467 残留溶剂 usp
USP(467)有机挥发性杂质—残留溶剂的限制

USP(467)有机挥发性杂质—残留溶剂的限制基于药典的要求,残留溶剂在此定义为有机挥发性化学物,它用于生产药物赋形剂,或药品制备过程中,通过规范的生产技术,不能将残留溶剂完全除去,在合成原料药或生产赋形剂过程中,选择适当的溶剂可提高产量或决定药物的外观,如晶型、纯度和溶解度。
因此,有机溶剂是合成过程的关键因素。
本章的指导原则不是指谨慎地用于赋形剂的溶剂,也不是溶剂化合物。
然而,在这样的产品中溶剂的含量应该被测定和判断。
因为残留溶剂不能提供药物功效,因此应去除所有的残留溶剂,更进一步的可能以符合产品规范,GMP或者基本质量要求。
原料药中残留溶剂的含量级别不能高于安全数据支持的残留溶剂级别。
已知溶剂可导致不可接受的毒性应避免在生产中使用,除非有特别的证明它的使用是合理的(一类溶剂)。
一些有较弱毒性的溶剂(二类)应限制使用,以保护病人出现潜在的不利影响。
低毒性溶剂清单不完整,其他可能使用的溶剂,以权威组织机构的批准为准,将会在清单中依次列入。
原料药,赋形剂和药品中残留溶剂的测试,应当在生产或者纯化过程中进行,它在生产或者纯化过程中出现的残留溶剂的测试才是必要的。
虽然生产商会选择性地测试药品,会使用各个成分中残留溶剂水平累积方法计算总残留溶剂水平,如果计算的结果等于或低于本章指导原则建议的溶剂水平,不用进行再测试。
如果高于规定的水平,应进行检测,以确定是否降低制备过程相关溶剂的量,使之在可接受量范围内。
如果一种残留溶剂在药品生产中出现,也应当进行检测。
一类一类残留溶剂(表1)不应该在原料药、赋形剂和药品生产中使用,因为这些溶剂是不可接受的毒性物质,且对环境有不良影响。
但是,如果使用一类溶剂,应根据表1限定它们的量,除非有独立专文支持。
表1中的三氯乙烷是环境有害物,≤1500PPM的限量是安全数据。
当在原料药、赋形剂和药品生产中使用一类溶剂,指导原则要求任何必要的地方,都要有对残留溶剂鉴别、控制和数量测试的方法的描述,除非引入适当的验证程序,这种程序应该符合在相关独立专文中的USP标准,见ICH。
467 残留溶剂 usp

<467> RESIDUAL SOLVENTS残留溶剂(Chapter under this new title—to become official July 1, 2008)(这个新名称下的通则----2008年7月1日起生效)(Current chapter title is <467> Organic Volatile Impurities)(当前通则名称是<467>有机挥发性杂质)INTRODUCTION介绍This general chapter applies to existing drug substances, excipients, and products. All substances and products are subject to relevant control of solvents likely to be present in a substance or product.本通则适用于现有原料药、辅助剂、成药。
所有原料药和成药均需对可能存在其中的溶剂进行控制。
Where the limits to be applied comply with those given below, tests for residual solvents are not generally mentioned in specific monographs because the solvents employed may vary from one manufacturer to another.在所适用的限度与下面所述相符的情况下,残留溶剂检测一般不在具体各论中提及,因为每个生产商所使用的溶剂可能都不相同。
The objective of this general chapter is to provide acceptable amounts of residual solvents in pharmaceuticals for the safety of the patient. The general chapter recommends the use of less toxic solvents and describes levels considered to be toxicologically acceptable for some residual solvents.本通则的目的是为了保障病人的安全,提供在药物中残留溶剂的可接受数量。
USP-467 溶剂残留

[467]有机挥发性杂质残留溶剂限度根据药典要求,药品中残留溶剂定义:在药物或赋形剂制造或使用过程中,或药物制剂生产过程中残存的有机挥发性物质。
目前制药技术不能完全去除残留溶剂。
药物或赋形剂合成中选择适当的溶剂可提高得率,或获得某些特性如晶型、纯度和溶解性。
因此,合成中所用溶剂有时可能是危险因素。
本章节不包括涉及用于组分的溶剂或溶剂化物。
尽管如此,以上产品仍应标明所含溶剂含量并证明对人体安全。
由于残留溶剂不用于治疗用途,因此应尽可能除去,以符合药物、赋形剂和产品规格要求、GMP或其他质量标准。
药物制剂中所含溶剂不得高于安全性评价规定限度。
众所周知,药物、赋形剂、药物制剂不应含能引起不可逆毒副作用(Ⅰ级,见表1)的残留溶剂,除非证明有很好的风险-效益比。
应对引起次级严重毒副作用的残留溶剂(Ⅱ级,见表2)规定限度,以防止病人发生潜在的不良反应。
理想状态下,应尽可能使用低毒性溶剂(Ⅲ级,见表3)。
本章节所有溶剂列表见附录1。
下述表格及限度并不代表全部。
当医药工业发展需使用其他溶剂情况下,应将新溶剂添加到列表中。
当药物、赋形剂、药物制剂生产和纯化过程中残留已知有机溶剂时,应依法检查溶剂限度。
本限度检查仅检查用于制造和纯化加工所用的溶剂。
虽然制造商可能选择测试药物,我们可采用累积法从制造工艺水平计算产品中残留溶剂。
如残留溶剂计算结果等于或低于本章节规定限度,可不进行残留溶剂检查。
如残留溶剂计算结果高于本章节规定限度,仍需检查残留溶剂限度以确定是否精加工降低了有关溶剂水平至可接受量。
如残留溶剂为制造所用溶剂,必须检查药物制剂残留溶剂限度。
附录2为有关残留溶剂的相关背景资料。
根据安全评估残留溶剂分类国际化学安全机构用术语“可忍受日摄食量”(TDI)描述有毒化学物质残留限度。
世界卫生组织和其他国家和国际卫生机构用术语“可接受日摄食量”(ADI)来描述残留限度。
术语“允许日接触量”(PDE)定义为根据药效学残留溶剂可接受摄入量,避免与ADIs混淆。
USP部分内容

麻烦帮忙翻译USP35中一下内容:(编号为页码)<11>USP对照标准品<21>温度计<31>容量仪器<41>重量与天平<429>粒度的光衍射测定<467>残留溶剂<616>松装密度/堆密度和振实密度<621>色谱分析法<641>溶液的澄清度<699>固体密度<724>药物释放度(P442)-2<786>通过分析筛选估计粒度分布/通过筛分法估算粒子分布<791>pH<811>粉体细度/粉剂细度<851>分光光度法与光散射(P577)-6<905>含量均匀度(P593)-3<921>水分测定<1051>清洗玻璃容器(P754)<1087>内部的溶出度(P871)-3<1092>溶出程序:开发与验证(P889)<1216>片剂的脆碎度(P1120)<1217>片剂破碎力<1251>在分析天平上称量(标红的部分为已翻译完成)<197>分光光度鉴别测试(P196)红外吸收干燥待测样品和分析用对照品的制备有7种方法。
各论中引用<197K>意味着待测物质与溴化钾完全混合。
各论中引用<197M>意味着待测物质经过精细的磨碎,并溶于矿物油中。
各论中引用<197F>意味着待测物质悬浮在不同的盘中(plate)(如,氯化钠或溴化钾)。
各论中引用<197S>意味着制备指定浓度的溶液,且溶解于各个各论中指定的溶剂中,除非各个各论中对光程长吸收池(cell path length)另有规定,否则溶液的检测应在0.1mm吸收池(cell)中进行。
各论中引用<197A>意味着待测物质与内反射元件亲密接触,用于衰减全反射(ATR)分析。
残留溶剂培训讲义USP467

• 2类混合A标准溶液:移取1.0ml2类标准贮备液A到合适的顶空瓶中,加
5.0ml水,密封,加盖 。 • 2类混合B标准溶液:移取5.0ml2类标准贮备液B到合适的顶空瓶中,加
1.0ml水,密封,加盖。
• 供试贮备液——精密称取供试品约250mg到25-ml容量瓶,用水溶解并稀 释到刻度,摇匀。
• 供试液——移取5.0ml供试贮备液到适宜的顶空瓶中,加入1.0ml水,
加盖密封,摇匀。 • 一类系统适应性溶液 ——移取1.0ml一类标准贮备液到合适的顶空瓶 中,加入5.0ml供试贮备液,加盖密封,摇匀。
气相色谱系统(见色谱法<621>)
• 色谱柱:0.32-mm × 30-m的弹性石英色谱柱,液膜厚1.8-µ m的 G43,或0.53-mm × 30-m的多孔性色谱柱,液膜厚3.0-µ m 的G43 • 载气:氮气(或氦气)线速度约为35cm/s • 分流比为1:5 (为了优化灵敏度,可适当调节) • 柱温:先在40℃维持20分钟,再以10℃/min的升温速率升至240,并 维持20分钟 • 进样口温度: 140℃ • FID检测器温℃:250℃
和在临床研究阶段研发的制剂,也不适用于已存
在的市售制剂。
残留溶剂按风险评估的分类 “ 每日允许接触量”(PDE)用于定义药物中可接
受的有机溶剂的最大摄入量。应采用多种灵敏度高、 选择性好、线形范围宽的检测器 按对人类健康的潜在危险分为如下三类 一类
应该避免使用
公认的对人体有致癌作用
非常可疑为对人体有致癌作用 对环境有危害作用
二类
应该限制使用 对动物无遗传毒性,但可能存在其他不可逆的毒性, 如神经毒性、致畸性。 非常可疑存在其他潜在的严重的可逆毒性。
USP30-NF25溶剂残留《467》(中文)20070703

残留溶剂的鉴别、控制、定量【注意-在下列各方法中指定的无有机物水在色谱上应没有明显的杂质干扰峰】1类和2类残留溶剂水溶性样品步骤A一类标准贮备溶液:转移1.0mL USP一类残留溶剂混合溶液标准品于100mL容量瓶中,加9mL二甲基亚砜,以水稀释至刻度,混匀。
转移1.0 mL此溶液于100mL容量瓶中,以水稀释至刻度,混匀。
转移1.0 mL该溶液于10mL容量瓶中,以水稀释至刻度,混匀。
一类标准溶液:转移1.0 mL一类标准贮备溶液于适合的顶空进样样品瓶中,加入5.0 mL 水,压上瓶塞和瓶盖,混匀。
二类标准贮备溶液:转移1.0mL USP二类残留溶剂混合溶液A标准品于100mL容量瓶中,以水稀释至刻度,混匀。
此为二类标准贮备溶液A。
转移1.0mL USP二类残留溶剂混合溶液B标准品于100mL容量瓶,以水稀释到刻度,混匀。
此为二类标准贮备溶液B。
二类混合溶液A标准溶液:转移1.0 mL二类标准贮备溶液A于适当的顶空进样样品瓶中,加入5.0 mL水,压上瓶塞和瓶盖,混匀。
二类混合溶液B标准溶液:转移5.0 mL二类标准贮备溶液B于适当的顶空进样样品瓶中,加入1.0 mL水,压上瓶塞和瓶盖,混匀。
供试贮备溶液:精确称取250mg供试样品,转引至25 mL容量瓶中,溶于水并以水稀释至刻度,混匀。
供试溶液:转移5.0 mL供试贮备溶液于适当的顶空进样样品瓶中,加入1.0 mL水,压上瓶塞和瓶盖,混匀。
一类系统适应性溶液:转移1.0 mL一类标准贮备溶液于适当的顶空进样样品瓶中,加入5.0 mL供试贮备溶液,压上瓶塞和瓶盖,混匀。
色谱系统(见色谱<621>):该气相色谱仪装备一个火焰离子化(FID)检测器,一个0.32mm ×30m 毛细管柱(fused-silica column)内部涂以1.8-µm 厚的phaseG43涂层或者一个0.53mm ×30m 大孔径毛细管柱以3.0-µm 厚的phaseG43为涂层。
使用 Agilent 7890A GC 和低热容 (LTM) 系统快速分析 USP 467 残留溶剂

使用Agilent 7890A GC 和低热容(LTM)系统快速分析USP 467 残留溶剂摘要使用安装在Agilent 7890A GC 系统上的低热容(LTM)系统根据USP 467(2008-09 修订版)进行双柱残留溶剂分析。
连接至挥发物接口的G1888 自动顶空进样器用于进样。
微板流路控制技术(CFT)双路分流器用于将样品等量地分流到5 英寸7 M x 0.25 mm x 1.4 µm Agilent J&W DB-624 色谱柱和5 英寸7 M x 0.25 mm x 0.25 µm Agilent J&WHP-INNOWax 色谱柱中。
每个柱模块通过保留间隙管柱连接至其相应的FID 。
已对1 级、2A 级和2B 级溶剂的水溶液进行了分析。
灵敏度、线性和分离度符合USP 467 的要求。
所有指定的1 级、2A 级和2B 级溶剂的总分析周期时间缩短至10 min 。
作者Roger L FirorAgilent Technologies, Inc.2850 Centerville Road Wilmington, DE 19808USA应用报告制药图1.在双LTM 柱配置中使用CFT 分流器的系统示意图图2.浓度范围为低于限值大约10 倍至高于限值6 倍的2A 级溶剂校准曲线。
(续)图2.浓度范围为低于限值大约10 倍至高于限值6 倍的2A 级溶剂校准曲线讨论在残留溶剂分析所要求的程序升温气相色谱中,柱温箱冷却时间对总周期时间有很大影响。
LTM 色谱柱模块与空气浴柱温箱相比冷却速率相当快,这要归功于其非常低的热容以及直接配置在色谱柱装置下方的冷却风扇。
LTM 色谱柱也可完成更高速率的程序升温,进一步缩短周期时间。
最大操作程序速率将取决于多个因素,包括:色谱柱尺寸、相比、载气以及所需的分离。
常规的空气浴方法转换至LTM 格式时,可使用安捷伦方法转换软件计算初始条件。
467_残留溶剂_usp

INTRODUCTION 介绍
This general chapter applies to existing drug substances, excipients, and products. All substances and products are subject to relevant control of solvents likely to be present in a substance or product. 本通则适用于现有原料药、辅助剂、成药。所有原料药和成药均需对可能存在其中的溶剂 进行控制。
<467> RESIDUAL SOLVENTS 残留溶剂
(Chapter under this new title—to become official July 1, 2008) (这个新名称下的通则----2008 年 7 月 1 日起生效)
USP30残留溶剂
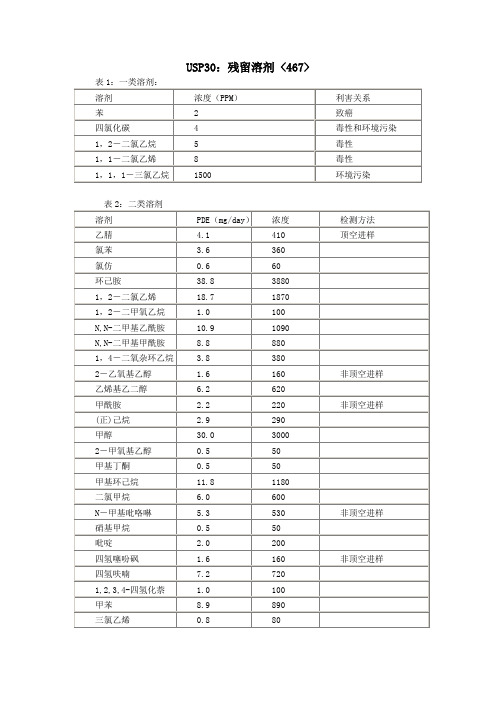
USP30:残留溶剂 <467>一类和二类溶剂(水溶性的物品)程序A:一类残留溶剂标准贮备液:取1.0ml的USP一类残留溶剂混合物RS到100ml的容量瓶中,加入9ml的二甲基亚砜,用水稀释到刻度,混匀。
取1.0ml该溶液到100ml的容量瓶中,用水稀释到刻度,混匀。
取1.0ml该溶液到10ml的容量瓶中,加水稀释到刻度,混匀。
一类残留溶剂标准液:移取1.0ml的一类残留溶剂标准贮备液到合适顶空安瓿瓶中,加5.0ml的水,加塞,加盖,混匀。
二类残留溶剂标准贮备液:取1.0ml的USP二类残留溶剂混合物A(RS)到100ml的容量瓶中,加水稀释到刻度,混匀,制得二类残留溶剂标准储备液A。
取1.0ml的USP二类残留溶剂混合物B(RS)到100ml 的容量瓶中,加水稀释到刻度,混匀,制得二类残留溶剂标准储备液B。
二类残留溶剂A标准液:移取1.0ml的二类残留溶剂标准贮备液A到合适顶空安瓿瓶中,加5.0ml的水,加塞,加盖,混匀。
二类残留溶剂B标准液:移取5.0ml的二类残留溶剂标准贮备液B到合适顶空安瓿瓶中,加1.0ml的水,加塞,加盖,混匀。
测试贮备液:取约250mg的测试品,准确称量,加入25ml的容量瓶中,加水溶解并稀释到刻度,混匀。
测试液:取测试贮备液5.0ml到合适顶空安瓿瓶中,加1.0ml的水,加塞,加盖,混匀。
一类系统适用性溶液:取1.0ml的一类残留溶剂标准贮备液到合适顶空安瓿瓶中,加入5.0ml的测试贮备液,加塞加盖,混匀。
色谱系统<见色谱621>:气相色谱,配制:FID检测器,0.32mm×30mm的炭化硅胶柱,涂布1.8µm厚的G43或0.53mm×30m的广口柱,涂布3µm厚的G43。
载气为氮气或氢气,线速约为35cm/秒,分流比为1:5。
柱温:40℃保持20分钟,然后以10℃/分钟的速度升温至240℃,保持20分钟;进样口和检测器的温度分别为140℃和250℃。
美国药典(USP)有机溶剂标准购买
search制药参考标准品中文版 | English公司简介新闻动态诚聘英才联系我们Restek GCRestek LCVarian GC资料产品类别现货速递(不断更新中)三氯乙烯DMSO0.4mg/mL MNK137016m-二甲苯DMSO 6.51mg/mL MNK137018o-二甲苯DMSO0.97mg/mL MNK137019p-二甲苯DMSO 1.52mg/mL MNK137020 DMSO = 二甲亚砜这些混合物反映了2008年7月1日生效的USP30/NF25中做出的改变。
残留溶剂 - 等级 1 (5种成分)苯 10mg/mL1,1-二氯乙烯 40四氯化碳 201,1,1-三氯乙烷 501,2-二氯乙烷 25溶剂为二甲亚砜, 1mL/安瓿货号 36279 (单件)残留溶剂等级 2 - 混合物 A (15种成分)乙腈 2.05mg/mL甲基环己烷 5.90氯苯 1.80二氯甲烷 3.00环己烷 19.40四氢呋喃 3.45顺-1,2-二氯乙烯 4.70甲苯 4.45反-1,2-二氯乙烯 4.70m-二甲苯 6.51 1,4-二氧己环 1.90o-二甲苯0.98乙基苯 1.84p-二甲苯 1.52甲醇 15.00溶剂为二甲亚砜, 1mL/安瓿货号 36271 (单件)残留溶剂等级 2 - 混合物 B (8种成分)氯仿60µg/mL硝基甲烷 501,2-二甲氧基乙烷 100嘧啶200n-己烷 (C6) 290萘满1002-己酮 50三氯乙烯 80溶剂为二甲亚砜, 1mL/安瓿货号 36280 (单件)残留溶剂等级 2 - 混合物 C (8种成分)2-乙二醇单乙醚800µg/mL2-甲氧基乙醇(甲基溶纤剂)乙烯乙二醇 3,100250甲酰胺 1,100N-甲基吡咯烷酮2,650N,N-二甲基乙酰胺 5,450环丁砜800N,N-二甲基甲酰胺 4,400溶剂为二甲亚砜, 1mL/安瓿货号 36273 (单件)第1级混合物反映了2000年1月1日生效的USP24/NF19与1995年1月1日到1999年12月31日之间有效的USP23/NF18之间的变化。
USP467残留溶剂分析-Agilent

前言残留溶剂分析是制药行业中的重要应用。
在生产过程中选择合适的溶剂可提高产量或通常影响所合成产品的化学特性。
但是,溶剂并不能增强产品的功效,所以必须尽可能完全将其去除以满足产品规格要求和药品生产质量管理规范1。
因此,在生产或纯化工艺中测试残留溶剂是生产过程的一个必要环节。
在配备有顶空进样器的 Agilent Intuvo 系统上评估根据 USP 467 对残留溶剂进行的分析。
Agilent Intuvo 9000 气相色谱仪与传统气相色谱系统相比具有多方面的优势:• 实现简化样品同时分流到两根色谱柱的模块化流路• 快速更换色谱柱,轻松实现方法开发• 体积更小巧USP 467残留溶剂分析如需了解更多信息,请访问:柱温箱85 °C定量环85 °C传输线100 °C样品瓶平衡40 分钟进样持续时间0.5 分钟样品瓶10 mL振荡开启,2 级样品瓶填充流速50 mL/min 样品瓶填充压力15 psi样品瓶压力平衡时间0.05 分钟定量环填充升压速率20 psi/min 定量环最终压力10 psi定量环平衡时间0.05 分钟实验部分Intuvo 9000 气相色谱仪配备有 Agilent 7697A 顶空进样器。
制备 1 类、2a 类和 2b 类标准溶液,并根据 USP 467 方法对其进行评估。
表 1. 1 类溶剂标准品的重现性1 类RSD%1,1-二氯乙烷 (1-1) 2.71,1,1-三氯乙烷 (1-2) 2.1四氯化碳 (1-3) 4.5苯 (1-4)1.91,2-二氯苯 (1-5)0.931,1-二氯乙烷 (1-6)2.7表3. 2b 类溶剂标准品的重现性2b 类RSD%己烷4.6硝基甲烷 6.7氯仿 4.21,2-二甲氧基乙烷 3.7三氯乙烯 4.6吡啶 2.82-己酮2.9四氢化萘3.6表 2. 2a 类溶剂标准品的重现性2a 类RSD%甲醇 1.3乙腈0.98二氯甲烷1.3反式-1,2-二氯乙烯2.4顺式-1,2-二氯乙烯 1.7四氢呋喃0.69环己烷2.5甲基环己烷 2.71,4-二氧六环1.1甲苯2.1氯苯 1.8乙苯2.3间、对二甲苯 2.3邻二甲苯2.1结果与讨论每种溶剂标准品(1 类、2a 类和 2b 类)准备八个顶空样品瓶,用于评估重现性。
USP30-NF25溶剂残留《467》(中文)20070703

残留溶剂的鉴别、控制、定量【注意-在下列各方法中指定的无有机物水在色谱上应没有明显的杂质干扰峰】1类和2类残留溶剂水溶性样品步骤A一类标准贮备溶液:转移1.0mLUSP一类残留溶剂混合溶液标准品于100mL容量瓶中,加9mL二甲基亚砜,以水稀释至刻度,混匀。
转移1.0mL此溶液于100mL容量瓶中,以水稀释至刻度,混匀。
转移1.0mL该溶液于10mL容量瓶中,以水稀释至刻度,混匀。
一类标准溶液:转移1.0mL一类标准贮备溶液于适合的顶空进样样品瓶中,加入5.0mL水,压上瓶塞和瓶盖,混匀。
二类标准贮备溶液转移l.OmLUSP二类残留溶剂混合溶液A标准品于100mL容量瓶中,以水稀释至刻度,混匀。
此为二类标准贮备溶液4。
转移l.OmLUSP二类残留溶剂混合溶液B标准品于100mL容量瓶,以水稀释到刻度,混匀。
此为二类标准贮备溶液B。
二类混合溶液4标准溶液转移1.0mL二类标准贮备溶液4于适当的顶空进样样品瓶中,加入5.OmL水,压上瓶塞和瓶盖,混匀。
二类混合溶液B标准溶液转移5.0mL二类标准贮备溶液B于适当的顶空进样样品瓶中,加入1.0mL水,压上瓶塞和瓶盖,混匀。
供试贮备溶液:精确称取250mg供试样品,转引至25mL容量瓶中,溶于水并以水稀释至刻度,混匀。
供试溶液:转移5.0mL供试贮备溶液于适当的顶空进样样品瓶中,加入1.0mL水,压上瓶塞和瓶盖,混匀。
一类系统适应性溶液转移1.0mL一类标准贮备溶液于适当的顶空进样样品瓶中,加入5.0mL供试贮备溶液,压上瓶塞和瓶盖,混匀。
色谱系统(见色谱<621>):该气相色谱仪装备一个火焰离子化(FID)检测器,一个0.32mm X30m毛细管柱(fused-silicacolumn)内部涂以1.8-pm厚的phaseG43涂层或者一个0.53mm X30m大孔径毛细管柱以3.0-pm厚的phaseG43为涂层。
载气为氮气或氦气,线速度为每秒35cm,分流比为1:5。
残留溶剂培训讲义——USP467

• 2类混合A标准溶液:移取1.0ml2类标准贮备液A到合适的顶空瓶中,加
5.0ml水,密封,加盖 。 • 2类混合B标准溶液:移取5.0ml2类标准贮备液B到合适的顶空瓶中,加
1.0ml水,密封,加盖。
• 供试贮备液——精密称取供试品约250mg到25-ml容量瓶,用水溶解并稀 释到刻度,摇匀。
• 供试液——移取5.0ml供试贮备液到适宜的顶空瓶中,加入1.0ml水,
残留溶剂 ——USP467
绪
言
——与国外的质控理念接轨
逐渐采用先进的分析方法和分析理念
专属的HPLC方法替代传统的容量法与生物法 重视对组分、相关物质(包括光学异构体)的控制 重视对残留溶剂的控制 重视对粒度的控制 重视对晶型的控制 用细菌内毒素检查法替代热原检查法 基本取消了异常毒性检查
气相色谱系统(见色谱法<621>) [注——若操作方法B得到的色谱图比操作方法A的色谱图好, 则采用操作方法B的色谱系统。]
• 色谱柱:0.32-mm × 30-m的弹性石英色谱柱,液膜厚1.8-µ m的 G43,或0.53-mm × 30-m的多孔性色谱柱,液膜厚3.0-µ m的 G43 • 载气:氮气(或氦气)线速度约为35cm/s • 分流比为1:5 (为了优化灵敏度,可适当调节) • 柱温:先在40℃维持20分钟,再以10℃/min的升温速率升至 240℃,并维持20分钟。 • 进样口温度: 140℃ • FID检测器温℃:250℃
在药品生产过程中使用过;
在药品生产过程中应当去除而未除尽; 特指有机挥发性化合物。
药品中残留溶剂的特点
种类相对固定,ICH规定了69种;
在具体样品中具有不确定性;
残留量相对较低,一般在痕量或微量范围
美国药典通则467残留溶剂的检测

美國藥典通則<467>殘留溶劑的檢測<467> RESIDUAL SOLVENTSSpectrum 质检部斯百全化学(上海)有限公司800.720.5720 通則適用範圍•所有藥品原料,輔料,製劑中可能存在的溶劑通則目標•為病人的安全,規定藥品中可接受的殘留溶劑量殘留溶劑分類•一類(class 1): 已知會產生不可接受毒性的一些溶劑(Solvents that are know to cause unacceptabletoxicities)•二類(class 2):與並不嚴重毒性相關的一些溶劑(Solvents associated with less severe toxicity)•三類(class 3):較少毒性的溶劑(Less toxic solvents )•其他殘留溶劑:無足夠毒理學數據(Other residual solvents)未列在本通則的溶劑怎麼處理?•當工廠使用經法定監管機構批准使用的未在本通則中羅列的新溶劑時,工廠有責任告知USP 在該品種標準中這一溶劑的鑒定、限度、測試方法,USP將會在個論中加上這一項。
•ICH 認可的新溶劑,USP將直接加在該通則中。
測試範圍•使用過的溶劑•生產或純化過程中產生的溶劑測試方式•直接測最終產品•或者各成份累加:如果各成分累加的值大于本通则的限度,则需测最终产品。
Colchicine秋水仙堿•Colchicine•C22H25NO6 399.44•Acetamide, N-(5,6,7,9-tetrahydro-1,2,3,10-tetramethoxy-9-oxobenzo[a]heptalen-7-yl)-, (S)-.Colchicine[64-86-8].•»Colchicine is an alkaloid contained in various species of Colchicum and in other genera. It contains not less than 94.0 percent and not more than 101.0 percent of C22H25NO6, calculated on the anhydrous, solvent-free basis.•Caution—Colchicine is extremely poisonous.•Packaging and storage—Preserve in tight, light-resistant containers.•USP Reference standards 11—USP Colchicine RS.•Identification, Infrared Absorption 197K—[note—Disregard any peak occurring at 1735 cm1.]•Specific rotation 781S: between 240 and 250, calculated on the anhydrous, solvent-free basis. Test solution: 10 mg per mL, in alcohol. •Water, Method I 921: not more than 2.0%.•Limit of colchiceine—To 5 mL of a solution (1 in 100) add 2 drops of ferric chloride TS: no definite green color is produced. •Limit of ethyl acetate—Internal standard solution—Dilute 0.5 mL of n-propyl alcohol with water to 100.0 mL. •Standard solution—Pipet1 mL of ethyl acetate and 0.5 mL of n-propyl alcohol into a 1000-mL volumetric flask, add water to volume, and mix. Each mL of Standard solution contains 0.90 mg of ethyl acetate.•Test solution—Place about 250 mg of Colchicine, accurately weighed, in a 10-mL volumetric flask, dissolve in about 8 mL of water, and add 1.0 mL of Internal standard solution. Add water to volume, and mix.•Procedure—Determine appropriate sensitivity settings on a gas chromatograph (see Chromatography 621) fitted with a 4-mm ×1.5-m column packed with 20% (w/v) phase G14 on support S1, maintaining the column temperature at75, using nitrogen as the carrier gas, and using a flame-ionization detector. Inject the Standard solution and the Test solution, determine the peak height for ethyl acetate relative to the peak height for n-propyl alcohol, and calculate the percentage, by weight, of ethyl acetate in the portion of Colchicine taken: not more than 8.0% is found.•Chromatographic purity—The sum of the responses of any peaks other than that due to colchicine, eluting within 1.5 times the retention time for colchicine, is not more than 5.0% of the sum of all responses, obtained as directed in the Assay.•Residual solvents 467: meets the requirements, except that the limit of chloroform is 100 ppm.二類溶劑計算選項•選項1:Dose ≤10g/dayConcentration(ppm)=(1000ug/mg ×PDE)/dose•選項2:根據藥品實際情況直接計算PDE是否符合要求,若超出限度則需測試。
467残留溶剂检查法(USP)

467有机挥发性杂质残留溶剂限度在药典中,药品中的残留溶剂系指在原料药或辅料的生产过程中,以及在制剂制备过程中使用过或产生的有机挥发物。
残留溶剂在实际的生产工艺中未能完全去除。
在原料药或辅料合成过程中选择适当的溶剂可以提高产量,或用于测定物质特性,如结晶形态、纯度、可溶性等。
所以,溶剂有时候是合成过程中的关键因素。
本章讨论的溶剂并非辅料,也不是溶剂化物。
但是,药品中的溶剂必须要评价并证明符合要求。
由于残留溶剂没有任何医疗效果,因此应尽可能地去除,使达到原料和成品的规定标准,生产工艺规定以及其它的质量要求。
药品中的残留溶剂的含量不得高于规定的安全范围。
已知会产生严重毒性的溶剂(表1,第一类)在药品原料、辅料和成品的生产中必须避免使用,除非它们严格符合毒性安全评估。
毒性稍低的溶剂(表2,第二类)必须限制含量,从而保护病患免除可能的副作用。
理论上,毒性最低的溶剂(表3,第三类)在实际需要的时候可以使用。
本章讨论的溶剂在附录1中已全部列出。
但这些表格和附录并不全面。
当有关调整部门正式批准同意使用其它溶剂时,这些溶剂可能就要添入表格和目录。
如果已知药品生产或净化过程中存在残留溶剂,那么原料、辅料和成品中必须检测残留溶剂。
但仅需检测在药品生产或净化过程中使用或产生的残留溶剂。
生产厂家可能会选择检测药品成品,但药品各成分的残留溶剂限度都可能需要逐步计算。
若计算的结果低于或在本章规定的范围内,那药品成品就没有必要检测残留溶剂。
但是若计算的结果高于规定范围,那成品就必须检测残留溶剂以确定处方在配制过程是否将有关的溶剂范围降低到可接受的量。
药品生产过程中如果使用了残留溶剂,那成品也必须检测。
见附录2有关残留溶剂的附加资料。
残留溶剂按毒性评估的分类国际化学安全会用“每日容许摄取量”(TDI)来衡量毒性化学药品的接触极限,而世界卫生组织(WHO)与其它国家、国际卫生机构和研究所则使用“每日允许摄取量”(ADI)。
在药品领域中,为了避免同一物质有不同的ADI S,将“每日安全接触量”(PDE)统一规定为残留溶剂的允许摄取量。
各国溶剂残留限量

USP[467]有机挥发性杂质残留溶剂限度Ⅰ级需避免的溶剂已知人体实验致癌物质;强烈疑似人体实验致癌物质;环境危害物质Ⅱ级需被限制的溶剂动物实验非生殖毒性(遗传);动物实验致癌物质或其它可能的非可逆致病因子;产生毒性如神经毒性或致畸性;其它疑似重大但可逆毒性Ⅲ级具有低潜在毒性溶剂对人体存在低毒性溶剂;无人体摄入量规定(注:Ⅲ级残留溶剂PDEs上限为≥50mg/天)表1 Ⅰ级残留溶剂溶剂浓度限度(ppm)不良反应苯 2 致癌物四氯化碳 4 中毒和外周脑组织损害1,2-二氯乙烷 5 中毒1,1-二氯乙烷8 中毒1,1,1-三氯乙烷1500 环境危害Ⅱ级残留溶剂表2 Ⅱ级残留溶剂溶剂PDE(mg/天)浓度限度(ppm)乙腈Acetonitrile 4.1 410氯苯Chlorobenzene 3.6 360氯仿Chloroform0.6 60环己烷Cyclohexane38.8 38801,2-二氯乙烯1,2-Dichloroethene18.7 18701,2-二甲氧乙烷 1.0 100N,N-二甲基乙酰胺N,N-Dimethylacetamide10.9 1090N,N-二甲基甲酰胺N,N-Dimethylformamide8.8 8801,4-二氧六环1,4-Dioxane3.8 3802-乙氧基乙醇 1.6 160乙二醇Ethylene glycol 6.2 620甲酰胺Formamide 2.2 220己烷Hexane 2.9 290甲醇Methanol30.0 30002-甲氧基乙醇0.5 50甲基丁基(甲)酮0.5 502-己酮甲基环己烷11.8 1180二氯甲烷 6.0 600N-甲基吡咯烷酮 5.3 530硝酸甲烷0.5 50吡啶 2.0 200环丁砜 1.6 160四氢呋喃7.2 7201,2,3,4-四氢化萘 1.0 100甲苯8.9 890三氯乙烯0.8 80二甲苯* 21.7 2170 *通常含有60%间-二甲苯,14%对-二甲苯,9%邻-二甲苯和17%乙苯Ⅲ级残留溶剂表3 Ⅲ级残留溶剂(GMP或其它原料药、赋形剂和药物制剂质量标准规定)醋酸Acetic acid 庚烷Heptane丙酮Acetone 乙酸异丁酯苯甲醚乙酸异丙酯正丁醇乙酸甲酯2-丁醇3-甲基-1-丁醇乙酸丁酯甲基乙基酮叔丁基甲基醚甲基异丁基酮异丙基苯2-甲基-1-丙醇二甲基亚砜戊烷乙醇1-戊醇乙酸乙酯1-丙醇乙醚2-丙醇甲酸乙酯乙酸丙酯甲酸表4 其它残留溶剂(缺乏足够的毒物学方面的资料)1,1-二乙氧基丙烷甲基异丙基酮1,1-二甲氧基甲烷甲基四氢呋喃2,2-二甲氧基丙烷溶剂己烷异辛烷三氯醋酸异丙醚三氟醋酸除另有规定外,样品中残留的有机挥发性杂质不得超过下表中规定限度:有机挥发性杂质限度(μg/g)氯仿601,4-二氧杂环己烷380二氯甲烷600三氯乙烷80附录1 本章节所述残留溶剂表溶剂别名化学结构式级别醋酸乙酸CH3COOH Ⅲ级CH3COCH3 Ⅲ级丙酮2-丙酮,丙醛-2-酮乙腈CH3CN Ⅱ级苯甲醚甲氧基苯Ⅲ级苯安息油Ⅰ级1-丁醇正丁醇,丁基-1-CH3(CH2)3OH Ⅲ级醇CH3CH2CH(OH)CH3 Ⅲ级2-丁醇2-丁基醇,丁基-2-醇醋酸丁酯醋酸丁基醚CH3COO(CH2)3CH3 Ⅲ级叔丁基甲基醚2-甲氧基-2-甲基(CH3)3COCH3 Ⅲ级丙烷四氯化碳四氯甲烷CCl4 Ⅰ级氯苯Ⅱ级氯仿三氯甲烷CHCl3 Ⅱ级枯烯异丙基苯,(1-甲Ⅲ级基乙基)苯环己烷Ⅱ级1,2-二氯乙烷均二氯乙烷CH2ClCH2Cl Ⅰ级1,1-二氯乙烷1,1-二氯乙烯H2C=CCl2 Ⅰ级1,2-二氯乙烷1,2-二氯乙烯ClHC=CHCl Ⅱ级1,2-二甲氧基乙二甲基溶纤剂H3COCH2CH2OCH3 Ⅱ级烷DMA CH3CON(CH3)2 Ⅱ级N,N-二甲基乙酰胺N,N-二甲基甲酰DMF HCON(CH3)2 Ⅱ级胺二甲亚砜DMSO (CH3)2SO Ⅲ级Ⅱ级1,4-二氧杂环己烷乙醇CH3CH2OH Ⅲ级2-乙氧基乙醇溶纤剂CH3CH2OCH2CH2OH Ⅱ级醋酸乙酯CH3COOCH2CH3 Ⅲ级乙二醇1,2-二羟基乙烷HOCH2CH2OH Ⅱ级乙醚二乙基醚CH3CH2OCH2CH3 Ⅲ级甲酸乙酯HCOOCH2CH3 Ⅲ级甲酰胺HCONH2 Ⅱ级甲酸HCOOH Ⅲ级庚烷正庚烷CH3(CH2)5CH3 Ⅲ级己烷正己烷CH3(CH2)4CH3 Ⅱ级醋酸异丁酯CH3COOCH2CH(CH3)2 Ⅲ级醋酸异丙酯CH3COOCH(CH3)2 Ⅲ级甲醇CH3OH Ⅱ级2-甲氧基乙醇甲基溶纤剂CH3OCH2CH2OH Ⅱ级醋酸甲酯CH3COOCH3 Ⅲ级3-甲基-1-丁醇异戊基醇(CH3)2CHCH2CH2OH Ⅲ级甲基丁基(甲)酮2-己酮CH3(CH2)3COCH3 Ⅱ级甲基环己烷Ⅱ级二氯甲烷CH2Cl2 Ⅱ级甲基乙基(甲)酮2-丁酮CH3CH2COCH3 Ⅲ级甲基异丁基(甲)MIBK CH3COCH2CH(CH3)2 Ⅲ级酮2-甲基-1-丙醇异丁基醇(CH3)2CHCH2OH Ⅲ级Ⅱ级N-甲基吡咯烷酮1-甲基-2-吡咯烷酮硝基甲烷CH3NO2 Ⅱ级戊烷正戊烷CH3(CH2)3CH3 Ⅲ级1-戊醇CH3CH2CH2OH Ⅲ级2-戊醇(CH3)2CHOH Ⅲ级醋酸丙酯CH3COOCH2CH2CH3 Ⅲ级吡啶Ⅱ级Ⅱ级环丁砜四氢噻吩1,1-二氧化物四氢呋喃环氧戊烷Ⅱ级Ⅱ级1,2,3,4-四氢化萘甲苯Ⅱ级1,1,1-三氯乙烷甲基氯仿CH3CCl3 Ⅰ级三氯乙烯1,1,2-三氯乙烯HClC=CCl2 Ⅱ级二甲苯* Ⅱ级*通常含有60%间-二甲苯,14%对-二甲苯,9%邻-二甲苯和17%乙苯EP残留溶剂表一.第一类残留溶剂溶剂极限浓度(ppm)涉及苯 2 致癌物质四氯化碳 4 有毒、对环境有害的1,2-二氯乙烷 5 有毒1,1-二氯乙烷8 有毒1,1,1-三氯乙烷1500 环境表二第二类残留溶剂溶剂PDE(mg/天)浓度极限(ppm)乙腈 4.1 410氯苯 3.6 360氯仿0.6 60环己烷38.8 3380 1,2-二氯乙烷18.7 18701,2-二甲氧基乙烷 1.0 100N,N-二甲基乙酰胺10.9 1090N,N-二甲基甲酰胺8.8 8801,4-二氧杂环己烷 3.8 380 2-乙氧基乙醇 1.6 160 乙二醇 6.2 620甲酰胺 2.2 220己烷 2.9 290甲醇30.0 3000 2-甲氧基乙醇0.5 50甲基丁基酮0.5 50甲基环己烷11.8 1180二氯甲烷 6.0 600 N-甲基吡咯烷酮 5.3 530硝基甲烷0.5 50 吡啶 2.0 200环丁砜 1.6 160四氢呋喃7.2 720四氢化奈 1.0 100 甲苯8.9 890 三氯乙烯0.8 80二甲苯* 21.7 2170 *通常含有60%间二甲苯,14%对二甲苯,9%邻二甲苯,和17%乙苯表三第三类残留溶剂(在药品、辅料、药物制剂中受到GMP或其它质量要求的限制)乙酸庚烷丙酮乙酸异丁酯苯甲醚乙酸异丙酯1-丁醇乙酸甲酯2-丁醇3-甲基-1-丁醇乙酸丁酯甲乙酮叔-丁基甲基醚甲异丁酮异丙基苯2-甲基-1-丙醇二甲基亚砜戊烷乙醇1-戊醇乙酸乙酯1-丙醇乙醚2-丙醇甲酸乙酯乙酸丙酯甲酸表四其它残留溶剂(没有充分毒物学数据)1,1-二乙氧基丙烷甲异丁酮1,1-二甲氧基甲烷甲基四氢呋喃2,2-二甲氧基丙烷溶剂已烷异辛烷三氯乙酸异丙醚三氟乙酸附录1 标准中所列的溶剂清单溶剂别名结构分类乙酸醋酸CH3COOH 第三类丙酮2-丙酮CH3COCH3第三类乙腈CH3CN 第二类苯甲醚茴香醚第三类苯安息油第一类1-丁醇丁-1-醇CH3(CH2)3OH 第三类2-丁醇丁-2-醇CH3CH2CH(OH)CH3第三类乙酸丁酯醋酸丁酯CH3CO(CH2)3CH3第三类叔丁基甲醚2-甲氧基-2-甲基(CH3)3COCH3第三类四氯化碳四氯甲烷CCl4第一类氯苯第二类氯仿CHCl3第二类异丙基苯异丙苯(1-甲基乙基)苯第三类环己烷环己胺1,2-二氯乙烷二氯化乙烯CH2ClCH2Cl 第一类1,1-二氯乙烷偏二氯乙烯H2C=CCl2第一类二氯乙烯二氯乙炔ClHC=CHCl 第二类二氯甲烷亚甲基氯CH2Cl21,2-乙二醇二甲醚H3COCH2CH2OCH3第二类NN-二甲基乙酰胺DMA CH3CON(CH3)2第二类NN-二甲基甲酰胺DMF HCON(CH3)2第二类二甲基亚砜DMSO HCON(CH3)2第三类1,4-二氧六环(1,4)二恶烷第二类乙醇CH3CH2OH 第三类二乙氧基乙醇溶纤剂CH3CH2OCH2CH2OH乙酸乙酯醋酸乙酯CH3COOCH2CH3第三类乙二醇1,2-乙二醇HOCH2CH2OH 第二类乙醚二乙醚CH3CH2OCH2CH3第三类甲酸乙酯HCOOCH2CH3第三类甲酰胺HCONH2第二类甲酸HCONH2第三类庚烷n-庚烷CH3(CH2)4CH3第三类正己烷n -正己烷CH3(CH2)4CH3第二类乙酸异丁酯醋酸异丁酯CH3COOCH2CH(CH3)2第三类乙酸异丙酯醋酸异丙酯CH3COOCH(CH3)2第三类甲醇CH3OH二甲醚CH3OCH2CH2OH 第二类乙酸甲酯醋酸甲酯CH3COOCH3第三类异戊醇3-甲基-1-丁醇(CH3)2CHCH2CH2OH 第三类甲丁酮2-己酮CH3(CH2)3COCH3第二类甲基环己烷环己基甲烷第二类丁酮2-丁酮 MEK CH3CH2COCH3第三类丙酮醛MIBK CH3COCH2CH(CH3)2第三类2-甲基-1-丙醇异丁醇(CH3)2CHCH2OH 第三类N-甲基吡咯烷酮第二类硝基甲烷CH3NO2第二类戊烷n-戊烷CH3(CH2)3CH3第三类正戊醇戊乙醇CH3(CH2)3CH2OH 第三类丙醇丙-1-醇CH3CH2CH2OH 第三类2-丙醇丙-2-醇(CH3)2CHOH 第三类丙基乙酸丙基醋酸CH3COOCH2CH2CH3第三类吡啶第二类环丁砜四亚甲基亚砜第二类四氢呋喃氧杂环戊烷第二类四氢萘1,2,3,4-四氢萘第二类甲苯第二类1,1,1-三氯乙烷CH3CCl3第一类1,1,2-三氯乙烯HClC=CCl2第二类二甲苯* 第二类*通常含有60%间二甲苯,14%对二甲苯,9%邻二甲苯,和17%乙苯限度标准一览表:药物中常见残留溶剂及其限度溶剂名称PDE值 (mg/天)限度(%)溶剂名称PDE值 (mg/天)限度(%)第一类溶剂(应该避免使用)第三类溶剂(GMP或其他质量要求限制使用)苯0.0 0.0002 醋酸50.0 0.5 四氯化碳0.0 0.0004 丙酮50.0 0.5 1, 2-二氯乙烷0.1 0.0005 甲氧基苯50.0 0.5CP限度标准一览表:。
USP方法467:有机溶液残余物测试方法说明书

Class 3 Solvents
• If only Class 3 solvents are present… • Solvent level is determined by <731> Loss on
Drying • The PDE limit, unless otherwise specified, is
Solvent
Conc. K Value*
1 Methylene Chloride 10mg/mL 5.65
3
Benzene
1mg/mL
2.9
5
1,4-dioxane
2mg/mL 1618
*Compounds dissolved in water at 40°C
Equilibration Time
Isopropyl Acetate • (25/26) Anisole/DMSO
Limitations of the Method <467>
• Long analysis time >30min • Poor resolution of some compounds • Long wait equilibration time for headspace samples • Poor detection of some compounds • No definitive identification of contaminates
• Addition of matrix modifiers:
– Ammonium Chloride – Ammonium Sulfate – Sodium Chloride – Sodium Citrate – Sodium Sulfate – Potassium Carbonate
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
<467> RESIDUAL SOLVENTS残留溶剂(Chapter under this new title—to become official July 1, 2008)(这个新名称下的通则----2008年7月1日起生效)(Current chapter title is <467> Organic Volatile Impurities)(当前通则名称是<467>有机挥发性杂质)INTRODUCTION介绍This general chapter applies to existing drug substances, excipients, and products. All substances and products are subject to relevant control of solvents likely to be present in a substance or product.本通则适用于现有原料药、辅助剂、成药。
所有原料药和成药均需对可能存在其中的溶剂进行控制。
Where the limits to be applied comply with those given below, tests for residual solvents are not generally mentioned in specific monographs because the solvents employed may vary from one manufacturer to another.在所适用的限度与下面所述相符的情况下,残留溶剂检测一般不在具体各论中提及,因为每个生产商所使用的溶剂可能都不相同。
The objective of this general chapter is to provide acceptable amounts of residual solvents in pharmaceuticals for the safety of the patient. The general chapter recommends the use of less toxic solvents and describes levels considered to be toxicologically acceptable for some residual solvents.本通则的目的是为了保障病人的安全,提供在药物中残留溶剂的可接受数量。
本通则建议使用毒性较低的溶剂,并描述了对于某些残留溶剂而言其毒性可接受的数量水平。
For pharmacopeial purposes, residual solvents in pharmaceuticals are defined as organic volatile chemicals that are used or produced in the manufacture of drug substances or excipients, or in the preparation of drug products. The residual solvents are not completely removed by practical manufacturing techniques. Appropriate selection of the solvent for the synthesis of a drug substance or an excipient may enhance the yield, or determine characteristics such as crystal form, purity, and solubility. Therefore, the solvent may sometimes be a critical element in the synthetic process. This general chapter does not address solvents deliberately used as excipients nor does it address solvates. However, the content of solvents in such products should be evaluated and justified.在药典中,药物中的残留溶剂被定义为有机挥发性化学品,其用于或产生于原料药或辅助剂的生产过程中,或成药的制备过程中。
残留溶剂并未通过实际生产技术完全去除。
适当选择用于合成原料药或辅助剂的溶剂可以增加得率,或确定特性,例如结晶状态、纯度、溶解性。
因此,溶剂有时可以成为合成工艺中的重要部分。
本通则并未涉及故意用于辅助剂的溶剂,也未涉及溶剂化物。
但是,在此类产品中的溶剂含量也应进行评估或论证。
Because residual solvents do not provide therapeutic benefit, they should be removed, to the extent possible, to meet ingredient and product specifications, good manufacturing practices, orother quality-based requirements. Drug products should contain no higher levels of residual solvents than can be supported by safety data. Solvents that are known to cause unacceptable toxicities (Class 1, Table 1) should be avoided in the production of drug substances, excipients, or drug products unless their use can be strongly justified in a risk-benefit assessment. Solvents associated with less severe toxicity (Class 2, Table 2) should be limited in order to protect patients from potential adverse effects. Ideally, less toxic solvents (Class 3, Table 3) should be used where practical. The complete list of solvents included in this general chapter is given in Appendix 1. These tables and the list are not exhaustive. For the purposes of this Pharmacopeia, when a manufacturer has received approval from a competent regulatory authority for the use of a new solvent not currently listed in this general chapter, it is the responsibility of that manufacturer to notify the USP regarding the identity of this solvent, the approved residual solvent limit in the article, and the appropriate test procedure for this residual solvent in the article. The USP will then address this topic in the individual monograph. When a new solvent has been approved through the ICH process, this new solvent will be added to the appropriate list in this general chapter. At that time consideration will be given for removal of the specific solvent test requirement in the individual monograph.由于残留溶剂不提供治疗作用,它们应当被尽可能地去掉,以达到原料药和成药质量标准、药品优良生产规范、和其他质量控制要求。
成药含有的残留溶剂应该不超过有安全数据所支持水平。
已知会产生不可接受的毒性(一类,表1)的溶剂应该避免用于原料药、辅助剂、成药的生产中,除非它们的使用得到了风险-收益评估的强力支持。
毒性严重程度稍低(二类,表2)的溶剂应当被限制使用,以使病人远离潜在的不良反映。
较理想的是,在可行的地方使用毒性较低(三类,表3)的溶剂。
在本通则中包含的完整溶剂清单在附件1中给出。
这些表格和溶剂清单并非全部。
对于本药典而言,当某个生产商从具备能力的监管当局收到了对使用目前本通则未列入的溶剂的许可时,该生产商有义务通知USP该溶剂的鉴别、在物品中准许的残留溶剂限度、对于物品中残留溶剂的适当检测规程。
然后,USP 将会在具体的各论中涉及此主题。
当新溶剂通过ICH程序被批准,此新溶剂将会被加入到本通则里的适当清单中。
到那时,将会考虑去掉在具体各论中对具体残留溶剂的要求。
Testing of drug substances, excipients, and drug products for residual solvents should be performed when production or purification processes are known to result in the presence of such residual solvents. It is only necessary to test for residual solvents that are used or produced in the manufacture or purification of drug substances, excipients, or products.应该在生产或纯化工艺已知会导致此类残留溶剂时,进行对原料药、辅助剂、成药的残留溶剂测试。