蛋白转染方法
细胞转染的原理操作步骤以及小技巧

细胞转染的原理操作步骤以及小技巧细胞转染是一种将外源DNA、RNA、蛋白质等分子导入到细胞内的实验技术。
这种技术可以用来研究基因功能、发现新的信号通路和治疗基因疾病等。
下面将介绍细胞转染的原理、操作步骤以及一些小技巧。
一、细胞转染的原理:细胞转染主要通过三种方法实现:物理法、化学法和生物学法。
1.物理法:通过高压、电穿孔、微射流等方式,使细胞膜发生瞬时破裂,从而使DNA、RNA等外源分子进入细胞。
常用的物理法有电穿孔法和基因枪法。
2.化学法:通过化学物质,如聚吡咯、脂质体等,使外源分子与细胞膜结合,从而实现转染。
常用的化学法有聚乙烯亚胺(PEI)法、磷酸钙共沉淀法等。
3.生物学法:通过利用病毒载体将外源基因导入目标细胞,实现基因的转移。
常用的生物学法有腺相关病毒(AAV)转染、逆转录病毒(RETRO)转染等。
二、细胞转染的操作步骤:1.细胞的预处理:根据细胞类型和实验要求,将细胞培养至合适的状态。
通常细胞应处于快速生长期,但还未达到接触抑制的阶段。
对于一些特定的细胞,如悬浮细胞,可能需要将其转接至适当的培养基中。
2.外源分子的准备:将外源DNA、RNA等转染载体制备好。
如将DNA克隆并纯化至高质量的质粒DNA,或将RNA合成或纯化。
根据实验要求选择合适的转染载体。
3.转染方法的选择:根据实验要求选择合适的转染方法,如物理法、化学法或生物学法。
一般情况下,物理法适用于悬浮细胞,化学法适用于贴壁细胞,而生物学法适用于大多数细胞类型。
4.细胞转染操作:a.物理法:i.电穿孔法:将细胞悬浮于含有外源分子的缓冲液中,然后通过电穿孔仪的电极或电穿孔板进行电穿孔。
ii. 基因枪法:使用基因枪将外源分子直接“枪”入目标细胞中。
b.化学法:i.PEI法:将PEI与外源DNA或RNA按一定比例混合,在适当条件下形成复合物,然后添加至目标细胞中。
ii. 磷酸钙共沉淀法:将外源DNA与磷酸钙按比例混合,并静置形成磷酸钙- DNA沉淀,然后加入至目标细胞中。
转染详细步骤大攻略

转染详细步骤大攻略范例----真核重组表达质粒pDsRed-N1-NS1在A549细胞中表达按上海索莱宝生物科技有限公司去内毒素质粒小提试剂盒说明书方法进行质粒抽提,测得质粒浓度为410.32ng/µl。
将培养的A549细胞铺板,待细胞密度长到90%左右时,按lipo2000说明书转染A549细胞,36h后于荧光显微镜下观察DsRed-NS1融合蛋白的表达情况。
具体操作步骤如下:1)质粒准备按上海索莱宝生物科技有限公司去内毒素质粒小提试剂盒说明书方法进行质粒抽提,具体步骤如下:(1) 取1-5ml细菌培养物,12000rpm离心1 min,尽量吸除上清(菌液较多时可以通过多次离心将菌体沉淀收集到一个离心管中)。
(2) 向留有菌体沉淀的离心管中加入200µl溶液P1(请先检查是否已加入RNaseA),使用移液器或涡旋振荡器彻底悬浮细菌细胞沉淀。
(注:如果菌块未彻底混匀,会影响裂解导致质粒提取量和纯度偏低)(3)向离心管中加入200µl溶液P2,温和地上下翻转6-8次使菌体充分裂解。
(注:混匀一定要温和,以免污染基因组DNA,此时菌液应变得清亮粘稠,所用时间不应超过5min,以免质粒受到破坏)(4)向离心管中加入250µl溶液P3,立即温和地上下翻转6-8次,充分混匀,此时会出现白色絮状沉淀。
12000rpm 离心10min,用移液器小心地将上清转移到另一个干净的离心管中,尽量不要吸出沉淀。
(注:溶液P3加入后应立即混合,避免产生局部沉淀。
如果上清中还有微小白色沉淀,可再次离心后取上清)(5)加入1/5体积冰预冷的去内毒素清除剂,振荡混匀,溶液变浑浊,冰浴2min 至溶液变清亮。
(6)37℃水浴5 min,不时振荡,溶液又变浑浊。
12000rpm室温离心5min,溶液应分为两相,上层水相含质粒DNA,下层油状相含内毒素。
(7)将质粒DNA上层水相转移至新管,弃下层油状相,注意不要吸入油状相,重复抽提三次,即重复步骤5-7三次。
转染的原理和应用

转染的原理和应用什么是转染?转染是将外源DNA、RNA或蛋白质等分子导入到细胞内的过程。
转染技术是现代生物学研究和应用中的重要工具,可以用于基因表达、细胞功能研究、药物筛选等领域。
转染的原理转染过程涉及将外源分子有效地导入到细胞内。
常用的转染方法包括物理法、化学法和生物法。
物理法物理法主要包括电穿孔、微射流、压力驱动、激光穿孔等。
这些方法通过破坏细胞膜的完整性,使外源分子能够进入细胞。
化学法化学法利用高分子化合物或化学试剂,如聚乙烯亚胺(PEI)、脂质体、钙磷共沉淀物等,在细胞膜上形成复合物来实现转染。
这些复合物能够与外源分子结合,并通过细胞膜的翻转、溶酶体逃逸等机制将其导入细胞。
生物法生物法主要通过利用病毒载体将外源分子导入细胞。
常用的病毒载体有腺病毒、腺相关病毒、逆转录病毒等。
病毒载体能够在细胞内复制和表达外源基因,并将其传递给后代细胞。
转染的应用转染技术在生物学研究和生物医学应用领域具有广泛的应用。
基因表达转染技术可以用于基因表达研究。
通过导入外源基因到细胞内,可以实现特定蛋白质的高水平表达。
这对研究特定基因的功能、调控机制以及疾病的发生机制具有重要意义。
细胞功能研究转染技术可以用于研究细胞的功能和调控机制。
通过转染特定基因,可以操纵细胞内蛋白质的表达水平,进而研究其对细胞功能的影响。
药物筛选转染技术可以用于药物筛选。
通过转染细胞系,可以高效地筛选和评估候选药物对特定靶点的活性和选择性。
基因治疗转染技术可以用于基因治疗。
通过导入正确的基因到患者体内,可以纠正某些基因缺陷导致的疾病。
转基因生物制造转染技术可以用于转基因生物的制造。
通过将外源基因导入植物、动物等生物体内,可以使其表达特定的蛋白质或产生特定的物质,用于工业生产或研发新药。
结论转染技术在生物学研究和应用中起着重要的作用。
以物理法、化学法和生物法为基础,转染技术提供了一种有效地将外源分子导入细胞的方法。
在基因表达、细胞功能研究、药物筛选、基因治疗和转基因生物制造等领域,转染技术都具有广泛的应用前景。
cho细胞表达重组蛋白方案

CHO (Chinese Hamster Ovary) 细胞是常用的哺乳动物细胞系统,用于表达重组蛋白的研究和生产。
以下是一般性的CHO 细胞表达重组蛋白的方案:
1. 购买表达载体:选择适合的表达载体,可以是质粒或病毒载体。
载体应包含适当的启动子、选择标记等。
2. 转染CHO 细胞:将表达载体导入CHO 细胞中。
转染方法可以选择经典的化学或电穿孔法,也可以选择使用特定的转染试剂或转染仪器。
3. 选择稳定转染株:在转染后,使用适当的选择剂(如抗生素) 处理细胞,以选择稳定表达重组蛋白的细胞株。
可通过单克隆分离等方法筛选和扩增单一细胞克隆。
4. 细胞培养条件优化:优化培养基配方和细胞培养条件,包括温度、pH 值、培养基组分等,以提高重组蛋白的产量和纯度。
5. 表达蛋白的诱导:使用适当的诱导剂或方法,例如添加诱导剂(如甲酪酸) 到培养基中,以启动重组蛋白的表达。
6. 重组蛋白的纯化和分析:通过细胞破碎和不同的纯化步骤(如亲和层析、离子交换层析、凝胶过滤等)从培养基或细胞提取物中纯化目标重组蛋白,并使用适当的分析方法验证表达的蛋白的纯度和功能。
在每个步骤中,需要根据具体的重组蛋白和研究目的进行优化和调整。
此外,合理的培养细胞和操作操作也至关重要,以确保产量和纯度的理想达到。
这些方案的细节将根据具体的实验目的和需要进行个体化定制。
重组蛋白转染实验步骤

重组蛋白转染实验步骤
重组蛋白转染实验的步骤包括以下几个部分:
1.重组蛋白的表达和纯化:首先,通过基因工程技术将目的基因克
隆到表达载体中,并在适当的宿主细胞中进行表达。
表达出的重组蛋白需要进行纯化,通常采用亲和层析、离子交换层析等方法进行分离纯化。
2.重组蛋白的转染:将纯化后的重组蛋白导入到细胞中,常用的转
染方法包括磷酸钙法、电穿孔法、脂质体法等。
转染时需要优化转染条件,如蛋白质浓度、转染时间、转染温度等,以提高转染效率和细胞存活率。
3.细胞培养和观察:将转染后的细胞放置在适当的培养条件下进行
培养,观察细胞的生长和形态变化。
可以通过荧光显微镜、免疫荧光等技术检测重组蛋白在细胞内的表达和定位。
4.数据分析:对实验数据进行统计分析,比较不同处理组之间的差
异,并评估重组蛋白对细胞生长、凋亡等生物学过程的影响。
需要注意的是,重组蛋白转染实验需要遵循相关的实验室安全规定,如穿戴防护服、手套等个人防护措施,以避免实验操作过程中可能产生的危险。
同时,实验前需要进行充分的文献调研和实验设计,确保实验的科学性和可行性。
CHO-S细胞蛋白表达操作规程

CHO-S细胞转染操作规程ExpiCHO™ Expression Medium不会抑制转染,因此转染时无需更换培养基。
一、实验材料:1、ExpiCHO-S™ cells cultured in ExpiCHO™ Expression Medium。
2、无菌、无苯酚、无氯化钠,主要是超螺旋结构的含目的基因的质粒。
N0te:推荐使用《PureLink™ HiPure Plasmid Ki t 》试剂盒抽提质粒。
为了保证无菌,可以使用0.22um的过滤器过滤质粒。
3、抗体表达阳性对照质粒(100 µL of the 1 mg/mL,由试剂盒提供)4、ExpiFectamine™ CHO Reagent(4℃冷藏使用,使用前无需预热)5、OptiPRO™ SFM complexation medium(4℃冷藏使用,使用前无需预热)6、ExpiCHO™ Expression Medium(使用前预热至37℃)Note:转染过程中不能添加抗生素,否则会影响转染效率。
7、Polycarbonate, disposable, sterile Erlenmeyer flasks。
8、37℃,8%CO2,可空气加湿的摇床。
9、测定活细胞密度和百分比的试剂和仪器。
(牛鲍计数板、台盼蓝染液)二、转染注意事项:1、新复苏的细胞在转染之前至少要传2代以上。
2、所有针对细胞的操作要严格无菌、动作轻柔,避免剧烈的震荡或移液,细胞的健康状态对最大程度的提高转染效率至关重要。
3、如果不使用ExpiFectamine™ CHO Reagent转染高密度的ExpiCHO-S™ 细胞,转染效率将会大幅度降低。
4、ExpiFectamine™ CHO Reagent使用之前柔和颠倒4-5次,确保试剂充分混匀。
5、质粒DNA与ExpiFectamine™ CHO Reagent的混合可在室温下进行,使用4℃冷藏试剂。
6、一旦ExpiFectamine™ CHO/DNA复合物形成,应立刻将该复合物加到细胞悬液中,5分钟之内不影响转染效率,但不能超过10分钟。
蛋白质转染技术制备方法

蛋白质转染技术是一种将表达出的目标蛋白直接转入细胞的过程。
这个过程中,通常会用到辅助蛋白或构建融合蛋白来帮助目标蛋白进入细胞质。
转染方法通常是共孵育。
具体制备方法如下:
1.准备实验材料,包括细胞(如鼠成纤维细胞)、试剂和试剂盒(如丙戊酸培养液)、仪器和耗材
(如培养皿、移液枪和枪头等)。
2.将目标蛋白(如重组重新编程蛋白质,包括Oct4-11R、Sox2-11R、Klf4-11R和c-Myc-11R)
与培养基混合,并可能添加其他辅助剂(如丙戊酸和HDAC抑制剂)来提高编程效率。
3.将处理过的细胞在特定条件下培养一段时间(如36小时),使目标蛋白有机会进入细胞并发挥
作用。
4.在完成多次(如四次)蛋白转导后,处理过的细胞可能需要进行后续处理,如转移到其他类型的
细胞上,并在特定培养基中保存和扩增。
另外,值得注意的是,由于蛋白质转染技术并不涉及基因的导入,因此它只适用于瞬时转染,即目标蛋白的作用会在一段时间后消失。
如果需要长期的蛋白表达,可能需要考虑其他方法,如基因转染。
此外,对于蛋白质转染,也有一些专门的试剂,如PULSinTM,这是一种由Polyplus Transfection公司开发的新型转染试剂,它可以通过正电荷外衣包裹许多蛋白质/抗体,使其能够有效地转染到活细胞中。
蛋白 共定位 步骤 质粒 转染

蛋白共定位步骤质粒转染
蛋白共定位是一种研究蛋白质相互作用的方法,其可以描述两个或多个蛋白质之间的关系,包括它们的结合位置、交互作用、功能等。
在进行蛋白共定位实验时,质粒转染是一个重要的步骤,下面我们来了解一下质粒转染的具体步骤。
首先,我们需要准备好需要转染的质粒和目标细胞。
质粒是一种可以被细胞摄取并在其中进行表达的DNA分子。
而目标细胞是我们希望进行共定位实验的细胞,通常选择表达目标蛋白质的细胞。
在准备好这些材料之后,我们可以进行质粒转染实验了。
第一步,我们需要将质粒与转染试剂(例如聚乙烯亚胺)混合均匀,并在室温下静置一段时间,使其形成复合物。
这种复合物可以帮助质粒进入细胞。
第二步,我们需要将转染复合物加入到目标细胞中。
在加入复合物之前,需要将细胞培养在适当的培养基中,并保证细胞处于生长状态。
第三步,我们需要对转染后的细胞进行处理,以保证蛋白质表达和共定位实验能够进行。
例如,可以使用药物或化合物来诱导或抑制蛋白质表达,或者对细胞进行特定的处理,以激活或抑制某些基因表达。
第四步,我们需要对转染后的细胞进行共定位实验。
在这一步中,我们可以使用各种分析方法,例如共免疫共沉淀、荧光共定位、双杂交等方法,来研究蛋白质之间的相互作用。
最后,在完成共定位实验后,我们可以通过分析和解释实验结果,来深入了解蛋白质之间的相互作用和功能,并为后续研究提供重要的信息和指导。
总之,质粒转染是蛋白共定位实验的重要步骤之一,其可以帮助我们将质粒有效地引入到目标细胞中,为后续的共定位实验提供必要的条件和保证。
Cas9 蛋白转染细胞方法
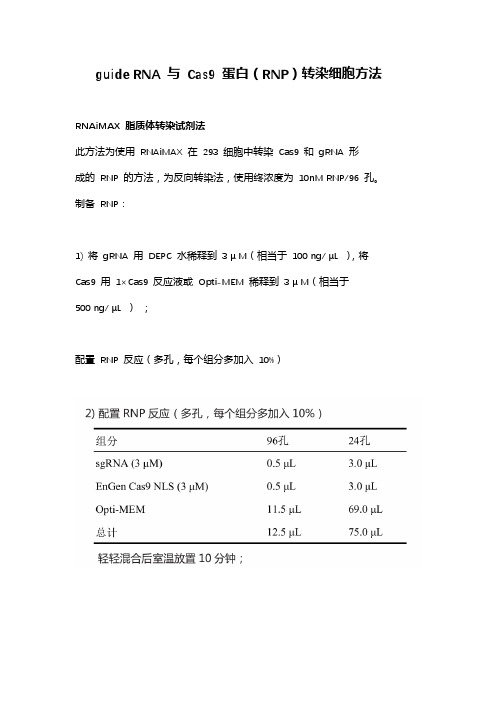
guide RNA 与Cas9 蛋白(RNP)转染细胞方法
RNAiMAX 脂质体转染试剂法
此方法为使用RNAiMAX 在293 细胞中转染Cas9 和gRNA 形
成的RNP 的方法,为反向转染法,使用终浓度为10nM RNP/96 孔。
制备RNP:
1) 将gRNA 用DEPC 水稀释到3 μ M(相当于100 ng/ μL ),将Cas9 用1×Cas9 反应液或Opti-MEM 稀释到3 μ M(相当于
500 ng/ μL );
配置RNP 反应(多孔,每个组分多加入10%)
准备转染的293 细胞:
1) 提前铺板,让转染时其汇合率70~90% 的293 细胞;
2) 在RNP/liposome 室温放置20 分钟期间,使用胰酶消化细胞,并清洗一次细胞,去除迹量胰酶(防止其对Cas9 蛋白的降解),重悬细胞到适量media 中并计数;
3) 加入media 时细胞终密度在3.2×10 5 cells/mL,每96 孔需要125 μL 细胞,每24 孔需要750 μL 细胞。
转染实验:
1) 向96 孔培养板中加入25 μL RNP/liposome 复合体,24 孔中加入150 μL RNP/liposome 复合体;
2) 96 孔板加入125 μL 细胞(3.2×10 5 cells/mL),24 孔加入
750 μL 细胞,并用枪轻轻吹打混合几次;
3) 将细胞放入细胞培养箱中培养48~72 小时后,收集部分细胞基因型鉴定。
细胞转染的方法和基本原理

细胞转染的方法和基本原理细胞转染是生物学研究中常用的实验技术,用于将外源DNA、RNA或蛋白质引入到靶细胞中。
本文将介绍细胞转染的方法和基本原理。
一、细胞转染的方法1. 化学法转染:化学法转染是最常用的细胞转染方法之一。
通过利用化学物质如聚乙烯亚胺(PEI)或脂质体等,将外源DNA或RNA 包裹成复合物,与细胞膜结合后进入细胞。
这种方法操作简单、成本低廉,适用于多种细胞类型。
但转染效率较低,对细胞有一定毒性。
2. 病毒载体转染:病毒载体转染是一种高效的细胞转染方法。
病毒载体可以将外源基因嵌入病毒基因组中,然后通过感染细胞的方式将基因导入细胞内。
常用的病毒载体有腺病毒、逆转录病毒等。
这种方法转染效率高,适用于多种细胞类型,但需要特殊设备和技术,同时也有一定的生物安全风险。
3. 电穿孔法转染:电穿孔法利用高压脉冲作用于细胞膜,破坏细胞膜结构,从而使外源DNA或RNA进入细胞。
这种方法操作简单,转染效率较高,但对细胞有一定的毒性,并且只适用于某些特定的细胞类型。
4. 基因枪法转染:基因枪法是一种生理穿孔法,通过利用高压气体或火药驱动基因枪,将外源DNA或RNA以微粒形式直接射入细胞。
这种方法适用于多种细胞类型,转染效率较高,但需要特殊设备和技术,并且对细胞有一定的毒性。
二、细胞转染的基本原理细胞转染的基本原理是通过一定的方法将外源DNA、RNA或蛋白质引入靶细胞,使其在细胞内表达或功能发挥。
转染后的细胞可以用于研究基因功能、蛋白质表达及相互作用等。
细胞转染的基本原理可以分为三个步骤:吸附、内化和表达。
1. 吸附:在化学法转染中,外源DNA或RNA会与载体相结合形成复合物,通过静电作用与细胞膜结合。
在病毒载体转染中,病毒载体会与细胞膜表面的受体结合。
吸附是转染的第一步,直接影响转染效率。
2. 内化:吸附后,外源DNA、RNA或蛋白质需要进入细胞内部。
在化学法转染中,复合物通过细胞膜的内吞作用或直接渗透进入细胞质。
两大蛋白质染色主流方法大比较

两大蛋白质染色主流方法大比较日期:2012-05-31 来源:未知标签:考马斯亮蓝银染法蛋白质染色摘要: 常用的蛋白质染色试剂分为已考马斯亮蓝为代表的有机试剂染色、银染、荧光染色及同位素显色。
其中考马斯亮蓝染色法的应用较为广泛,现将其与其他的蛋白质染色方法(主要是银染法)作一比较,帮助大家更好地去选择合适的蛋白质染色方法。
蛋白质的染色常用的有4类:有机试剂染色、银染、荧光染色及同位素显色。
其中有机试剂染色以考马斯亮蓝染色法(Coomasie brilliant blue,汉恒生物--慢病毒腺病毒免费试用申请入口汉恒生物--腺相关病毒(动物体内转染神器)免费试用申请入口恩必美生物新一轮2-5折生物试剂大促销!常用的蛋白质染色试剂分为已考马斯亮蓝为代表的有机试剂染色、银染、荧光染色及同位素显色。
其中考马斯亮蓝染色法的应用较为广泛,现将其与其他的蛋白质染色方法(主要是银染法)作一比较,帮助大家更好地去选择合适的蛋白质染色方法。
蛋白质的染色常用的有4类:有机试剂染色、银染、荧光染色及同位素显色。
其中有机试剂染色以考马斯亮蓝染色法(Coomasie brilliant blue,CBB)为代表,在蛋白质分析中常用,但对低丰度蛋白质的显现较差;银染灵敏度虽高,却常与质谱不兼容;荧光染色以SYPRO 试剂为主,蛋白质检测灵敏度高,能兼容质谱,但由于需要配备特殊的检测仪器及试剂的昂贵,未被作为常规方法使用;而同位素显色则存在安全性和操作局限性等问题。
因此,筛选简便、节约、检测灵敏度高、质谱兼容的蛋白质着色法是蛋白质组研究所需。
由于考马斯亮蓝染色法的广泛运用,近年来就考马斯亮蓝染色法在提高其灵敏性方面研究者们作了许多改进,方法众多,评价不一。
我们在作双向凝胶电泳时,将常用的几种考马斯亮蓝染色法及银染进行了比较,并就其染色影响因素作出分析。
试剂固相pH梯度干胶条(IPG strip pH3-10NL,IPG strip pH5-8,17cm)、urea、CHAPS、TBP、DTT、Bio-Lyte 3/10 Ampholyte、IAM、SDS、Tris、丙烯酰胺、甲叉双丙烯酰胺、甘氨酸、CBB-G250均为BIO-RAD公司产品。
几种转染方法的比较

几种转染方法的比较
目前,基因转染技术已经发展为分子生物学、生物工程和基因治疗等
领域的重要实用工具,它可以极大地提高研究的效率和准确度,是许多重
要的基础实验的重要手段。
基因转染,就是把DNA片段植入受体细胞,使
其形成完整的外源基因,从而使其编码的蛋白质可以表达出来。
其中,质
粒转染(经典CaCl2转染)、电穿孔转染、膜融合转染、磁珠转染、膜膜
转染、管状细胞转染(cylinder-mediated gene transfer)、病毒转染
和小肠转染等转染方法,是目前比较常用的基因转染方法。
一、质粒转染
质粒转染是将外源DNA片段载体在质粒上,用极低的浓度CaCl2诱导
细胞膜的瞬时通透性,使外源基因可以通过通透的细胞膜进入细胞,这是
质粒转染的原理,也是最常用的一种质粒转染方法。
质粒转染的优点:
(1)操作简单,易于大批量高效率的实验。
(2)操作条件宽松,不受受体细胞类型的限制,可以适应多种宿主
细胞。
(3)转染效率高,可以达到百分之九十以上。
(4)可以通过有效的筛选系统,有效控制外源DNA的插入和表达量。
质粒转染的缺点:
(1)转染过程对细胞毒性较大,转染效率有限。
丁酸钠促进转染蛋白表达的原理

在细胞学和分子生物学领域,转染蛋白表达是一项非常重要的技术。
当我们想要研究特定的蛋白质在细胞内的功能、机制或定位时,常常需要将外源的DNA或RNA转染入细胞内,使其表达或干预细胞内的生物过程。
为了提高转染效率,常常需要借助一些辅助剂,其中丁酸钠就是一种常用的转染增效剂。
本文将对丁酸钠促进转染蛋白表达的原理进行详细解析。
1. 丁酸钠的作用机制丁酸钠,又称丁酸氢钠,是一种带正电荷的化合物。
在转染过程中,它的作用主要是通过中和DNA或RNA的负电荷,帮助其穿过细胞膜进入细胞内。
通常情况下,细胞膜是一层由脂质组成的疏水屏障,对于带有负电荷的核酸分子来说,很难自行穿越细胞膜,因此需要丁酸钠等转染增效剂的辅助。
2. 丁酸钠的增效作用丁酸钠的带正电荷可以与DNA或RNA的负电荷相吸引,形成复合物。
这些复合物在与细胞膜接触时,可以改善核酸分子的穿膜能力,从而增加转染效率。
丁酸钠还可以促进转染后的内吞作用,帮助DNA或RNA更快速地进入细胞质,并避免被溶酶体降解,从而提高外源基因的表达水平。
3. 丁酸钠的使用注意事项尽管丁酸钠在转染过程中可以起到促进作用,但过量使用也可能对细胞产生毒性。
在进行转染实验时,需要根据细胞类型、转染试剂的浓度和转染时间等因素进行调整,以确保最佳的转染效果。
4. 个人观点和理解从我个人的实验经验来看,丁酸钠的确能够显著增加外源基因的表达水平。
然而,在使用过程中也发现,不同细胞株对丁酸钠的耐受性存在差异,需要根据具体情况进行优化。
丁酸钠也可能影响细胞的生长和代谢,因此在实验设计和数据解读时需要考虑到这一点。
总结回顾,本文详细介绍了丁酸钠促进转染蛋白表达的原理,包括其作用机制、增效作用以及使用注意事项。
通过本文的阅读,相信读者能够更全面、深刻地理解丁酸钠在转染实验中的作用,为今后的实验设计和数据解读提供有益的参考。
在文章中多次提及了“丁酸钠促进转染蛋白表达”的关键词,并以从简到繁、由浅入深的方式探讨了这一主题。
LIPOFECTAMINE2000转染试剂转染步骤

LIPOFECTAMINE2000转染试剂转染步骤转染是一种将外源DNA或RNA导入到细胞内的技术,以研究基因功能、蛋白质表达、细胞信号转导等方面的问题。
LIPOFECTAMINE2000是一种常用的转染试剂,广泛应用于多种细胞系中。
以下是LIPOFECTAMINE2000转染试剂转染步骤的详细介绍。
一、细胞种植与处理准备1.1细胞传代:将细胞进行传代,以保证其在良好的状态下进行实验。
1.2细胞密度调整:将细胞于适宜培养皿中培养至60-80%的密度,以保证细胞的适宜转染。
1.3细胞处理准备:在转染前,将细胞用无酶EDTA或胰酶剥离并重新悬浮在适宜的培养基中,以保持细胞的完整性和适宜的状态。
二、试剂配制2.1DNA或RNA的制备:将外源DNA或RNA在无菌条件下制备,并使用纯化试剂进行纯化和浓缩。
2.2转染试剂配制:将冻干的LIPOFECTAMINE2000转染试剂通过加入合适的无菌水,稀释成适宜浓度的转染试剂。
三、转染操作3.1转染试剂与DNA/RNA的混合:将适量的LIPOFECTAMINE2000转染试剂与DNA或RNA混合在无菌的管中,轻轻混合均匀。
注意,避免过量试剂和核酸的使用,以减少细胞的毒性和副作用。
3.2孵育混合物:将混合物在常温条件下孵育15-30分钟,以促使脂质体与核酸形成稳定的复合体。
四、转染过程4.1转染试剂与细胞的混合:将混合物缓慢滴加到处理好的细胞培养基上,缓慢摇晃培养皿以使混合物均匀分布。
4.2转染时间及培养条件:将细胞放置在转染液中,保持静止状态,同时将培养皿放回培养箱中,在37℃、5%CO2的恒温恒湿条件下进行转染。
转染时间需要根据细胞系和转染试剂的要求进行优化,一般为4-6小时。
4.3转染液的去除:将转染液小心去除,并将细胞用含有适宜抗生素或筛选剂的培养基洗涤一次,以去除残留的转染试剂。
五、细胞处理及分析5.1细胞培养:将细胞放回恒温恒湿培养箱中,用适宜培养基进行细胞的培养。
蛋白核酸杂交转印膜(PVDF膜和尼龙膜)介绍

蛋白、核酸杂交转印膜(PVDF、尼龙膜)技术介绍转印膜:用于蛋白质和核酸的转移和检验过程的微孔膜。
Southern blot:又称Southern 印迹杂交,是研究DNA 图谱的基本技术。
Northern blot:又称Northern 印迹杂交,与Southern blot 方法类似,是检测RNA的基本技术。
Western blot:又称Western 印迹杂交,与Southern blot 方法类似,是蛋白质检测分析的基本技术。
转印膜在蛋白质和核酸的转移和检测过程中应用广泛,不同的转印膜规格和参数不同,选择正确、均一稳定的转印膜对达到最佳的实验结果起到关键性的作用。
常用的转印膜对比:常用的三种转印膜:硝酸纤维素膜(NC 膜)、带正电的尼龙膜(N66膜)、聚偏氟乙烯膜(PVDF膜)。
C ob e t t er三种转印膜结合DNA或RNA的能力:带正电尼龙膜>PVDF膜>硝酸纤维素膜。
三种转印膜机械强度比较:硝酸纤维素膜较脆,易破碎,不能重复使用;尼龙膜和PVDF 膜机械强度较高,可重复使用。
带正电的尼龙膜可结合短至10bp核酸片段,因此是最理想的核酸杂交转印膜。
PVDF膜机械强度高、耐高温,是蛋白印迹最理想的转印膜。
Cobetter专为蛋白核酸杂交研发了性能优良的转印膜,Cobetter杂交转印膜的种类如下:retteboC⏹N66尼龙转印膜⏹N66尼龙带正电荷转印膜⏹PVDF疏水转印膜⏹PVDF亲水转印膜Cobetter转印膜孔径有0.45um、0.22um 两种。
CobetterN66尼龙转印膜:相比起硝酸纤维素膜而言,其机械强度高,在经历多次杂交、洗脱和标记后不易产生破损和变形,可重复使用,非常适用于核酸检测。
制模过程中通过特殊的表面改性方法嫁接了亲水性的基团,使用过程中不需要再做预润湿处理。
通过在膜表面嫁接羟基基团,改变其表面电荷性能(正电),使得其能紧密结合DNA 而降低背景,快速结合DNA。
转染效率的测定方法

转染效率的测定方法
一、实时定量PCR检测
实时荧光定量PCR (Quantitative Real-time PCR)是一种在DNA扩增反应中,以荧光化学物质测每次聚合酶链式反应(PCR)循环后产物总量的方法。
通过内参或者外参法对待测样品中的特定DNA序列进行定量分析的方法。
Real-time PCR是在PCR扩增过程中,通过荧光信号,对PCR进程进行实时检测。
由于在PCR扩增的指数时期,模板的Ct值和该模板的起始拷贝数存在线性关系,所以成为定量的依据。
但是,荧光定量PCR所检测的是转染后细胞中待测基因的mRNA的表达水平,对于目的基因的蛋白表达水平不能够检测;
二、Wester Blot检测
Western Blot又称蛋白质免疫印迹(免疫印迹实验),其基本原理是通过特异性抗体对凝胶电泳处理过的细胞或者生物组织样品进行着色,并通过分析着色的位置和着色的深度获得特定蛋白在所分析的细胞或者组织中表达情况的信息。
Western Blot所检测的是组织或者细胞中蛋白质的表达水平。
三、流式细胞术
流式细胞术(Flow Cytometry,FCM)是一种在功能水平上对细胞或者其他生物粒子进行定量检测和分析的检测手段,它可以高速分析上万个细胞,并能同时从一个细胞中检测得到多个参数,与传统检测方法相比具有更加快速、准确以及定量等特点。
使用流式细胞术检测转染效率可以更加精que的确定转染的效率,对转染效率进行量化。
四、荧光显微镜观察
当我们转染的质粒DNA含有荧光蛋白基因是,可以通过荧光显微镜观察的方法来确定转染的效率,在荧光显微镜下,可以观察到荧光的强弱以及荧光的效率。
70kd蛋白转膜时间

70kd蛋白转膜时间一、什么是70kd蛋白转膜时间转膜时间是指蛋白质从细胞内转移到细胞外的时间。
70kd蛋白是一种分子量为70千道尔顿(kd)的蛋白质,其转膜时间是指这种蛋白质从细胞内转移到细胞外所需的时间。
二、70kd蛋白转膜的重要性70kd蛋白在细胞功能调控中起着重要的作用。
它参与了细胞信号传导、细胞黏附和细胞凋亡等多种生物学过程。
因此,了解70kd蛋白的转膜时间对于研究细胞功能和疾病发生机制具有重要意义。
三、研究70kd蛋白转膜时间的方法研究70kd蛋白转膜时间的方法有多种,下面列举了几种常用的方法:1. 免疫印迹免疫印迹是一种常用的蛋白质检测方法,可以用于检测70kd蛋白在细胞内外的表达水平。
通过比较不同时间点的免疫印迹结果,可以推测出70kd蛋白的转膜时间。
2. 转染法转染法是将外源基因导入细胞的一种常用方法。
研究者可以将70kd蛋白的编码基因转染到细胞中,然后观察蛋白在细胞内外的表达情况,进而推测出70kd蛋白的转膜时间。
3. 转位实验转位实验是一种通过标记蛋白质的方法研究蛋白质转位的技术。
研究者可以将70kd蛋白与荧光标记结合,然后观察标记蛋白在细胞内外的分布情况,进而推测出70kd蛋白的转膜时间。
四、影响70kd蛋白转膜时间的因素70kd蛋白的转膜时间受到多种因素的影响,下面列举了几个常见的因素:1. 细胞类型不同类型的细胞对于70kd蛋白的转膜时间可能存在差异。
不同细胞类型具有不同的细胞膜通透性和分子运输机制,这可能会影响70kd蛋白的转膜时间。
2. 细胞状态细胞的生理状态和外界环境条件也会对70kd蛋白的转膜时间产生影响。
例如,细胞处于应激状态或缺氧状态时,可能会导致70kd蛋白的转膜时间延长。
3. 分子量分子量较大的蛋白质通常转膜时间较长。
70kd蛋白的分子量较大,因此其转膜时间可能相对较长。
五、应用前景和意义研究70kd蛋白的转膜时间对于深入了解细胞功能和疾病发生机制具有重要意义。
荧光素酶实验步骤

荧光素酶实验步骤
荧光素酶实验是一种常用的生物学实验,用于检测蛋白质的表达和定量。
下面是荧光素酶实验的步骤。
1. 转染细胞
首先需要将目标蛋白质的编码基因转染到细胞中。
转染可以通过多种方法实现,如化学转染、电穿孔转染、病毒载体转染等。
转染后需要等待一定时间,让细胞表达目标蛋白质。
2. 加入底物
荧光素酶实验的底物是荧光素,它可以被荧光素酶催化产生荧光。
将荧光素加入培养基中,使其与细胞接触。
3. 加入荧光素酶
荧光素酶可以从多种来源中获得,如贝类、昆虫等。
将荧光素酶加入培养基中,使其与荧光素反应产生荧光。
4. 检测荧光
使用荧光显微镜或荧光分析仪检测细胞中的荧光强度。
荧光强度越高,说明细胞中的目标蛋白质表达量越高。
需要注意的是,荧光素酶实验的结果受到多种因素的影响,如细胞状态、转染效率、荧光素酶活性等。
因此,在进行实验前需要进行充分
的优化和控制,以确保实验结果的准确性和可靠性。
总之,荧光素酶实验是一种常用的生物学实验,可以用于检测蛋白质
的表达和定量。
实验步骤包括转染细胞、加入底物、加入荧光素酶和
检测荧光。
在进行实验前需要进行充分的优化和控制,以确保实验结
果的准确性和可靠性。
转染实验方法(全)

转染实验方法(全)转染实验方案1.lb培养基的配制:500ml(5皿+2锥形瓶)准备工作:500ml玻璃瓶1个,250ml玻璃瓶2个,500ml容量瓶一个,平皿5个称取:酵母金属粉末液5.0g蛋白胨10.0g氯化钠10.0g实验操作方式:混合后加去离子水(或注射用水)水400ml,待充分溶解后加水定容至500ml,取100ml做固体培养基用装入a瓶,余下装入500mlb瓶中(400ml/瓶)称取1.5g琼脂粉加入a瓶中另挑一瓶(c瓶)接100ml水(吸收氨苄西林用)将以上3瓶高压杀菌:121°c,30min挑氨苄西林一支:规格:1g/支加水于安瓶中溶解后,移入c瓶中,浓度:10mg/ml按照50μg/ml氨苄西林的浓度加到abc三瓶中a瓶:0.5ml(即为lb液态培养基)――僵硬型100μg/ml,世俗性型50μg/mlb瓶:2ml(即为lb液体培养基)将a瓶按20ml/平皿,分别移到5个平皿中,待冷却后即为固体培养基。
用膜封口放入4℃保存。
2.感受态转变与培育(top10)准备:37℃摇床,37℃烘箱,10cm培养皿(lb琼脂凝胶+氨苄=50mllb+0.75g琼脂+50μl氨苄),冰盒,42℃水浴,无菌牙签,氨苄溶液40~60μg/ml实验前:水浴调到42℃,soc培养液放入室温,lb提氨苄培养皿于37度烘箱中冷却半小时实验操作:1)将质粒较长时间Vergt后快速放进冰浴中2)冰上鞭叶top103)吸取1到10μl质粒加入top10中,轻轻敲打混匀(切记不可用移液枪混匀)。
剩余的质粒可储存于-20℃中4)冰上孵育top10离心管30min。
5)42℃水浴30s(精确),不要晃动离心管6)快速转至冰浴2-3min,重新加入250μl预演的soc溶液,确保过程无菌7)37℃来靠水平225rpm挥1h8)吸取200μl用三角耙铺板(最好同时做不同加入量的2个皿,la培养皿预热),余下溶液储存于4℃9)正置20min,倒置37℃培养过夜10)第二天抽出平皿,观测菌落,挑选很大菌落,用牙签挑取菌落放进对应的试管中,加入15μl氨苄西林(c瓶母液),最后加3mllb液体培养基,试管放架子上37℃摇4-6h(预培养)11)按照培养基:菌液=200ml:2ml展开转菌,余下菌液放进ep管中-20℃留存(衰退加时100μl菌液预培养)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Journal of Biotechnology 208(2015)44–53Contents lists available at ScienceDirectJournal ofBiotechnologyj o u r n a l h o m e p a g e :w w w.e l s e v i e r.c o m /l o c a t e /j b i o t ecRapid and highly efficient mammalian cell engineering via Cas9protein transfectionXiquan Liang 1,Jason Potter ∗,1,Shantanu Kumar,Yanfei Zou,Rene Quintanilla,Mahalakshmi Sridharan,Jason Carte,Wen Chen,Natasha Roark,Sridhar Ranganathan,Namritha Ravinder,Jonathan D.ChesnutThermo Fisher Scientific,5781Van Allen Way,Carlsbad,CA 92008,USAa r t i c l e i n f o Article history:Received 19February 2015Received in revised form 18April 2015Accepted 27April 2015Available online 21May 2015Keywords:CRISPR Cas9gRNAGene editing Transfection Multiplexa b s t r a c tCRISPR-Cas9systems provide a platform for high efficiency genome editing that are enabling innovative applications of mammalian cell engineering.However,the delivery of Cas9and synthesis of guide RNA (gRNA)remain as steps that can limit overall efficiency and ease of use.Here we describe methods for rapid synthesis of gRNA and for delivery of Cas9protein/gRNA ribonucleoprotein complexes (Cas9RNPs)into a variety of mammalian cells through liposome-mediated transfection or ing these methods,we report nuclease-mediated indel rates of up to 94%in Jurkat T cells and 87%in induced pluripotent stem cells (iPSC)for a single target.When we used this approach for multigene targeting in Jurkat cells we found that two-locus and three-locus indels were achieved in approximately 93%and 65%of the resulting isolated cell lines,respectively.Further,we found that the off-target cleavage rate is reduced using Cas9protein when compared to plasmid DNA transfection.Taken together,we present a streamlined cell engineering workflow that enables gRNA design to analysis of edited cells in as little as four days and results in highly efficient genome modulation in hard-to-transfect cells.The reagent preparation and delivery to cells is amenable to high throughput,multiplexed genome-wide cell engineering.©2015The Authors.Published by Elsevier B.V.This is an open access article under the CC BY-NC-NDlicense (/licenses/by-nc-nd/4.0/).1.IntroductionCRISPR-Cas9mediated genome engineering enables researchers to modify genomic DNA in vivo directly and efficiently (Cho et al.,2013a;Mali et al.,2013;Jiang et al.,2013;Wang et al.,2013).Three components (Cas9,mature crRNA and tracrRNA)are essential for functional activity.Although the mature crRNA and tracrRNA can be synthesized chemically,the quality of the synthetic RNA is not sufficient for in vivo cell engineering due to the presence of trun-cated by-products (data not shown).Therefore,templates for the mature crRNA and tracrRNA or a combined single gRNA are often cloned into a Cas9expression plasmid or built into separate plas-mids driven by either U6or H1promoters for transcription after transfection of mammalian cells (Cong et al.,2013;Mali et al.,2013).However the plasmids have been shown to have toxicity in someAbbreviations:CRISPR,clustered regularly interspaced short palindromic repeats;CAS9,CRISPR associated protein;gRNA,guide RNA;crRNA,CRISPR RNA;tracrRNA,trans-activating crRNA.∗Corresponding author.Tel.:+17604766068;fax:+17602687477.E-mail address:jason.potter@thermofi (J.Potter).1Contributed equally to this work.cell lines (Kim et al.,2014).Recently,the use of Cas9delivered as mRNA has led to increases in the rate of genomic cleavage in some cells.For example,a mixture of Cas9mRNA and a single species of gRNA were co-injected into mouse embryonic stem (ES)cells resulting in biallelic mutations in 95%of newborn mice (Wang et al.,2013).To make guide RNA,often a linearized plasmid containing the T7promoter and the gRNA sequence is used directly or a linear template is created via PCR amplification of the targeting sequence from a plasmid.If a 5′T7promoter does not appear in the plas-mid,it is often added at this step and the resulting PCR product can be used in an in vitro transcription reaction (Jinek et al.,2012;Wang et al.,2013).Alternatively,a synthetic DNA fragment contain-ing a T7promoter,crRNA and tracrRNA can be used as a template to prepare a gRNA by in vitro transcription.Overall,these repre-sent a labor-intensive and time-consuming workflow,which led us to seek a simpler method to synthesize high quality gRNA.To that,we describe here a streamlined modular approach for gRNA production in vitro .Starting with two short single stranded oligos,the gRNA template is assembled in a ‘one pot’PCR reaction.The product is then used as template in an in vitro transcription (IVT)reaction which is followed by a rapid purification step,yielding transfection-ready gRNA in as little as four hours./10.1016/j.jbiotec.2015.04.0240168-1656/©2015The Authors.Published by Elsevier B.V.This is an open access article under the CC BY-NC-ND license (/licenses/by-nc-nd/4.0/).X.Liang et al./Journal of Biotechnology208(2015)44–5345To streamline the cell engineering workflow further,we sought to eliminate any remaining cellular transcription or translation by directly introducing Cas9protein/gRNA ribonucleoprotein(RNP) complexes directly to the cells.Microinjection of Cas9protein and gRNA complexes into C.elegans wasfirst described in2013 (Cho et al.,2013b)and was subsequently used to generate gene-knockout mice and zebrafish with mutation rates of up to93% in newborn mice(Sung et al.,2014).Following that report,Cas9 protein/gRNA RNP complexes were delivered into cultured human fibroblasts and induced pluripotent stem cells(iPSC)via elec-troporation with high efficiency and relatively low off-target effects(Kim et al.,2014).In that study,a large amount of Cas9 protein(4.5–45g)and gRNA(6–60g)were necessary for effi-cient genome modification(up to79%indel efficiency).Another recent study delivered Cas9/gRNA RNPs along with donor DNA for homology directed repair into HEK293T,human primary neona-talfibroblasts and human ESCs cells via electroporation(Lin et al., 2014).Here also,large amounts of Cas9protein(4.8–16g)were necessary for efficient modification.Most recently,delivery of Cas9 protein-associated gRNA complexes via liposomes was reported,in which RNAiMAX was used to deliver Cas9:sgRNA nuclease com-plexes into cultured human cells and into the mouse inner ear in vivo with up to80%and20%genome modification efficiency, respectively(Zuris et al.,2015).The CRISPR/Cas system has been demonstrated as an efficient gene-targeting tool for multiplexed genome editing(Wang et al., 2013;Kabadi et al.,2014;Sakuma et al.,2014;Cong et al.,2013). For example,co-transfections of mouse ES cells with constructs expressing Cas9and three sgRNAs targeting Tet1,2,and3resulted in20%of cells having mutations in all six alleles of the three genes based on restriction fragment length polymorphism(RFLP) assay(Wang et al.,2013).Lentiviral delivery of a single vector expressing Cas9and four sgRNAs into primary human dermal fibroblasts resulted in about30%simultaneous editing of four genomic loci among ten clonal populations based upon genomic cleavage detection assays(Kabadi et al.,2014).In one recent study,‘all-in-one’expression vectors containing seven guide RNA expres-sion cassettes and a Cas9nuclease/nickase expression cassette were delivered into293T cells with genome cleavage efficiency ranging from4to36%for each individual target(Sakuma et al., 2014).In general,the efficiency of editing multiple genes in the human genome using plasmid-based delivery methods remains relatively low which subsequently increases the workload for downstream clonal isolation.In this study,we developed an in vitro gRNA production sys-tem and used a systematic approach to optimize the conditions for delivery of Cas9:gRNA complexes via lipid-mediated transfec-tion or electroporation.A variety of mammalian cell lines were tested,including primary cells and other hard-to-transfect cells. Plasmid DNA,mRNA and Cas9protein transfections were evaluated side by ing Cas9protein transfection via electroporation, we achieved superior genome editing efficiencies even in hard-to-transfect cells.In addition,we explored the genome editing of multiple targets simultaneously using the Cas9RNPs delivery sys-tem described here.We found that delivery of Cas9RNPs not only led to high indel production at single locus,but supports highly efficient biallelic modulation of at least two genes in a single trans-fection.2.Materials and methods2.1.Materials293FT cells,Gibco®Human Episomal iPSC line,mouse E14Tg2a.4embryonic stem cells,primary human keratinocytes cells neonatal,inactivated embryonicfibroblasts,DMEM medium, RPMI1640medium,IMDM,McCoy5A modified medium,DMEM/F-12,KnockOut TM DMEM,Fetal Bovine Serum(FBS),Knockout TM Serum Replacement,Non-Essential Amino Acid solution,basic fibroblast growth factor,Collagenase IV,TrypLE TM Express Enzyme, Geltrex,Opti-MEM Medium,Essential8TM medium,StemPro®-34SFM Complete Medium,FluoroBrite TM DMEM,recombinant human leukemia inhibitory factor,GeneArt®Genomic Cleav-age Detection Kit(GCD),Lipofectamine®2000,Lipofectamine®3000,Lipofectamine®RNAiMAX,Lipofectamine®MessengerMAX, GeneArt®CRISPR Nuclease Vector with OFP Reporter,2%E-Gel®EX Agarose Gels,PureLink®PCR Micro Kit,TranscriptAid T7High Yield Transcription Kit,MEGAclear TM Transcription Clean-Up Kit,Zero Blunt®TOPO®PCR Cloning Kit,PureLink®Pro Quick96Plasmid Purification Kit,Qubit®RNA BR Assay Kit,TRA-1-60Alexa Fluor®488conjugated antibodies,SSEA4Alexa Fluor®647,and Phusion Flash High-Fidelity PCR Master Mix were from Thermo Fisher Scien-tific.Jurkat T cells and K562cells were obtained from the American Type Culture Collection(ATCC).CD34+cord blood cells were pur-chased from AllCells.A549cells,U-2OS cells,Neuro-2a(N2A)cells were purchased from ATCC.MEF feeder cells and ROCK inhibitor Y-27632were purchased from EMD Millipore.Monoclonal Cas9 antibody was ordered from Diagenode.Recombinant Cas9protein with a NLS was initially purified as described(Kim et al.,2014)and later obtained from Thermo Fisher Scientific.All oligonucleotides used for gRNA synthesis and genomic cleavage detection were from Thermo Fisher Scientific(Table S1).2.2.One-step synthesis of gRNA templateThe80bp cr/tracrRNA constant region was PCR amplified from the GeneArt®CRISPR Nuclease Vector(1ng)using the Constant Forward and Universal Reverse oligos(10M)and purified via agarose gel extraction.The concentration of PCR product was mea-sured by Nanodrop(Thermo Fisher Scientific)and the molarity was calculated based on the molecular weight of49.6kDa.To prepare a mixture of oligonucleotides,the80bp cr/tracrRNA PCR product(0.15M)was mixed with universal forward and reverse oligos(10M)as well as target-specific forward and reverse oligos (0.3M).For each target locus,2oligonucleotides that recreate the target sequence and share complementary with the bordering T7pro-moter and80bp cr/tracr constant region were designed(Fig.2A). The forward oligo(Target F1)contains the18base T7promoter sequence as well as thefirst16bases of the target and the reverse oligo(Target R1)contains the reverse complement of thefirst 15bases of the cr/tracr constant region and the last19bases of the target(Table1s).To set up the synthesis of gRNA template, aliquots of the pooled oligonucleotides were added to a Phusion Flash High-Fidelity PCR Master Mix and amplified using manufac-turer’s recommended reaction conditions.The PCR product was analyzed by a2%E-Gel®EX Agarose Gel,followed by purifica-tion using Purelink®PCR micro column.The gRNA template was eluted with13l water and the concentration was determined by Nanodrop instrument.To determine the error rate,the PCR product was cloned into Zero Blunt®TOPO®vector,followed by plasmid DNA isolation and sequencing with a3500xl DNA analyzer(Thermo Fisher Scientific).2.3.In vitro transcriptionThe in vitro transcription of gRNA template was carried out using TranscriptAid T7High Yield Transcription Kit using the manufac-turer’s recommended conditions.The gRNA product was purified using MEGAclear TM Transcription Clean-Up kit as described in the46X.Liang et al./Journal of Biotechnology208(2015)44–53Table1Comparison of plasmid DNA,Cas9mRNA/gRNA and Cas9RNP transfection and resulting editing efficiencies as measured by GCD assay in a variety of cell lines. Cell lines Plasmid mRNA ProteinLipid Electro Lipid Electro Lipid Electro HEK293FT494970405188U2OS155021241870 Mouse ESCs304545202570 Human ESCs(H9)082050064 Human iPSCs020********N2A667566806682 Jurkat063042094*K562045027072A549154423292066 Human Keratinocytes(NHEK)030050035 Human Cord BloodCells CD34+n/a0n/a0n/a24Note:(1)gRNA targets are HPRT for human cell lines and Rosa26for mouse cell lines.(2)*Confirmed by sequencing.manual.The concentration of RNA was determined using Qubit®RNA BR Assay Kit.2.4.Mammalian cell cultureHEK293FT,A549and N2A cells were maintained in DMEM medium supplemented with10%FBS.U2OS cells were main-tained in McCoy5A modified medium supplemented with25mM HEPES and10%FBS.Jurkat T cells were propagated in RPMI medium containing10%FBS.K562cells were cultured in IMDM medium supplemented with10%FBS.Human ESCs(H9)were cul-tured in Essential8TM medium on tissue culture dishes coated with Geltrex®hESC-qualified reduced growth factor basement membrane matrix.After thawing,cells were passaged2–3times before using for transfection.Feeder-dependent human episo-mal iPSC were cultured on mitotically inactivated MEF feeder cells in human ESC(hESC)media containing20%Knockout TM Serum Replacement,10M Non-Essential Amino Acid solution, 55M2-Mercaptoethanol,and4ng/ml basicfibroblast growth factor in DMEM/F-12.iPSC cultures were maintained with daily media changes and were passaged regularly using Collagenase IV.Mouse E14Tg2a.4embryonic stem cells were cultured on mouse(strain ICR)inactivated embryonicfibroblasts in the pres-ence of recombinant human leukemia inhibitory factor(LIF) in mouse ESC medium consisting of KnockOut TM DMEM15% embryonic stem cell–qualified Fetal Bovine Serum,1×MEM Non-Essential Amino Acids Solution,1×GlutaMAX TM Supplement, 1×2-mercaptoethanol,and10ng/mL LIF.Before transfection, cells were adapted to feeder-free conditions and maintained on attachment-factor-coated plates in mouse ESC-conditioned medium.When setting up the experiments for transfections, 1×105cells were plated per well in a24-well tissue culture dish coated with attachment factor.CD34+cord blood cells were cul-tured using StemPro®-34SFM Complete Medium supplemented with100ng/mL of SCF,50ng/mL of IL-3and25ng/mL of GM-CSF. Primary human keratinocytes cells neonatal(HEKn)were grown in a tissue culture dish treated with Coating Matrix Kit in EpiLife medium containing60M of calcium chloride.Cells were grown for three passages before using for experiments.All cultures were maintained in5%CO2at37◦C in a humidified incubator.Prior to transfection,adherent cells were detached with Gibco TrypLE Select Enzyme and then resuspended in the appropriate growth media.2.5.Lipid-mediated cell transfectionOne day prior to transfection,the cells were seeded in a24-well plate at a cell density of1–2×105cells per well.For plasmid DNA transfection,0.5g DNA was added to25l of Opti-MEM medium,followed by addition of25l of Opti-MEM containing2l of Lipofectamine2000.The mixture was incubated at room temperature for15min and then added to the cells.For Cas9mRNA transfection,0.5g Cas9mRNA(Thermo Fisher Scientific)was added to25l of Opti-MEM,followed by addition of50–100ng gRNA.Meanwhile,2l of either Lipofectamine3000, MessengerMax or RNAiMAX was diluted into25l of Opti-MEM and then mixed with mRNA/gRNA sample.The mixture was incu-bated for15min prior to addition to the cells.For Cas9protein transfection,500ng of purified Cas9protein was added to25l of Opti-MEM medium,followed by addition of 120ng gRNA.The molar ratio of gRNA to Cas9protein was kept at approximately1to1.2:1.The sample was mixed by gently tapping the tubes a few times and then incubated at room temperature for 10min.To a separate test tube,2l of Lipofectamine RNAiMAX, MessengerMax,or3000was added to25l of Opti-MEM medium. The diluted transfection reagent was transferred to the tube con-taining Cas9protein/gRNA complexes,followed by incubation at room temperature for15min and then added to the cells.In each case,the entire solution was added to the cells in a24-well plate and mixed by gently swirling the plate.The plate was incubated at37◦C for48h in a5%CO2incubator.The percentage of locus-specific indel formation was measured by GeneArt®Genomic Cleavage Detection Kit(GCD).The band inten-sities were quantitated using the Alpha Imager software(Bio-Rad). Each cell line was tested with each version of Lipofectamine(3000, RNAiMAX,and MEssengerMAX).The lipid that resulted in highest cleavage efficiency is listed in Table S2.2.6.ElectroporationFor suspension cells,such as Jurkat T cells,K562cells or CD34+ human cord blood cells,1–2×105cells were used per electropo-ration using Neon®Transfection System10L Kit(Thermo Fisher Scientific).For adherent A549,U2OS and N2A cells,5×104cells were used per electroporation.For adherent HEK293FT,mESC, hESC,human iPSC,A549,and NHEK cells,1×105were used per electroporation.To maximize the genome cleavage efficiency,the Neon24opti-mization protocol was applied according to the manufacturer’s instruction.To set up a master mix,24g of purified Cas9pro-tein was added to240l of Resuspension Buffer R provided in the kit,followed by addition of4.8g of gRNA.The mixture was incu-bated at room temperature for10min.Meanwhile,4.8×106cells were transferred to a sterile test tube and centrifuged at500×g for 5min.The supernatant was aspirated and the cell pellet was resus-pended in1ml of PBS without Ca2+and Mg2+.Upon centrifugation, the supernatant was carefully aspirated so that almost all the PBS buffer was removed with no or minimum loss of cells.The Resus-pension Buffer R containing the Cas9protein/gRNA complexes was then used to resuspend the cell pellets.A10l cell suspension was used for each of the24optimization conditions,which varied in pulse voltage,pulse width and the number of pulses.The electro-porated cells were transferred immediately to a24well containing 0.5ml of the corresponding growth medium for each cell line and then incubated for48h in a5%CO2incubator.The cells were har-vested by centrifugation and then washed once with PBS,followed by GCD assay.Upon optimization of electroporation condition,a higher amount of Cas9protein(1.5–2g)and gRNA(300–400ng) could be applied to further increase the genome editing efficiency if needed.X.Liang et al./Journal of Biotechnology208(2015)44–5347Each cell line was tested for with the Neon24optimization protocol.Specific electroporation conditions that resulted in the highest cleavage efficiency are listed in Table S2.For each target in the multiplexing assays,1–2g of Cas9 protein and200–400ng of gRNA(maintaining a1:1ratio)were pre-incubated separately in Resuspension Buffer R for10min at room temperature prior to mixing with the cell pellet for electroporation.For clonal isolation,the cell number of transfected cells was counted upon48h incubation,followed by a serial of dilution to 96well plates with a cell density of10–20cells per plate based on the cell count.After clonal expansion for three weeks,cells from each individual well were harvested,followed by PCR ampli-fication of the target locus.The PCR fragments were then cloned using a TOPO vector and transformed into TOP10competent cells. Approximately8E.coli colonies were randomly picked for sequenc-ing for each individual target locus.The single cell population was determined by the homogeneity of sequences for each allele.Sin-gle cells containing bi-allelic mutations on all desired targets were considered homozygotic indels.Downstream sequence analysis to confirm frame-shift induced stop codon introduction was not done.For transfection of feeder free adaptation of iPSC,feeder depend-ent iPSC were grown to80%confluence prior to harvest with collagenase.Following removal of the cell clusters from the feeder layer,they were gravity sedimented to prevent MEF contamina-tion.The cell clusters were then seeded on to tissue culture dishes coated with Geltrex®in MEF conditioned media supplemented with4ng/ml bFGF.MEF conditioned media was produced using inactivated feeder cells,which was harvested on7continuous days, sterilefiltered and frozen until usage.The cultures were allowed to reach80–90%confluence.The day prior to transfection,the cultures were pretreated with5M ROCK inhibitor Y-27632.On the day of harvest the cultures were inspected for signs of differentiation and any contamination differentiated cells were removed via micro-dissection.The cultures were washed once with DPBS and then harvested using TrypLE TM Express Enzyme.Single cells suspensions were counted using the Countess®automated cell counter.Follow-ing transfections,the cells were seeded onto multi-well(24well) tissue culture dish coated with Geltrex®and incubated overnight with MEF conditioned media containing5M ROCK.Media was replaced daily,without ROCK inhibitor,prior to analysis.2.7.Cell surface immunostainingTo ensure maintenance of pluripotency post transfection and genome editing,iPSC cells were tested for expression of cell sur-face markers of self-renewal.The wells to be probed were washed with DMEM/F12basal media.TRA-1-60Alexa Fluor®488conju-gated antibodies and SSEA4Alexa Fluor®647were multiplexed in basal DMEM/F-12media.Both antibodies were added at a con-centration of2l of each antibody into0.5ml of pre-warmed DMEM/F-12media and incubated at37◦C for45min.Following the incubation,the antibody solution was removed and the wells were washed twice with DMEM/F-12.Prior to observation the media was exchanged with pre-warmed FluoroBrite TM DMEM.Images were taken using a Zeiss Axiovision microscope using a FITC and Cy5 laser/filter combination.2.8.Analysis of pluripotency markersCultures were detached and dissociated using TrypLE TM Select and trituration.Single cell suspensions were incubated with TRA-1-60Alexa Fluor®488conjugated antibodies and SSEA4Alexa Fluor®647for1h at room temperature with gentle agitation. Two microliters(50×concentration,as supplied)of each antibody were added to0.5mL of DMEM/F-12.Following the incubation, the cells were centrifuged and washed once with Dulbecco’s Phosphate-Buffered Saline(DPBS).After the removal of the DPBS wash,the pelleted cells were gently re-suspended in1ml of DPBS and stained through a strainer capped tube.The cells were then measured for the expression of both markers using the Attune®Acoustic Focusing Cytometer and the data was analyzed using FlowJo software.2.9.Western Blot analysisHEK293FT cells were transfected with either Cas9plasmid DNA, mRNA or protein as described above.Mouse ESCs were electropo-rated with Cas9RNPs as described above.Cells were harvested at indicated times to perform both GCD assay and Western Blot analy-sis.The cell lysate was fractionated using a4–12%Novex Bis-tris gel. The proteins were transferred to a PVDF membrane using an iBlot following the manufacturer’s protocol.Upon blocking,the mem-brane was incubated for2h with monoclonal mouse Cas9antibody at1:3000dilution.After washing,the membrane was incubated for 1hour with rabbit anti-mouse antibody-HRP conjugate at1:2000 dilution.Upon extensive washing,the membrane was developed with Pierce ECL reagent,followed by imaging using a Fuji imager LAS4000instrument.3.Results3.1.Three day cell engineering workflowTo streamline the genome engineering workflow,we sought to simplify the gRNA synthesis procedure and shorten the time from experimental design to initial analysis as much as possible.We present a process where on day1,the researcher designs and orders short DNA oligonucleotides and seeds the cells of interest for next day transfection(Fig.1).Upon receiving the oligonucleotides on day 2,the researcher assembles the gRNA template in less than1h by ‘one pot’PCR.The resulting PCR product is then subjected to in vitro transcription to synthesize gRNA in approximately3h.Upon asso-ciation of gRNA with purified Cas9protein,the Cas9RNPs are used to transfect cells via lipid-mediated delivery or electroporation.As early as day3(24h post transfection),the cells can be harvested for analysis of locus-specific genome modification efficiency.We used an online web tool to identify candidate20nt gRNA target sequences for each loci(Beta Testing Version,Thermo Fisher Scientific).A pair of34nt forward and reverse oligonucleotides comprising each target sequence were designed as described.To assemble the DNA template for gRNA production,we combined the 2target specific oligos(Target F1/R1)with a mixture of the2univer-sal oligos(Universal Forward/Reverse)and a purified PCR product representing the constant non-targeting region of the full gRNA (Fig.2A).The oligonucleotide pool concentrations as well as the PCR conditions were optimized such that the template was ampli-fied in less than40min in a single tube with(Fig.2B).The gRNA template was used directly to prepare gRNA via in vitro transcrip-tion(IVT).The resulting gRNA was purified,yielding high levels of gRNA with low amounts of detectable by-products(Fig.2C).This approach was validated by synthesis of more than96distinct gRNAs (unpublished results).To determine the error rate in the synthetic gRNA DNA template,we cloned and sequenced the PCR fragments and found that using this design approximately7%of gRNA tem-plates harbored mutations,mainly small deletions occurring at the extreme3′end and5′ends of the mature template compared to 21%mutants with an alternative design of using long overlapping oligonucleotides(Fig.2D).Since all the mutations observed using our design appeared to be due to errors in the oligonucleotides, we next tried HPLC-purified universal forward and reverse primers which further decreased the error rate to3.6%with no mutations48X.Liang et al./Journal of Biotechnology 208(2015)44–53Fig.1.Cell engineering workflow .On day 1,the researcher designs CRISPR targets and seeds cells.Synthesis of gRNA and cell transfection with Cas9protein/gRNA complex (Cas9RNP)are performed on day 2.Genome cleavage assays carried out on days 3–4.detected in the target region,which was similar to the 2%error rate observed with the control template prepared from an ‘all-in-one’plasmid.Taken together,this optimized process facilitates the con-version of a small set of DNA oligonucleotides into purified gRNA in approximately 4h with an accuracy of up to 96%and no errors detected in the targeting or Cas9complexing (cr/tracrRNA)regions.This level of accuracy should be sufficient for routine screening of gRNAs,but if a sequence verified gRNA is required the gRNA template can easily be Topo cloned.Given that the process con-sists solely of liquid handling PCR,transcription,and RNA isolation steps,it is well suited for high throughput gRNA production and screening.3.2.Liposome-mediated Cas9protein transfectionTo examine the activity of synthetic gRNA,we pre-complexed purified synthetic IVT gRNA with Cas9protein,hypothesizing that creating complexes of purified gRNAs with Cas9protein prior to delivery to the cells might lead to higher genome editing efficiency due to the protection of the gRNA as it transits to the nucleus during the transfection process.To examine in vivo functionality of the sys-tem,human embryonic kidney (HEK293FT)cells were transfected with pre-complexed Cas9/gRNA ribonucleoproteins (Cas9RNPs)using a set of cationic lipid reagents,followed by a GCD assay.The commonly-used plasmid DNA or RNA lipofectamine transfection reagent had been shown to be able to deliver many proteins into cells,so we tested several lipofectamine variants for their ability to efficiently deliver Cas9RNPs (Sells et al.,1995).Lipofectamine 3000and RNAiMAX outperformed Lipofectamine 2000in HEK 293cells (Figure S1),which is in agreement with the recent finding that RNAiMAX performed better than Lipofectamine 2000for delivery of Cas9RNPs with low cell toxicity (Zuris et al.,2014).We tested the molar ratio of cas9protein to gRNA and generally observed that cleavage activity plateaued at a 1:1ratio (data not shown).For protein transfection,serum-free medium is generally used to avoidserum protein inference.In this study however,we observed that the complete medium containing 10%FBS could facilitate increased protein transfection and genome modification (Fig.3A,panel a).The efficiencies of genome editing via plasmid DNA,mRNA and Cas9RNP transfection were evaluated using three different target loci,HPRT,AAVS and RelA.Plasmid DNA and mRNA were deliv-ered into HEK293cells by Lipofectamine 3000,whereas Cas9RNPs were delivered with RNAiMAX.The efficiencies of genome modi-fication were similar among three target loci in DNA,mRNA and Cas9protein-transfected cells (Fig.3A).Next we examined the kinetics of genome cleavage by transfect-ing cells with either plasmid DNA,mRNA,or Cas9RNPs,followed by GCD assays and western blot analysis of cell lysates.We observed similar cleavage kinetics between Cas9delivered as plasmid DNA,mRNA and protein with efficient cleavage seen at 24h and plateau-ing at 48–72h post-transfection in HEK293cells (Fig.3B).However,the kinetics of Cas9RNP and mRNA encoded Cas9appearance and turnover inside the transfected cells was quite different from that seen with Cas9delivered via plasmid DNA.Measuring by western blot (Fig.3C),we found that Cas9protein accumulated over time in plasmid DNA-transfected cells,whereas the relatively low expres-sion of Cas9in mRNA-transfected cells seemed to peak as early as four hours post transfection and remained relatively stable for approximately 48h before diminishing.In the Cas9RNP transfected cells,the level of Cas9protein peaked in at the first time point,then rapidly decreased and was barely detectable in our assay at 48h.As a control,the blot membrane was stripped and re-probed with anti-actin antibody.Similar levels of actin expression were observed among samples (data not shown).Because of the observed faster protein depletion,we hypoth-esized that the off-target cleavage activity for Cas9RNP and mRNA/gRNA transfection would be lower than that of plasmid DNA transfection (Fu et al.,2013;Pattanayak et al.,2013;Hsu et al.,2013).To test this,we targeted a locus (target site 3)in the VEGFA gene which has been identified as having several high activity。