版药典》限量检测标准
国家中药重金属及农药残留残留标准
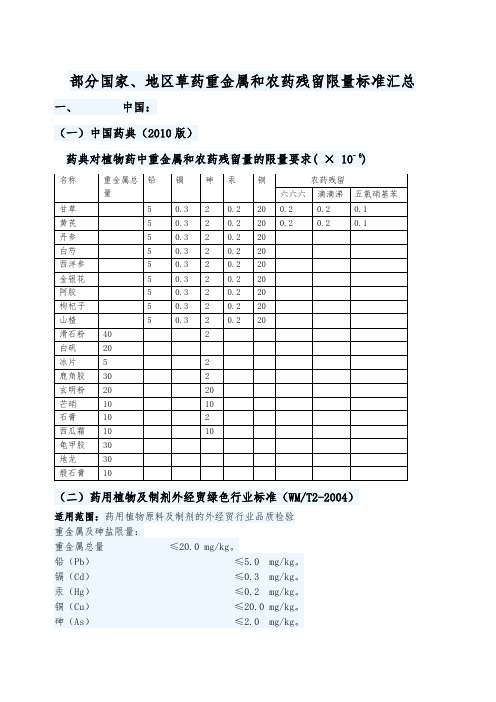
部分国家、地区草药重金属和农药残留限量标准汇总一、中国:(一)中国药典(2010版)药典对植物药中重金属和农药残留量的限量要求( × 10- 6)(二)药用植物及制剂外经贸绿色行业标准(WM/T2-2004)适用范围:药用植物原料及制剂的外经贸行业品质检验重金属及砷盐限量:重金属总量≤20.0 mg/kg。
铅(Pb)≤5.0 mg/kg。
镉(Cd)≤0.3 mg/kg。
汞(Hg)≤0.2 mg/kg。
铜(Cu)≤20.0 mg/kg。
砷(As)≤2.0 mg/kg。
农药残留限量:六六六(BHC) ≤0.1 mg/kg。
DDT ≤0.1 mg/kg。
五氯硝基苯(PCNB) ≤0.1 mg/kg。
艾氏剂(Aldrin) ≤0.02 mg/kg。
二、香港:(香港中药材标准第一册)表1:药材中重金属限度三、澳门:(技術性指示第02/2003號)重金属种类上限砷(无机) 每日1500.00微克镉(水溶性) 每剂3500.00微克铅每日179.00微克汞每日36.00微克重金属种类上限砷 5.00 ppm铜 150.00 ppm铅 20.00 ppm汞 0.50 ppm四、新加坡:(1995年药物决议(禁止销售及供应)(修正案))重金属及砷盐限量:铅(Pb)≤20 mg/kg。
汞(Hg)≤0.5 mg/kg。
铜(Cu)≤150 mg/kg。
砷(As)≤5 mg/kg。
镉(Cd)≤5 mg/kg。
五、马来西亚:重金属及砷盐限量::铅(Pb)≤10 mg/kg。
汞(Hg)≤0.5 mg/kg。
砷(As)≤5 mg/kg。
六、泰国:重金属及砷盐限量:适用范围:草药原料及产品铅(Pb)≤10 mg/kg。
镉(Cd) ≤0.3 mg/kg。
砷(As)≤ 4 mg/kg。
七、韩国:重金属限量(药品安全厅公示第2005-62号):1、植物性生药:铅(Pb)≤5 mg/kg。
汞(Hg ) ≤0.2 mg/kg。
镉 (Cd) ≤0.3 mg/kg。
版药典三部

15版药典三部含细菌内毒素热原的品种细菌内毒素检查(通则1143)54个品种125个样品Ⅰ预防类A群脑膜炎球菌多糖疫苗P69原液检定细菌内毒素检查应不高于25EU/μg;也可采用热原检查法检查,注射剂量按家兔体重每1kg注射0.05μg多糖。
细菌内毒素检查,每一次人用剂量应不高于1250EU。
稀释剂细菌内毒素检查,应不高于0.25EU/ml.A群C群脑膜炎球菌多糖疫苗P71原液检定细菌内毒素检查A群、C群多糖均应不高于12EU/ug细菌内毒素检查每1次人用剂量应不超过1250EU稀释剂细菌内毒素检查,应不高于0.25EU/ml.A群C群脑膜炎球菌多糖结合疫苗(新增)多糖原液检定细菌内毒素检查,A群、C群多糖均应不高于25EU/ug细菌内毒素检查,每1次人用剂量应不高于500EU。
稀释剂细菌内毒素检查,应不高于0.25EU/ml.ACYW135群脑膜炎球菌多糖疫苗P77(新增)原液检定细菌内毒素检查,A群、C群、Y群、W135群多糖均应不高于12.5EU/μg.细菌内毒素检查,每1次人用剂量应不超过1500EU。
稀释剂细菌内毒素检查,应不高于0.25EU/ml.b型流感嗜血杆菌结合疫苗P81多糖检定细菌内毒素检查,应不高于25EU/μg.结合物原液检定细菌内毒素应不高于5EU/μg。
细菌内毒素检查每1次人用剂量应不超过25EU乙型脑炎减毒活疫苗P124成品检定异常毒性细菌内毒素检查(通则1143凝胶限度试验),应不高于50EU/剂疫苗稀释剂细菌内毒素检查(通则1143凝胶限度试验),应不高于0.25EU/ml。
冻干乙型脑炎灭活疫苗(Vero细胞)P129成品检定异常毒性细菌内毒素检查应不高于50EU/ml(通则1143凝胶限度试验)森林脑炎灭活疫苗P133成品检定异常毒性细菌内毒素检查应不高于100EU/ml(通则1143凝胶限度试验)双价肾综合征出血热灭活疫苗(Vero细胞)P137成品检定异常毒性细菌内毒素检查应不高于50EU/剂(通则1143凝胶限度试验)双价肾综合征出血热灭活疫苗(地鼠肾细胞)P140成品检定异常毒性细菌内毒素检查应小于50EU/剂(通则1143凝胶限度试验)双价肾综合征出血热灭活疫苗(沙鼠肾细胞)P144成品检定异常毒性细菌内毒素检查应小于50EU/ml(通则1143凝胶限度试验)冻干人用狂犬病疫苗(Vero细胞)P147成品检定异常毒性细菌内毒素检查应不高于25EU/剂(通则1143凝胶限度试验)冻干甲型肝炎减毒活疫苗P152成品检定异常毒性细菌内毒素检查应不高于50EU/剂(通则1143凝胶限度试验)甲型肝炎灭活疫苗(人二倍体细胞)P155成品检定异常毒性细菌内毒素检查应不高于10EU/ml(通则1143凝胶限度试验)重组乙型肝炎疫苗(酿酒酵母)P158原液细菌内毒素检查应小于10EU/ml(通则1143凝胶限度试验)半成品检定细菌内毒素检查应小于5EU/ml(通则1143凝胶限度试验)成品检定异常毒性细菌内毒素检查应小于5EU/ml(通则1143凝胶限度试验)重组乙型肝炎疫苗(CHO细胞)P161纯化产物检定细菌内毒素检查每10ug蛋白质应小于10EU。
【2020版中国药典】通则-非无菌微生物限度检查

【2020版中国药典】通则-非无菌微生物限度检查1105非无菌产品微生物限度检查:微生物计数法
微生物计数法系用于能在有氧条件下生长的嗜温细菌和真菌的计数。
当本法用于检查非无菌制剂及其原、辅料等是否符合规定的微生物限度标准时,应按下述规定进行检验,包括样品的取样量和结果的判断等。
除另有规定外,本法不适用于活菌制剂的检查。
学习:将旧版的“相应”更换为“规定”,更便于按照1107进行判定执行。
微生物计数试验环境应符合微生物限度检查的要求。
检验全过程必须严格遵守无菌操作,防止再污染,防止污染的措施不得影响供试品中微生物的检出。
洁净空气区域、工作台面及环境应定期进行监测。
学习:将旧版的“单向流空气区域”更换为“洁净空气区域”。
计数方法
……供试品检查时,应根据供试品理化特性和微生物限度标准等因素选择计数方法,检测的样品量应能保证所获得的试验结果能够判断供试品是否符合规定。
所选方法的适用性须经确认。
……
提示:后文增加了对于“贵重药品、微量包装药品”的检验量的更全面的表述,因注意结合此处的要求。
计数培养基适用性检查和供试品计数方法适用性试验
……菌液制备……取黑曲霉的新鲜培养物加人适量含0.05%(ml/ml)聚山梨酯80的pH7.0无菌氯化钠-蛋白胨缓冲液或……
学习:此处改动同无菌检查法,将“3~5ml”的具体量调整为“适量”,便于根据孢子的量灵活掌握菌液制备方法。
培养基适用性检查
微生物计数用的商品化的预制培养基、由脱水培养基或按处方配制的培养基均应进行培养基适用性检查。
学习:类似于无菌检查法,此处将“成品培养基”修改为“商品。
2023年版《中国药典》通则调整—9101 药品质量标准分析方法验证指导原则

(蓝色字体表示新增内容,红色字体表示删减内容)药品质量标准分析方法验证(analytical method validation)的目的是证明采用建立的方法适合于相应检测要求。
在建立药品质量标准时,分析方法需经验证;在药品生产工艺变更、制剂的组分变更、原分析方法进行修订时,则质量标准分析方法也需进行验证。
在建立药品质量标准、变更药品生产工艺或制剂组分、修订原分析方法时,需对分析方法进行验证。
方法验证的理由、过程和结果均应记载在药品质量标准起草说明或修订说明中。
生物制品质量控制中采用的方法包括理化分析方法和生物学测定方法,其中理化分析方法的验证原则与化学药品基本相同,所以可参照本指导原则进行,但在进行具体验证时还需要结合生物制品的特点考虑;相对于理化分析方法而言,生物学测定方法存在更多的影响因素,因此本指导原则不涉及生物学测定方法验证的内容。
验证的分析项目有:鉴别试验、限量或定量检查、原料药或制剂中有效成分含量测定,以及制剂中其他成分(如防腐剂等,中药中其他残留物、添加剂等)的测定。
药品溶出度、释放度等检查中,其溶出量等的测定方法也应进行必要验证。
鉴别试验、杂质测定(限度或定量分析)、含量测定和特性参数(如:药物溶出度、释放度等)。
验证的指标有:专属性、准确度、精密度(包括重复性、中间精密度和重现性)、专属性、检测限、定量限、线性、范围和耐用性。
在分析方法验证中,须用标准物质进行试验。
由于分析方法具有各自的特点,并随分析对象而变化,因此需要视具体情况拟订验证的指标。
表 1 中列出的分析项目和相应的验证指标可供参考。
表 1 检验项目和验证指标项目杂质测定鉴别定量限度+-含量测定及溶校正因子指标专属性① 出量测定+++-+++准确度精密度++①③ +--+++-+重复性中间精密度--+--++①③ +-③② +--++-专属性② 检测限定量限线性范围耐用性--++++--+++++++① 已有重现性验证,不需验证中间精密度。
2015版中国药典微生物限度

1.4.2供试品检查
• 供试液制备
– ⑵ 水不溶性非油脂类供试品
• 取供试品, 用 pH7.0 无菌氯化钠-蛋白胨 缓冲液,或 pH7.2 磷酸盐缓冲液,或胰酪 大豆胨液体培养基制备成 1:10 供试液。 分散力较差的供试品,可在稀释液中加入 表面活性剂如 0.1%的聚山梨酯 80,使供 试品分散均匀。若需要,调节供试液 pH 值至 6~8。必要时,用同一稀释液将供 试液进一步 10倍系列稀释。
1.3.3计数方法适用性试验
1. 供试液制备 2. 接种和稀释 3. 抗菌活性的去除与灭活 4. 供试品中微生物的回收
– 平皿法 – 薄膜过滤法 – 最可能数法(MPN 法)
5. 结果判断
1.4 供试品检查
• 1.4.1检验量
– 检验量即一次试验所用的供试品量(g、ml
或cm²)。
– 除另有规定外,一般供试品的检验量为10g 或
• 需氧菌总数是指胰酪大豆胨琼脂培养基上生长的 总菌落数(包括真菌菌落数);
• 霉菌和酵母菌总数是指沙氏葡萄糖琼脂培养基上 生长的总菌落数(包括细菌菌落数)。
• 若因沙氏葡萄糖琼脂培养基上生长的细菌使霉菌 和酵母菌的计数结果不符合微生物限度要求,可 使用含抗生素(如氯霉素、庆大霉素)的沙氏葡 萄糖琼脂培养基或其他选择性培养基(如玫瑰红 钠琼脂培养基)进行霉菌和酵母菌总数测定。
1.4.2供试品检查
• 供试液制备
– ⑷需用特殊方法制备供试液的供试品
• 膜剂供试品 • 肠溶及结肠溶制剂供试品 • 气雾剂、喷雾剂供试品 • 贴膏剂供试品
1.4.2供试品检查
1. 平皿法
– 平皿法包括倾注法和涂布法。 – 除另有规定外,取规定量供试品,按方法适用性
试验确认的方法进行供试液制备和菌数测定,每 稀释级每种培养基至少制备2个平皿。 – 培养和计数 除另有规定外,胰酪大豆胨琼脂培养 基平板在30~35℃培养3~5天,沙氏葡萄糖琼脂 培养基平板在20~25℃培养5 ~7天, 观察菌落 生长情况,点计平板上生长的所有菌落数,必要时 可适当延长培养时间至7 天进行菌落计数并报告 。菌落蔓延生长成片的平皿不宜计数。点计菌落 数后,计算各稀释级供试液的平均菌落数,按菌 数报告规则报告菌数。 – 若同稀释级两个平皿的菌落数平均值不小于15, 则两个平皿的菌落数不能相差1 倍或以上。
2020版《中国药典》三部通则3204收载的戊二醛残留量测定法(高效液相色谱法)

戊二醛残留量测定法(高效液相色谱法)《中国药典》(2020版)的三部通则3204中收载了戊二醛残留量测定法,采用了高效液相色谱法(HPLC)进行检测。
本文将对该测定法进行详细介绍。
戊二醛是一种常用的消毒剂,也被广泛应用于医疗、制药和食品工业等领域。
然而,由于其具有一定的毒性和致癌性,对其残留量进行监测和控制非常重要。
《中国药典》(2020版)的三部通则3204中收录的戊二醛残留量测定法就是用于这一目的。
该测定法采用高效液相色谱法((HPLC)进行分析。
HPLC是一种广泛应用于药物分析和质量控制的分析技术,其特点是分离效果好,灵敏度高。
在戊二醛残留量的测定中,HPLC 技术可以准确、快速地分离和测定药物样品中的戊二醛。
以下是该测定法的主要步骤和操作流程:1.(样品准备:将待测样品进行合适的前处理,例如提取、固相萃取等,以获得戊二醛的纯化样品。
2.(色谱条件设置:根据具体仪器和柱的性能,优化色谱条件,包括流动相、柱温、检测波长等参数的选择与调整。
3.(样品注射:将纯化的样品注入到色谱仪中,一般采用自动进样器进行精确控制。
4.(色谱分离:在设定的色谱条件下,样品中的戊二醛与色谱柱发生相互作用,分离出各组分。
5.(检测和定量:利用紫外可见光检测器检测样品中戊二醛的吸收峰,并根据标准曲线或外标法进行定量分析。
6.(结果分析:计算出样品中戊二醛的浓度,并与相应的规定限度进行比较,判断样品是否符合要求。
通过上述步骤,该测定法能够准确、可靠地测定样品中的戊二醛残留量,为药物生产等领域的质量控制提供重要参考。
需要注意的是,该测定法的操作人员应具备一定的理论知识和实验技能,确保操作的准确性和可重复性。
同时,还需要严格控制实验条件和各项参数,以确保测试结果的准确性和可信度。
综上所述,《中国药典》(2020版)部通则3204中收载的戊二醛残留量测定法采用了高效液相色谱法((HPLC),该方法具有准确、灵敏的特点,对戊二醛残留量进行测定具有重要意义。
重金属检查法

中药限量检测——重金属检查法2015年版《药典》四部通则0821本法所指的重金属系指在规定实验条件下能与硫代乙酰胺或硫化钠作用显色的金属杂质。
标准铅溶液的制备称取硝酸铅0.1599g,置1000ml量瓶中,加硝酸5ml 与水50ml溶解后,用水稀释至刻度,摇匀,作为贮备液。
精密量取贮备液10ml,置100ml量瓶中,加水稀释至刻度,摇匀,即得(每1ml相当于10μg的Pb)。
本液仅供当日使用。
配制与贮存用的玻璃容器均不得含铅。
第一法除另有规定外,取25ml纳氏比色管三支,甲管中加标准铅溶液一定量与醋酸盐缓冲液(pH3.5)2ml后,加水或各品种项下规定的溶剂稀释成25ml,乙管中加入按各品种项下规定的方法制成的供试品溶液25ml,丙管中加入与乙管相同重量的供试品,加配制供试品溶液的溶剂适量使溶解,再加与甲管相同量的标准铅溶液与醋酸盐缓冲液(pH3.5)2ml后,用溶剂稀释成25ml;若供试品溶液带颜色,可在甲管中滴加少量的稀焦糖溶液或其他无干扰的有色溶液,使之与乙管、丙管一致;再在甲、乙、丙三管中分别加硫代乙酰胺试液各2ml,摇匀,放置2分钟,同置白纸上,自上向下透视,当丙管中显出的颜色不浅于甲管时,乙管中显示的颜色与甲管比较,不得更深。
如丙管中显出的颜色浅于甲管,应取样按第二法重新检查。
如在甲管中滴加稀焦糖溶液或其他无干扰的有色溶液,仍不能使颜色一致时,应取样按第二法检查。
供试品如含高铁盐影响重金属检查时,可在甲、乙、丙三管中分别加入相同量的维生素C 0.5~1.0g,再照上述方法检查。
配制供试品溶液时,如使用的盐酸超过1ml,氨试液超过2ml,或加入其他试剂进行处理者,除另有规定外,甲管溶液应取同样同量的试剂置瓷皿中蒸干后,加醋酸盐缓冲液(pH3.5)2ml与水15ml,微热溶解后,移置纳氏比色管中,加标准铅溶液一定量,再用水或各品种项下规定的溶剂稀释成25ml。
第二法除另有规定外,当需改用第二法检查时,取各品种项下规定量的供试品,按炽灼残渣检查法(通则0841)进行炽灼处理,然后取遗留的残渣;或直接取炽灼残渣项下遗留的残渣;如供试品为溶液,则取各品种项下规定量的溶液,蒸发至干,再按上述方法处理后取遗留的残渣;加硝酸0.5ml,蒸干,至氧化氮蒸气除尽后(或取供试品一定量,缓缓炽灼至完全炭化,放冷,加硫酸0.5~1ml,使恰湿润,用低温加热至硫酸除尽后,加硝酸0.5ml,蒸干,至氧化氮蒸气除尽后,放冷,在500~600℃炽灼使完全灰化),放冷,加盐酸2ml,置水浴上蒸干后加水15ml,滴加氨试液至对酚酞指示液显微粉红色,再加醋酸盐缓冲液(pH3.5)2ml,微热溶解后,移置纳氏比色管中,加水稀释成25ml,作为乙管;另取配制供试品溶液的试剂,置瓷皿中蒸干后,加醋酸盐缓冲液(pH3.5)2ml 与水15ml,微热溶解后,移置纳氏比色管中,加标准铅溶液一定量,再用水稀释成25ml,作为甲管;再在甲、乙两管中分别加硫代乙酰胺试液各2ml,摇匀,放置2分钟,同置白纸上,自上向下透视,乙管中显出的颜色与甲管比较,不得更深。
中国药品检验标准操作规范2010年版中药补充部分20铅、镉、砷、汞、铜测定法---原子吸收分光光度法

铅、镉、砷、汞、铜测定法---原子吸收分光光度法1 简述本法系采用原子吸收分光光度法对中药材中的铅、镉、砷、汞、铜进行限量检查。
2 仪器与用具2.1 原子吸收分光光度计应配备有火焰原子化器、石墨炉原子化器和适宜的氢化物发生装置,并具有氘灯或塞曼效应背景校正功能;铅、镉、砷、汞、铜等元素的空心阴极灯;普通或热解涂层石墨管;乙炔气、高纯氩气或高纯氮气;空气压缩机及冷却循环水泵等。
2.2 微波消解仪内罐为聚四氟乙烯材料制成,具有适宜的耐压密封装置和过压安全保护装置;具有程序控制、功率可调的微波发生装置;可采用适宜的方式监控反应罐内的温度和压力。
2.3 电热板应具有温度均匀的加热表面和温度控制装置。
2.4 纳氏比色管或量瓶应尽可能使用耐腐蚀的塑料器具,以聚四氟乙烯材料制成的为好,玻璃器皿易吸附或吸收金属离子,因此仅适于短时间内对溶液的容量使用。
3 试药与试液3.1 铅、镉、砷、汞、铜单元素标准溶液及国家一级标准物质杨树叶中国剂量科学研究院提供,单元素标准溶液用于制备标准曲线,杨树叶或茶树叶可作为工作对照物质,检查方法的可靠性。
3.2 硝酸、高氯酸应采用高纯试剂,盐酸、硫酸、磷酸二氢铵、硝酸镁为优级纯,碘化钾、抗坏血酸、盐酸羟胺为分析纯,使用前应检查各试剂中的相关金属元素含量符合测定的要求。
3.3 水去离子水或用石英蒸馏器蒸馏的超纯水,使用前应检查其中的相关金属元素含量符合测定的要求。
3.4 25%碘化钾溶液取碘化钾25g,加水100ml使溶解,即得。
本液应临用新制。
3.5 10%抗坏血酸溶液取抗坏血酸10g,加水100ml使溶解,即得。
本液应临用新制。
3.6 含1%磷酸二氢铵溶液和0.2%硝酸镁溶液的混合溶液取磷酸二氢铵1g,硝酸镁0.2g,加水100ml使溶解,即得。
3.7 1%硼氢化钠和0.3%氢氧化钠混合溶液取氢氧化钠3g,加水1000ml使溶解,加入硼氢化钠3g,使溶解,即得。
本液应临用新制。
3.8 4%硫酸溶液取硫酸4ml,加入水中稀释,并加水至100ml,即得。
部分国家、区草药重金属和农药残留限量标准汇总

部分国家、地区草药重金属和农药残留限量标准汇总发布时间:2010-05-24一、中国:(一)中国药典(05版)甘草重金属及有害元素:铅、镉、砷、汞、铜含量限定如下:铅不得过百万分之五,镉不得过千万分之三,砷不得过百万分之二,汞不得过千万分之二,铜不得过百万分之二十。
有机氯农药残留量:六六六(总BHC)不得过千万分之二,滴滴涕(总DDT)不得过千万分之二,五氯硝基苯(PCNB)不得过千万分之一。
黄芪重金属及有害元素:铅、镉、砷、汞、铜含量限定如下:铅不得过百万分之五,镉不得过千万分之三,砷不得过百万分之二,汞不得过千万分之二,铜不得过百万分之二十。
有机氯农药残留量:六六六(总BHC)不得过千万分之二,滴滴涕(总DDT)不得过千万分之二,五氯硝基苯(PCNB)不得过千万分之一。
丹参重金属及有害元素:铅、镉、砷、汞、铜含量限定如下:铅不得过百万分之五,镉不得过千万分之三,砷不得过百万分之二,汞不得过千万分之二,铜不得过百万分之二十。
白芍重金属及有害元素:铅、镉、砷、汞、铜含量限定如下:铅不得过百万分之五,镉不得过千万分之三,砷不得过百万分之二,汞不得过千万分之二,铜不得过百万分之二十。
西洋参重金属及有害元素:铅、镉、砷、汞、铜含量限定如下:铅不得过百万分之五,镉不得过千万分之三,砷不得过百万分之二,汞不得过千万分之二,铜不得过百万分之二十。
金银花重金属及有害元素:铅、镉、砷、汞、铜含量限定如下:铅不得过百万分之五,镉不得过千万分之三,砷不得过百万分之二,汞不得过千万分之二,铜不得过百万分之二十。
石膏重金属:含重金属不得过百万分之十;含砷量不得过百万分之二。
芍药 0.118) 三泰芬(Triadimefon)芍药 0.0119) 赛福宁(Triforine)芍药 0.120) 赛福唑(Triflumizole)黄芪 0.1 芍药 1.021) 芬瑞莫(Fenarimol)黄芪 0.522) 二甲戊乐灵(Pendimethalin)当归 0.2 麦门冬 0.2 柴胡 0.2芍药 0.2 红花 0.123) 芬普宁(Fenpropathrin)当归 0.224) 福赛绝(Fosthiazate)柴胡 0.0225) 甲基锌乃浦(Propineb)芍药 0.226) 派灭净(Pymetrozine)红花 0.05 黄芪 0.0527) 勿落菌恶(Fludioxonil)芍药 0.1八、日本:重金属及砷盐限量:铅(Pb)≤20PPM砷As2O3 ≤ 2PPM农药残留限量:1、中药材:(生药农药残留量的行业标准)适用范围:黄芪、远志、甘草、桂皮、细辛、山茱萸、苏叶、大枣、陈皮、枇杷叶、牡丹皮BHC总量≤0.2 mg/kgDDT总量≤0.2 mg/kg2、中药制剂:(汉方及生药制剂农药残留量的行业标准)1)有机氯类农药:适用范围:含有黄芪、远志、甘草、桂皮、细辛、山茱萸、苏叶、大枣、陈皮、枇杷叶、牡丹皮、人参、红参、番泻叶的汉方及生药制剂BHC总量≤0.2 mg/kgDDT总量≤0.2 mg/kg2)有机磷类农药:适用范围:含有远志、山茱萸、苏叶及陈皮的汉方制剂对硫磷≤0.5 mg/kg甲基对硫磷≤0.2 mg/kg杀扑磷≤0.2 mg/kg马拉硫磷≤1.0 mg/kg3)菊酯类农药适用范围:含有远志、苏叶、大枣、陈皮及枇杷叶的汉方制剂氰戊菊酯≤1.5 mg/kg氯氰菊酯≤1.0 mg/kg九、德国:重金属限量:铅(Pb)≤5 mg/kg。
2025版药典新增标准

2025版药典新增标准一、中药材与饮片中农药、植物生长调节剂等残留的检测方法及限量标准在2025版药典中,新增了中药材与饮片中农药、植物生长调节剂等残留的检测方法及限量标准。
这一标准的制定和实施,旨在保障中药材与饮片的安全性和质量,进一步推动中药产业的健康发展。
在检测方法方面,药典采用了多种现代分析技术,如高效液相色谱、气相色谱等,对中药材与饮片中的农药、植物生长调节剂等残留进行定性和定量分析。
这些技术的引入,提高了检测的准确性和灵敏度,有助于更好地控制中药材与饮片的质量。
在限量标准方面,药典根据大量的研究和实验数据,对中药材与饮片中的农药、植物生长调节剂等残留量制定了相应的标准。
这些标准的制定,充分考虑了中药材与饮片的实际情况和安全性要求,有助于保障中药材与饮片的质量和安全。
二、基于中成药中马兜铃酸不得检出的检测方法在2025版药典中,新增了基于中成药中马兜铃酸不得检出的检测方法。
这一标准的制定和实施,旨在保障中成药的安全性和质量,进一步推动中医药的发展。
马兜铃酸是一种具有肾毒性的物质,长期摄入会对人体健康造成严重危害。
因此,对于含有马兜铃酸成分的中成药,必须严格控制其含量。
在药典中,采用了高效液相色谱等技术,对中成药中的马兜铃酸进行定性和定量分析。
这些技术的引入,提高了检测的准确性和灵敏度,有助于更好地控制中成药的质量。
三、中药材野生品和栽培品质量一致性的基础研究在2025版药典中,新增了中药材野生品和栽培品质量一致性的基础研究。
这一标准的制定和实施,旨在深入探讨中药材野生品和栽培品的质量差异和一致性,为中药材的合理开发和利用提供科学依据。
中药材野生品和栽培品的质量差异一直是中医药研究关注的焦点之一。
为了深入了解这种差异,药典采用了多种现代科学技术手段,如基因组学、蛋白质组学等,对中药材野生品和栽培品的化学成分、生物活性等方面进行了深入研究。
这些研究的结果将有助于更好地认识中药材野生品和栽培品的本质特征和差异,为中药材的合理开发和利用提供科学依据。
2015年版中国药典微生物限度检查法

1.1 总则:
• 环境: – 微生物计数试验环境应符合微生物限度检查的要 求。(在不低于GMP 现行版要求的D 级洁净环境 、局部洁净度不低于B 级的单向流空气区域内进 行)【10版:在环境洁净度10000级下的局部洁净 度100级的单向流空气区域内】。 – 检验全过程必须严格遵守无菌操作,防止再污染 ,防止污染的措施不得影响供试品中微生物的检 出。 – 单向流空气区域、工作台面及环境应定期进行监 测。
菌数报告规则
– 需氧菌总数测定宜选取平均菌落数小于 300cfu 的稀释级、霉菌和酵母菌总数测定宜 选取平均菌落数小于100cfu 的稀释级,作为 菌数报告(取两位有效数字)的依据。取最 高的平均菌落数,计算1g、1ml 或10 cm² 供 试品中所含的微生物数。 – 如各稀释级的平皿均无菌落生长,或仅最低 稀释级的平板有菌落生长,但平均菌落数小 于1 时,以﹤1 乘以最低稀释倍数的值报告菌 数。
1.4.2供试品检查
• 供试液制备 – ⑴ 水溶性供试品
• 取供试品,用 pH7.0 无菌氯化钠-蛋白胨 缓冲液,或pH7.2 磷酸盐缓冲液,或胰酪 大豆胨液体培养基溶解或稀释制成 1:10 供试液。若需要,调节供试液 pH 值至 6 ~8。必要时,用同一稀释液将供试液进 一步 10倍系列稀释。水溶性液体制剂也 可用混合的供试品原液作为供试液。
1.3.1菌液制备及使用
试验菌株 试验菌液的制备
金黄色葡萄球菌 〔CMCC(B) 26 003)〕
铜绿假单胞菌 〔CMCC(B)10 104〕 枯草芽孢杆菌 〔CMCC(B) 63 501〕 白色念珠菌 〔CMCC(F) 98 001〕
胰酪大豆胨琼脂培养基或胰酪大豆胨液体培 养基 【10版:营养肉汤或营养琼脂培养基】 30~35℃,18~24小时
2020版中国药典】通则-非无菌微生物限度检查

2020版中国药典】通则-非无菌微生物限度检查本文介绍了非无菌产品微生物限度检查中的微生物计数法。
该方法适用于能在有氧条件下生长的嗜温细菌和真菌的计数。
在检查非无菌制剂及其原、辅料等是否符合规定的微生物限度标准时,应严格遵守无菌操作,防止再污染。
计数方法的选择应考虑供试品理化特性和微生物限度标准等因素,并经过确认。
此外,商品化的预制培养基和由脱水培养基或按处方配制的培养基都应进行培养基适用性检查。
在供试液制备过程中,应根据供试品的理化特性与生物学特性,采取适宜的方法制备供试液,并在加温时均匀加热,且温度不应超过45°C。
供试液从制备至加人检验用培养基,不得超过1小时。
在进行多种品种计数方法的适用性试验时,需要注意时效问题。
在“供试液制备”这一节中,水溶性供试品、水不溶性非油脂供试品、油脂类供试品的制备方法没有变化。
在2020版药典中,将原来的“需用特殊方法制备供试液的供试品”中的小类(四种)提升至与上述三类并列,除气雾剂供试品外,制备方法没有变化。
具体是膜剂供试品、肠溶及结肠溶制剂供试品、气雾剂供试品(2015版:气雾剂、喷雾剂供试品)、贴剂、贴膏剂供试品(2015版:贴膏剂供试品)。
需要注意的是,气雾剂供试品的变化较大,需要特别注意。
在进行供试液接种后的抗菌活性去除或灭活时,可以使用中和剂或灭活剂(表2)来消除干扰物的抑菌活性。
最好在稀释液或培养基灭菌前加入。
如果使用中和剂或灭活剂,试验中应设中和剂或灭活剂对照组,即取相应量含中和剂或灭活剂的稀释液替代供试品同试验组操作。
需要注意的是,在方法学中,这个对照组的稀释液中显然要含有相应的中和剂或者灭活剂,进一步明确,以防止错误套用。
在供试品中微生物的回收中,涂布法中“取15~20ml”修订为“取适量(通常为15~20ml)”。
在薄膜过滤法中,所采用的滤膜孔径应不大于0.45μm,直径一般为50mm。
如果采用其他直径的滤膜,冲洗量应进行相应的调整。
2020中国药典通则1108

2020年中国药典通则1108是关于中药饮片微生物限度检查的标准,主要包括以下内容:
对中药饮片进行精准分类:新版药典按照服用方式来界定中药材和中药饮片的微生物限度规定。
在通则1107-非无菌药品微生物限度标准中,规定了直接口服及泡服的中药饮片微生物限度标准。
样品取样过程有待进一步明晰:通则1108-中药饮片微生物限度检查法规定,中药饮片抽样参照通则0211-药材和饮片取样法抽取试验样品。
由于中药饮片的取样是拆开包装抽取样品,大包装饮片每批抽取100~500g,混匀;独立小包装饮片安装量抽取100~500g 的包装数。
抽样时存在中药饮片直接接触取样环境空气、抽样工具和包装袋的过程。
药材和饮片取样法只规定了常规理化检验用样品抽样时的原则、步骤、取样比例和取样量,但对于微生物限度检查用样品,在抽取样品过程中对取样环境、取样工具及包装容器等如何防范微生物污染却未有明确说明。
中国药典成品检验标准

艾司唑仑片本品含艾司唑仑(C16H11ClN4)应为标示量的90.0%- 110.0%。
[性状] 本品为白色片。
[鉴别]取本品的细粉适量(约相当于艾司唑仑10mg),加乙醇10mI,振摇使艾司唑仑溶解,滤过,滤液蒸干,残渣照艾司唑仑项下的鉴别(1)、(2)项试验,显相同的反应。
[检查]有关物质取本品细粉适量(约相当于艾司唑仑2mg),加流动相制成每1m1中约含0.2mg的溶液,滤过,取续滤液作为供试品溶液;精密量取适量,加流动相稀释制成每1m1中约含2ug的溶液,作为对照溶液。
照高效液相色谱法(附录V D)测定。
用十八烷基硅烷键合硅胶为填充剂;以甲醇—水(65:35)为流动相;检测波长为223nm。
理论板数按艾司唑仑峰计算不低于2000。
取对照溶液20uI注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高为满量程的20%~25%;再精密量取供试品溶液与对照溶液各20UI,分别注入液相色谱仪,记录色谱图至主成分色谱峰保留时间的3倍。
供试品溶液色谱图中如有杂质峰,各杂质峰面积的和不得大于对照溶液主峰面积的1/2。
[含量均匀度]取本品l片,置100mI(1mg规格)或200ml(2mg规格)量瓶中,加盐酸溶液(9—1000)适量,充分振摇,使艾司唑仑溶解,加盐酸溶液(9一1000)稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液,照含量测定项下的方法测定含量,应符合规定(附录X E)。
[溶出度] 取本品,照溶出度测定法(附录X C第三法),以盐酸溶液(9—1000)100mI(1mg规格)或200ml(2mg规格)为溶出介质,转速为每分钟100转,依法操作,经30分钟时,取溶液10mI,滤过,取续滤液,照紫外—可见分光光度法(附录ⅣA),在268nm的波长处测定吸光度,按C16'H11C1N4的吸,收系数(E1CM1%)为352计算每片的溶出量。
限度为标示量的80%,应符合规定。
其他应符合片剂项下有关的各项规定(附录IA)。
中国药典中限量法测定杂质的品种

中国药典中限量法测定杂质的品种全文共四篇示例,供读者参考第一篇示例:中国药典是我国药品质量标准的权威性规范,其制订和实施对确保我国药品质量安全至关重要。
在中国药典中,对药物质量的要求不仅仅包括药物的主要成分,还包括对杂质的要求。
杂质是药物中非药用成分或者其他成分的总称,它们的存在会影响药物的质量和安全性。
在中国药典中限量法测定杂质的品种是非常重要的。
中国药典中对药物质量的要求是非常严格的,除了对药物的主要成分进行质量控制外,还对药物中的杂质进行了详细的规定。
杂质是指药物中与原料无关或与原料相关但不是原料中本身成分的任何不期望或不想要的物质。
杂质的存在会影响药物的有效性和安全性,因此需要对其进行严格控制。
在中国药典中,对药物中的杂质进行测定的方法主要有限量法。
限量法是指根据药物中杂质的种类和浓度,通过合适的方法测定其含量,然后根据规定的限量标准判定其合格性。
通过限量法测定杂质的品种多种多样,主要包括以下几类:一、重金属类杂质:重金属是指相对密度大于4g/cm3的金属元素,包括铅、汞、砷、镉等。
重金属类杂质在药物中的存在可能来源于原料、生产过程或包装材料等,对人体健康造成潜在危害。
中国药典对药物中的重金属类杂质的限量标准进行了详细规定,要求进行严格的检测和控制。
二、有机溶剂残留物:有机溶剂是一种常用的药物生产工艺辅助剂,但在药物中长时间的残留可能对人体健康造成影响。
中国药典规定了对药物中有机溶剂残留物的测定方法和限量标准,要求对其进行严格的监测和控制。
五、其他杂质:除了以上几类常见的杂质外,中国药典还对药物中的其他杂质进行了多方面的规定,包括不符合规范的原料、过程中产生的不期望物质等,都需要进行严格的测定和控制。
中国药典对药物中的杂质进行了详细的规定,通过限量法测定其含量并对其进行严格控制,保证了药物的质量和安全性。
在药物的生产和使用过程中,制药企业和药品监管部门都要严格遥遥符合中国药典的要求,确保药物的质量安全,保障人民群众的用药安全。
版《药典》限量检测标准

新药典“检查”修订情况
▪二氧化硫残留量
通则规定,除另有规定外,中药材及饮片(矿物类除外)的二氧化硫残留量不得超过150mg/kg。
正文规定,山药、天冬、天花粉、天麻、牛膝、白及、白术、白芍、党参、粉葛10味中药及其饮片的二氧化硫残留量不得超过400mg/kg。
▪农药残留量
注:
•总六六六(α-BHC、β-BHC、γ-BHC、δ-BHC之和)不得过0.2mg/kg;
•总滴滴涕(pp’-DDE、pp’-DDD、op’-DDT、pp’-DDT之和)不得过0.2mg/kg;
•五氯硝基苯不得过0.1mg/kg;
•六氯苯不得过0.1mg/kg;
•七氯(七氯、环氧七氯之和)不得过0.05mg/kg;
•艾氏剂不得过0.05mg/kg;
•氯丹(顺式氯丹、反式氯丹、氧化氯丹之和)不得过0.1mg/kg。
注注:
人参、西洋参为15版药典新增。
▪黄曲霉毒素
▪重金属及有害元素
10版药典原有重金属限量品种:
山楂、丹参、甘草、石膏、煅石膏、白芍、白矾、玄明粉、地龙、芒硝、西瓜霜、西洋参、冰片、龟甲胶、阿胶、金银花、枸杞子、黄芪、鹿角胶、滑石粉。
中国药典二部附录通用限度

(
按 第丸 一数 法服
用
)
0.3g<W≤1g 1g<W≤2g W>2g
第服( 二用按 法)重
量
±9% ±8% ±7% ±6% ±5% ±8% ±7% ±6%
温度℃
5 10 15
20 25 30 35
40
不同温度下各种标准缓冲液 PH 值一览表
草酸盐标准缓 邻苯二甲酸氢 磷酸盐标准缓 硼砂标准缓冲 氢氧化钙标准
二、结果判定
样品
检查用水 V≥100ml V<100ml、静脉注射用无 菌粉末或注射用浓溶液
10um <10(粒/10ml)
≤25(粒/ml)
≤6000(粒/瓶)
25um <2(粒/10ml) ≤3(粒/ml)
≤600(粒/瓶)
供试品(可见异物) 无色溶液 透明塑料容器包装或有色溶液 混悬型溶液
光照度 1000~1500lx 2000~3000lx
4000lx
胶囊剂(二部)
不少于标示装量
V ≥V 标;V≥V 标×97% ±15% ±10% ±7% ±5%
片剂(一部)
W<0.3g W≥0.3g 原粉片 浸膏(半浸膏)、糖衣片 薄膜衣片
±7.5% ±5%
30min 60min 60min
散剂
≤0.1g 0.1g<W≤0.5g 0.5g<W≤1.5g 1.5g<W≤6g >6g
冲液
钾标准缓冲液 冲液(pH6.8)
液
缓冲液(25℃)
1.67 1.67 1.67
1.68 1.68 1.68 1.69
1.69
4.00 4.00 4.00
4.00 4.01 4.02 4.02
4.04
6.95 6.92 6.90
gmp限量指标

gmp限量指标
GMP限量指标是指制药行业中,药品生产质量管理规范(GMP)对药品生产的各个环节设定的一系列限制和标准,以确保药品的安全性和有效性。
以下是GMP限量指标的一些主要方面:
1. 原料和辅料:GMP要求药品生产所使用的原料和辅料必须是合格的,并且符合相关质量标准。
这些标准包括化学性质、物理性质、微生物限度等方面的要求。
2. 生产环境:GMP对药品生产环境的要求非常严格,包括空气洁净度、温度、湿度、压力等参数。
生产环境必须保持清洁、卫生,并定期进行消毒。
3. 设备设施:GMP要求药品生产所使用的设备、仪器、容器等必须符合相关标准和规定,并且定期进行校准和维护。
4. 工艺控制:GMP要求药品生产的工艺必须经过验证,并且符合相关规定。
生产过程中的关键工艺参数必须进行控制和监控,以确保药品的质量和安全性。
5. 质量控制:GMP要求药品生产过程中必须进行严格的质量控制,包括原辅料检验、中间体控制、成品检验等环节。
质量控制人员必须具备相应的资质和经验。
6. 文件记录:GMP要求药品生产过程中必须建立完整的文件记录体系,包括生产记录、检验记录、质量监控记录等。
这些记录必须真实、准确、完整,并且能够追溯到生产全过程。
总之,GMP限量指标是确保药品安全性和有效性的重要保障措施,也是制药行业的重要法规和标准之一。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
七氯(七氯、环氧七氯之和)不得过0.05mg/kg;
艾氏剂不得过0.05mg/kg;
氯丹(顺式氯丹、反式氯丹、氧化氯丹之和)不得过0.1mg/kg。
注注:
人参、西洋参为15版药典新增。
黄曲霉毒素
15版药典仅对以下19种药材做黄曲霉毒素限量规定。
品种
限量
大枣、水蛭、地龙、肉豆蔻、全蝎、决明子、麦芽、远志、陈皮、使君子、柏子仁、胖大海、莲子、桃仁、蜈蚣、槟榔、酸枣仁、僵蚕、薏苡仁
黄曲霉毒素B1不得过5μg/kg;
黄曲霉毒素G2、黄曲霉毒素G1、黄曲霉毒素B2总量不得过10μg/kg。
重金属及有害元素
15版药典新增以下8种药材的重金属限量规定。
品种
重金属
Pb
(mg/kg)
Cd
(mg/kg)
As
(mg/kg)
Hg
(mg/kg)
Cu
(mg/kg)
水蛭
10
1
5
1
-
牡蛎
5
0.3
2
0.2
新药典“检查”修订情况
二氧化硫残留量
通则规定,除另有规定外,中药材及饮片(矿物类除外)的二氧化硫残留量不得超过150mg/kg。
正文规定,山药、天冬、天花粉、天麻、牛膝、白及、白术、白芍、党参、粉葛10味中药及其饮片的二氧化硫残留量不得超过400mg/kg。
农药残留量
15版药典对以下4中药材做农药残留量规定。
品种
限量
总六六六
总滴滴涕
五氯
六氯
七氯
艾氏剂
氯丹
人参
√
√
√
√
√
√
√
西洋参
√
√
√
√
√
√
√
甘草
√
√
√
黄芪
√
√
√
注:
总六六六(α-BHC、β-BHC、γ-BHC、δ-BHC之和)不得过0.2mg/kg;
总滴滴涕(pp’-DDE、pp’-DDD、op’-DDT、pp’-DDT之和)不得过0.2mg/kg;
五氯硝基苯不得过0.1mg/kg;
20
昆布
5
4
-
0.1
20
珍珠
5
0.3
2
0.2
20
海螵蛸
5
5
10
0.2
20
海藻
5
4
-
0.1
20
蛤壳
5
0.3
2
0.2
20
蜂胶
8Hale Waihona Puke ----
10版药典原有重金属限量品种:
山楂、丹参、甘草、石膏、煅石膏、白芍、白矾、玄明粉、地龙、芒硝、西瓜霜、西洋参、冰片、龟甲胶、阿胶、金银花、枸杞子、黄芪、鹿角胶、滑石粉。