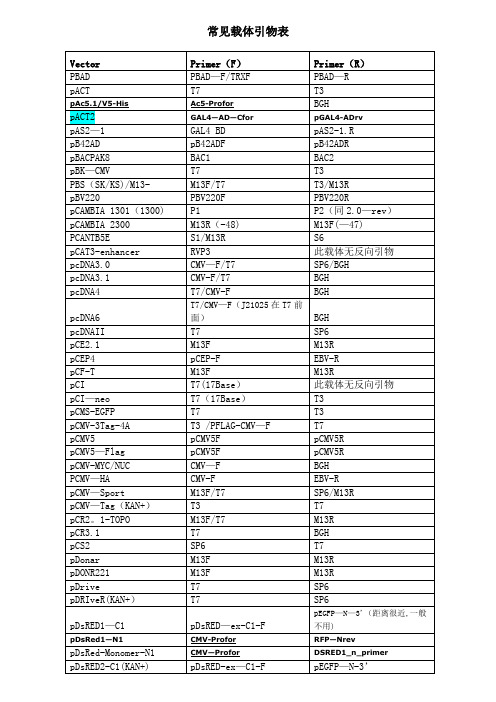
Vector
Primer(F)
Primer(R)
PBAD
PBAD—F/TRXF
PBAD—R
pACT
T7
T3
pAc5.1/V5-His
Ac5-Profor
BGH
pACT2
GAL4—AD—Cfor
pGAL4-ADrv
pAS2—1
GAL4 BD
pAS2-1.R
pB42AD
pB42ADF
pB42ADR
M13F(-47)
3。0REV
pSILENCEV2。0-U6
T7
2.0REV
pSK01-T
M13F
M13R
pSK—CMV(KAN+)
T7
T3
pSP72
T7
SP6
pSport1
SP6/M13F(—47)
T7/M13R
pSUPER
T7
T3
pTA2
M13F/T7(优先使用M13F)
T3/M13R
pT—Adv
DuetDOWN1
pDSRED2—N1(KAN+)
pEGFP—N-5
DsRed1—N
pFastBac Dual (MCSI)
BaculoVfor/PH
pFastBac-R/SV40-pArev
pBSN—T
T7
T3
pHISi-1
PHISi—1
无
pRSV—Rev(根据酶切位点选择不同引物)
pREPF/M13F
pLVX-IRES—ZsGreen1
PIRES2—EGFP-F
PIRES2-EGFP-R
pDC316
pDC316F