电子能谱定量分析和深度分析
XPS定量分析例子

1、一级原理模型(First Principle Model)
l
l
从光电子发射的“三步模型”出发,将所观测到的谱线强 度和激发源,待测样品的性质以及谱仪的检测条件等统 一起来考虑,形成一定的物理模型。 由于模型涉及较多的因素,目前还缺乏必要精度的实验 数据,因此一级原理模型计算还未得到真正应用。
I ij = K ⋅ T ( E ) ⋅ Lij (γ ) ⋅ σ ij ⋅ ∫ ni ( z ) ⋅ e
谱峰 C 1s C 1s C 1s C 1s O 1s O 1s O 1s EB (eV) 285.0 286.6 289.0 291.6 532.1 533.7 538.7 面积 2000 700 700 100 1600 1685 85
*
*
〖一个定量分析例子〗
l
解:
nC SC 1 s = = nO I O SO1s
C1s Sat C1s =OOH C1s -OH
峰面积 归一化峰面积
原子浓度
灵敏度因子
传输函数
原子百分数的精确度如何?
定量计算结果中存在几个不确定性的来源,它们来自于: l 峰强度的测定? 如何测量峰面积, 从什么位置到什么位置, 包括什么, 什么 形状背底等 l 灵敏度因子数据库的精确度? 不同的数据库给出不同的结果 – 哪个更好? l 传输函数的精确度? 对特定仪器传输函数定义的准确程度如何 l 其它因素包括λ的测定?
厚度测量 - ARXPS
AR-XPS – 检测相对于表面不同角度的电子来自于样品中不 同深度。
电子来自于A和B 电子仅来自于A Apparent depth 表观分析深度 of analysis
角分辨XPS (ARXPS) 用于小于XPS 分析深 度的分析 可用数学方法计算出 各层的成分、厚度和 分布。
现代材料分析方法——四大分析方法的应用论文

四大分析方法及应用摘要:本文论述材料的X射线粉末衍射分析(XRD)、电子显微分析、能谱分析(XPS,UPS,AES)和热分析(TG,DTA, DSC)等测试原理、制样技术、影响因素、图谱解析以及它们在材料研究中的具体应用。
以一些常见的化合物为基质的各类复合或是掺杂的材料为例,来重点介绍XRD、电镜、热分析等在研究材料物相组成、结构特征、形貌等方面的应用。
关键词:TiO2,XRD,SEM,XPS,TG,DTA前言由于铝等一些金属和无机物的优良的性质,如铝的密度很小,仅为2.7 g/cm3,虽然它比较软,但可制成各种铝合金,如硬铝、超硬铝、防锈铝、铸铝等。
.铝的导电性仅次于银、铜,虽然它的导电率只有铜的2/3,但密度只有铜的1/3,所以输送同量的电,铝线的质量只有铜线的一半铝是热的良导体,它的导热能力比铁大3倍,工业上可用铝制造各种热交换器、散热材料和炊具等。
铝有较好的延展性(它的延展性仅次于金和银),在100 ℃~150 ℃时可制成薄于0.01 mm 的铝箔。
铝的表面因有致密的氧化物保护膜,不易受到腐蚀,常被用来制造化学反应器、医疗器械、冷冻装置、石油精炼装置、石油和天然气管道等。
铝热剂常用来熔炼难熔金属和焊接钢轨等。
铝还用做炼钢过程中的脱氧剂。
铝粉和石墨、二氧化钛(或其他高熔点金属的氧化物)按一定比率均匀混合后,涂在金属上,经高温煅烧而制成耐高温的金属陶瓷,它在火箭及导弹技术上有重要应用。
所以工业上应用非常广泛。
1 X射线衍射分析(XRD)1.1 X射线衍射仪仪器核心部件:光源---高压发生器与X 光管、精度测角仪、光学系统、探测器、控测,数据采集与数据处理软件、X射线衍射应用软件。
定性相分析(物相鉴定):目的:分析试样属何物质,那种晶体结构,并确定其化学式。
原理:任何结晶物质均具有特定结晶结构(结晶类型,晶胞大小及质点种类,数目分布)和组成元素。
一种物质有自已独特衍射谱与之对应,多相物质的衍射谱为各个物相行对谱的叠加。
能谱分析是测什么的

能谱分析是测什么的做SEM或者TEM的时候,一般都会进行EDS打点或者区域扫描,这时软件会给出一个峰谱图,选择想要参与计算的元素及相应的线系后,就能得到各个元素的原子百分比。
今天给大详细介绍一下EDS能谱仪。
能谱分析是测什么的 1EDS能谱仪,又名显微电子探针,是一种分析物质元素的仪器,常与扫描电镜或者透射电镜联用,在真空室下用电子束轰击样品表面,激发物质发射出特征x射线,根据特征x射线的波长,定性与半定量分析元素周期表中Be以上的物质元素,检测流程包括电镜样品制备,上机操作分析,后提供成份分析谱图与半定量成份组成比等数据。
能谱分析是测什么的 21、EDS测试与扫描电镜或者透射电镜联用,选定微小位置区域,探测元素成份与含量;2、EDS测试是失效分析当中对于微小痕量金属物质检测的重要的检测手段;3、EDS测试是区分有机物与无机物的简便的手段,对于有机物只要发现检出大量碳和氧元素,基本可以断定含有大量有机物。
能谱分析是测什么的 31.如果不需要将样本切片,直接观察对话,用电子显微镜准备样本一般需要半个小时。
在电脑上观察前必须保持机器处于真空状态,半小时内即可获得图像数据。
一般你喜欢怎么看就怎么看。
2.需要液氮冷却探头。
如果不加液氮,要等一个小时才能冷却。
能谱分析是测什么的 4对于非金属样品,为了提高放大倍率,需要镀金,样品原貌会有一定改变;对于金属样品,不用镀金就可以进行元素分析;EDS的结构1、探测头:把x射线光子信号转换成电脉冲信号,脉冲高度与x射线光子的能量成正比。
2、放大器:放大电脉冲信号。
3、多道脉冲高度分析器:把脉冲按高度不同编入不同频道,也就是说,把不同的特征X射线按能量不同进行区分。
4、信号处理和显示系统:鉴别谱、定性、定量计算;记录分析结果。
EDS的分析技术1、定性分析:EDS的谱图中谱峰代表样品中存在的元素。
定性分析是分析未知样品的第一步,即鉴别所含的元素。
如果不能正确地鉴别元素的种类,最后定量分析的精度就毫无意义。
X射线光电子能谱(XPS)谱图分析

一、X光电子能谱分析的基本原理X光电子能谱分析的基本原理:一定能量的X光照射到样品表面,和待测物质发生作用,可以使待测物质原子中的电子脱离原子成为自由电子。
该过程可用下式表示:hn=Ek+Eb+Er (1)其中:hn:X光子的能量;Ek:光电子的能量;Eb:电子的结合能;Er:原子的反冲能量。
其中Er很小,可以忽略。
对于固体样品,计算结合能的参考点不是选真空中的静止电子,而是选用费米能级,由内层电子跃迁到费米能级消耗的能量为结合能Eb,由费米能级进入真空成为自由电子所需的能量为功函数Φ,剩余的能量成为自由电子的动能Ek,式(1)又可表示为:hn=Ek+Eb+Φ(2) Eb=hn-Ek-Φ(3)仪器材料的功函数Φ是一个定值,约为 4 eV,入射X光子能量已知,这样,如果测出电子的动能Ek,便可得到固体样品电子的结合能。
各种原子,分子的轨道电子结合能是一定的。
因此,通过对样品产生的光子能量的测定,就可以了解样品中元素的组成。
元素所处的化学环境不同,其结合能会有微小的差别,这种由化学环境不同引起的结合能的微小差别叫化学位移,由化学位移的大小可以确定元素所处的状态。
例如某元素失去电子成为离子后,其结合能会增加,如果得到电子成为负离子,则结合能会降低。
因此,利用化学位移值可以分析元素的化合价和存在形式。
二、电子能谱法的特点(1)可以分析除H和He以外的所有元素;可以直接测定来自样品单个能级光电发射电子的能量分布,且直接得到电子能级结构的信息。
(2)从能量范围看,如果把红外光谱提供的信息称之为“分子指纹”,那么电子能谱提供的信息可称作“原子指纹”。
它提供有关化学键方面的信息,即直接测量价层电子及内层电子轨道能级。
而相邻元素的同种能级的谱线相隔较远,相互干扰少,元素定性的标识性强。
(3)是一种无损分析。
(4)是一种高灵敏超微量表面分析技术,分析所需试样约10-8g即可,绝对灵敏度高达10-18g,样品分析深度约2nm。
能谱仪面扫描定量分析

能谱仪面扫描定量分析什么是能谱仪?能谱仪是一种可以根据物质的放射性衰变和能量分布特性,通过测量样品射出的电子或光子能谱来对样品进行分析和表征的仪器。
它可以测量样品中放射性核素的种类、含量以及其能量分布情况。
能谱仪的结构和原理能谱仪由探测器、放大器、多道分析器等组成。
其原理是将样品置于放射性源中,放射性核素经过衰变放出α、β 射线和γ 射线。
这些射线经过样品后,与探测器相互作用,通过探测器转换成电信号,并经过放大器进行电信号放大,然后由多道分析器进行多道计数,最后形成一个完整的能谱图。
面扫描定量分析面扫描定量分析技术是通过能谱仪对样品表面进行一定深度范围内的扫描测量,然后计算出样品中放射性核素的数量浓度。
其原理是将样品表面与探测器保持一定距离,通过扫描的方式测量样品表面上的放射线计数率,然后根据放射性核素的半衰期和相对照射强度进行定量测量分析。
面扫描定量分析有以下三种方法:1. 面积扫描面积扫描是通过能谱仪对样品表面上一定面积区域内的放射线进行测量,然后计算出该区域内放射性核素的数量浓度。
这种方法适用于比较均匀的样品。
2. 垂直式线扫描垂直式线扫描是在样品表面上横向扫描一定长度的线,并测量线上的放射线计数率。
然后根据扫描线的长度和扫描速度来计算出单位长度内的放射性核素数量浓度。
这种方法适用于比较分散的样品。
3. 水平式线扫描水平式线扫描是在样品表面上纵向扫描一定长度的线,并测量线上的放射线计数率。
然后根据扫描线的长度和扫描速度来计算出单位长度内的放射性核素数量浓度。
这种方法同样适用于比较分散的样品。
面扫描定量分析的优点和应用面扫描定量分析技术具有下列优点:1.操作简便,不需要对样品进行特殊处理。
2.可同时对样品中多种放射性核素进行定量分析,可广泛应用于核辐射环境监管、环境污染控制、地质勘探等领域。
3.可快速获得样品表面的放射分布情况,并能够进行三维重建和定量分析。
面扫描定量分析技术是一种非常重要的分析手段,在核科学、环境安全、医学诊断等领域有着广泛的应用前景。
俄歇电子能谱

通过俄歇电子能谱的深度剖析,可以研究离子注入元素 沿深度方向的分布,还可以研究注入元素的化学状态。
注入Sb元素后,Sn元素 MNN俄歇动能发生变化, 介于Sn和SnO2之间。说 明Sn外层获得部分电子。
由于俄歇电子能 谱具有很高的表 面灵敏度,采样 深度为1-3nm, 因此非常适用于 研究固体表面的 化学吸附和化学 反应。
二、基础知识
1 . 俄歇效应 (1925年, 法国人 Pierre Auger) 用某种方法使原子内层电子(如K层)电离出去,内
层出现空位。电离原子去激发可采用如下两种形式:
Δ 辐射跃迁:
一外层电子填充空位后,发射出特征X射线
(例L3上电子填充K能级上空位,发出X射线Kα 1)
Δ 无辐射过程(即Auger过程): 一外层电子填充空位,使 另一个电子脱离原子发
俄歇电子能量与激发源的种类和数量无关,与元素的存在量有关,还与原子的电 离截面、俄歇电子产率以及逃逸深度有关。
特点: Δ一种原子可能产生几组不同
能级组合的俄歇跃迁,因而 可以有若干不同特征能量的 俄歇电子。 Δ可能出现的俄歇跃迁数随原 子序数增大(壳层数增多)而 迅速增加。 Δ 俄歇电子的能量大多在502000eV (不随入射电子能量改变) Δ主峰
在低氧分压的情况下,只有部分Zn被 氧化为ZnO,而其他的Zn只与氧形成 吸附状态。
俄歇电子能谱在研究固体化学反应上也有着重要的作用。
金刚石耐磨颗粒通 常在表面进行预金 属化,以提高与基 底金属的结合强度。 图中看出界面层有 两层。结合其他方 法分析得出,分别 为CrC和Cr3C4。
• 4 表面元素的化学价态分析
射 出去 (例L1上电子填充K能级空位,同时L3上的电 子发射出去, 称KL1L3俄歇跃迁)。 标记: WXY来标记
电子能谱分析edx
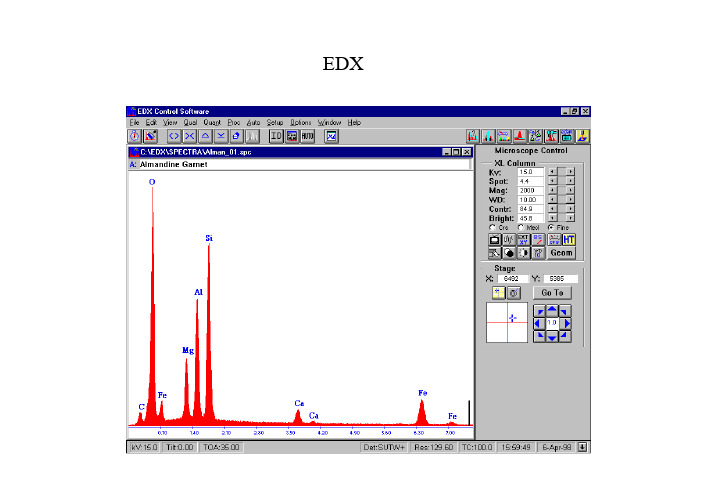
EDXX射线能谱一介绍EDS(energy dispersive spectro-scopy)能量色散谱EDX (Energy Dispersive X-ray)WDS(wavelength dispersive spectro-scopy)波长色散谱EDX:X射线强度和能量曲线,定量分析样品的化学成份主要用途:•1) 非均匀样品的局部化学成份•2) 较少量材料或小颗粒材料的化学成份•3) 非均匀样品种一维或二维的成份分布•4) 沉积在任意衬底上的薄膜成份特点1)铍以上元素2)最小能探测到的重量比:0.1 wt% ——1 wt%3)定量结果的相对误差:2-20%(取决于校正方法等)4)在计算机控制下,1分钟以内可分析16种元素5)空间分辨率取决于平均原子序数、样品密度、束能量等(SEM中0.2——10微米)获得可靠的分析结果要求样品:·1) 样品平整光滑(尤其对定量分析,样品要抛光)2) 可以分析表面粗糙的样品,但仅限于定性和半定量分析3) 样品必须导热导电,必要的时候表面需要喷炭或金推荐书目:Scanning Electron Microscopy and X-ray microanalysisNew York 1992 (生物学、材料科学、地质学)Scanning Electron Microscopy,X-ray microanalysisand Anlytical Electron MicroscopyNew York 1990二定量分析Fig.2: Schematic diagram showingwhere 29<Z<37.Detected Energy (E)Fig.5: Schematic diagram of the intensity variation of the continuum backgroundwith energy, showing the generated and detected background energy.Intensity (I)Generated A )背底和特征峰(二)影响X 射线强度的几种因素B)原子序数对X射线强度的影响Variation in fluorescence yield with atomic number.C )荧光产生率E )Mass absorption coefficient of Fe, for X-rays of varying energyD)X 射线的吸收探测角度:角度越小,X射线吸收越强。
三、EDS定量分析方法与分析误差

P/B-ZAF无标样定量分析方法
P/B-ZAF无标样定量,是不需要标样和标准 物质,也不需要标样X射线强度数据库的定量分 析方法,不需要测量系统校正因子(system factor calibration),是根据改进的ZAF基体校正 方法、利用基本参数(FP)进行校正的定量分 析方法。定量结果可为非归一化结果和归一化 结果,根据“能谱法定量分析”国家标准规定 ,正规定量分析报告建议用归一化定量结果。
I = I0 exp(-µ/t)
特别是由于标样和试样所组成
EDS
E0
的元素种类和含量不同,对X射
线的吸收程度也不同,必须加以 试样
强度I
校正。在定量分析中这是一项
主要校正。
强度I0
11
3、荧光(F)校正
X射线荧光效应 X-ray fluorescence effect
二次荧光 secondary fluorescence
9
1、原子序数(Z)校正
原子序数(Z)校正:对分析体积中所有元素的
电子背散射及阻止本领引起某一元素X射线强
度变化进行的基体校正。
特别是试样和标样的平均原子序数
X-射线 入射电子束 背散射电子
不同时,卢瑟福散射产生的背散射
电子数不同、电子被减速的程度不
样品
同,所以入射电子进入试样和标样
中激发X射线的电子数不同,应校正。
i
Nch d i t q c ZAFch
i
4 0
ii ii
i
br Bremsstrahlung ch characteristic radiation
论计算结果导出。 1951年提出,可分析金属、氧化物等,对平的抛光
试样从Na-U元素的相对误差为~2% 。
X射线光电子能谱(XPS)

另外,原子中的电子既有轨道运动又有自旋运动。它们之间存在着耦合(电磁相
互)作用,使得能级发生分裂。对于ι >0的内壳层,这种分裂可以用内量子数j来
表示。其数值为:
j=
l + ms
=
l±
1 2
所以:对于ι =0,j=1/2。对于ι >0,则j= ι +½或者ι -½。也就是说,除了s能
级不发生分裂外,其他能级均分裂为两个能级:在XPS谱图中出现双峰。
3
电子能谱的基本原理
基本原理就是光电效应。
能量关系可表示:
hv = Eb + Ek + Er
电子结合能 电子动能
原子的反冲能量
Er
=
1(M
2
− m)υa*2
忽略 Er (<0.1eV)得
hv = Ek + Eb
4
对孤立原子或分子,Eb 就是把电子从所在轨道
移到真空需的能量,是以真空能级为能量零点的。
S能级的内量子数½通 常省略。如:C的1s 能级没有分裂,在 XPS谱图上只有一个 峰,表示为:C1s。
C1s
14
基本原理
4、电子结合能Eb: 一个自由原子或者离子的结合能,等于将此电子从所在的能级转移到无限远处所 需要的能量。对于气体样品,如果样品室和谱仪制作材料的影响可以忽略,那么 电子的结合能Eb可以从光子的入射能量hν以及测得的电子的动能Ek求出,即:
21
X射线光电子能谱分析的基本原理
5、XPS信息深度: 在XPS分析中,一般用能量较低的软X射线激发光电子(如:Al 和Mg的Kα线)。虽然软X射线的能量不高,但是仍然可以穿透 10nm厚的固体表层,并引起那里的原子轨道上的电子光电离。 产生的光电子在离开固体表面之前,要经历一系列的弹性(光 电子与原子核或者其他电子相互作用时不损失能量)和非弹性 散射(光电子损失能量)。弹性散射的光电子形成了XPS谱的 主峰;非弹性散射形成某些伴峰或者信号的背底。 一般认为:对于那些具有特征能量的光电子穿过固体表面时, 其强度衰减遵从指数规律。假设光电子的初始强度为I0,在固体 中经过dt距离,强度损失了dI,有:
电子能谱分析范文

电子能谱分析范文电子能谱分析是一种通过分析物质中电子能级的特征,来研究物质的结构和化学性质的方法。
它是一种非常重要的分析方法,广泛应用于有机化学、物理化学、材料科学等领域。
在本文中,我们将介绍电子能谱分析的原理、常用的实验技术和应用。
电子能谱分析的原理是基于能级的分布和电子能量的定理。
在原子、分子或固体中,电子存在不同的能级,每个能级上的电子具有不同的能量。
当物质处于激发态时,电子会从低能级跃迁到高能级,吸收一定的能量;当物质处于基态时,电子会从高能级跃迁到低能级,释放出一定的能量。
这些能量的变化可以通过测量电子发射或吸收的能量谱来获得,从而推断出物质的能级分布和电子结构。
电子能谱分析有多种实验技术,其中最常用的是X射线光电子能谱(XPS)和紫外光电子能谱(UPS)。
XPS是利用X射线激发物质表面的电子,测量其能量分布和强度的变化。
它可以提供物质表面的元素组成、化学状态和价态信息,并且具有非常高的表面灵敏度。
UPS则是利用紫外光激发物质中的电子,测量其能量分布和强度的变化。
相比XPS,UPS可以提供更多关于电子能级和束缚态的信息,对于研究分子和固体的电子结构非常有用。
电子能谱分析在许多领域有着广泛的应用。
在有机化学领域,它可以用于研究有机分子的结构和化学反应过程。
通过测量电子能谱,可以确定有机分子的键合和取代基团的位置,揭示分子的电子结构和反应机理。
在物理化学领域,电子能谱分析对于研究材料的电子结构、能带与导电性质有着重要意义。
它可以用于表征材料的表面态、表面吸附和氧化还原反应等。
在材料科学领域,电子能谱分析可以用于研究新型材料的电子结构和光电性质。
通过对材料中电子能级和能带结构的分析,可以为设计和开发新型功能材料提供有价值的信息。
除了XPS和UPS,还有其他一些电子能谱分析的技术,如电子能量损失谱(EELS)和光电子能谱显微镜(PEEM)。
EELS是利用电子束与物质相互作用而损失能量的原理,测量被探测物质中电子能量的变化。
电子能谱定量分析和深度分析

l 实用XPS定量方法可以概括为标样法,元素灵敏度因子法 和一级原理模型。
l 标样法需制备一定数量的标准样品作为参考,且标样的 表面结构和组成难于长期稳定和重复使用,故一般实验 研究均不采用。目前XPS定量分析多采用元素灵敏度因子 法。该方法利用特定元素谱线强度作参考标准,测得其 它元素相对谱线强度,求得各元素的相对含量。
Elemental Symbol
2.1.1 X射线电离截面
X射线电离截面项 (特定跃迁将产生多少光电子)
一般使用下列两个数据库: (客户也可产生其自己的数据库, 大多数不必)
Scofield – 理论灵敏度因子数据库, 基于 C1s = 1 (即一定量的光子作用到样品上后所产生光电子数目的 一个相对计算值)
Scofield – 理论灵敏度因子数据库, 基于 C1s = 1 为此我们需要增加一项来说明分析的深度 (即 λ 并通常取 KE0.6)
Wagner -实验灵敏度因子数据库, 基于 F1s = 1 为此我们需要增加一项来修正不同(仪器)类型分析器 产生的因子 (A CMA rather than our HSA). 这可通过乘 以峰动能来实现。(λ项已包含在里面)
Wagner – 实验灵敏度因子数据库, 基于 F1s = 1
1 1 3 (即在某种谱仪上真实测量大量的已知化合物并计算出
相对灵敏度因子)
电离截面计算值(Scofield)
U的相关结合能和电离截面
Scofield vs. Wagner?
两数据库都在用 – 但对同一套数据可能有不同结果!! 它们必须以稍有不同的方式来使用
SIMS溅射深度剖析的定量分析

书山有路勤为径,学海无涯苦作舟SIMS 溅射深度剖析的定量分析本文综述了二次离子质谱(Secondary溅射就是用具有一定能量的一次性粒子轰击样品表面(粒子的动能通常在0.1~5 keV 之间),通过样品发射二次粒子而使材料表面原子或分子剥离的一个过程。
深度剖面分析(简称深度剖析)是指对分析样品元素的组分含量随深度变化的二维分析。
目前,有两种不同的深度剖析方法:非破坏性和破坏性的方法。
通常的非破坏性深度剖析技术只提供间接的信息,需通过定量分析才能得到浓度深度谱,如卢瑟福背散射(Rutherford Backscattering Spectrometry,RBS) 或者角分辨的X-射线光电子能谱(Angle Resolved X-ray Photoelectron Spectroscopy,AR-XPS)。
而破坏性的溅射深度剖析的原始数据是元素强度相对溅射时间的关系溅射深度剖析是将离子溅射与表面元素成分表征结合在一起的一种测量分析技术,其主要目的是为了获得薄膜材料(从几纳米到几百微米)中元素成分的深度分布。
按对元素表征方式的不同,溅射深度剖析可分为两类:一类是分析溅射出来的元素,如二次离子质谱(Secondary深度分辨率的提出标志着深度剖析定量分析的开始。
深度分辨率表示了深度剖析谱的失真程度,即由于离子束和样品的相互作用在表面区域产生的成分与形貌的改变,使得实际测得的深度剖析谱与真实的成分深度分布之间产生的偏差程度,它是表征深度剖析实验优劣的一个主要指标。
深度分辨率Δz 的定义如下:假设一理想的、原子单层的界面A/B,当测量信号的归一化强度从84% 降到16% 或从16% 上升到84% 所对应的溅射深度Δz。
Δz 愈小意味着深度剖析的分辨率愈大,测量的成分深度分布就愈接近真实的成分深度分布,深度剖析的质量就愈高1970-1990。
电子能谱分析edx
EDXX射线能谱一介绍EDS(energy dispersive spectro-scopy)能量色散谱EDX (Energy Dispersive X-ray)WDS(wavelength dispersive spectro-scopy)波长色散谱EDX:X射线强度和能量曲线,定量分析样品的化学成份主要用途:•1) 非均匀样品的局部化学成份•2) 较少量材料或小颗粒材料的化学成份•3) 非均匀样品种一维或二维的成份分布•4) 沉积在任意衬底上的薄膜成份特点1)铍以上元素2)最小能探测到的重量比:0.1 wt% ——1 wt%3)定量结果的相对误差:2-20%(取决于校正方法等)4)在计算机控制下,1分钟以内可分析16种元素5)空间分辨率取决于平均原子序数、样品密度、束能量等(SEM中0.2——10微米)获得可靠的分析结果要求样品:·1) 样品平整光滑(尤其对定量分析,样品要抛光)2) 可以分析表面粗糙的样品,但仅限于定性和半定量分析3) 样品必须导热导电,必要的时候表面需要喷炭或金推荐书目:Scanning Electron Microscopy and X-ray microanalysisNew York 1992 (生物学、材料科学、地质学)Scanning Electron Microscopy,X-ray microanalysisand Anlytical Electron MicroscopyNew York 1990二定量分析Fig.2: Schematic diagram showingwhere 29<Z<37.Detected Energy (E)Fig.5: Schematic diagram of the intensity variation of the continuum backgroundwith energy, showing the generated and detected background energy.Intensity (I)Generated A )背底和特征峰(二)影响X 射线强度的几种因素B)原子序数对X射线强度的影响Variation in fluorescence yield with atomic number.C )荧光产生率E )Mass absorption coefficient of Fe, for X-rays of varying energyD)X 射线的吸收探测角度:角度越小,X射线吸收越强。
Avantage 数据处理方法

[FO]
元素 Nb Ni Pb Pd Re Rh Sb Sc Se Si Sn Ta Te Tl V W Y Zn Zr
C (1s) BE of Hydrocarbons
自然氧化物 离子刻蚀金属
285.1
284.9
285.4
284.9
285.6
285.2
285.3
284.2
284.5
285.0
284.6
Savitzky-Golay,其次可采用高斯函数。 当平滑点数取谱图中可分辨的最窄峰的FWHM
所含的数据点数时,Savitzky-Golay函数效 果最佳,失真低。
2、本底去除
在XPS谱中,通常为较小的谱峰叠加在大的 本底之上。如果要检查谱峰的细节,在某些 情况下就需要进行本底去除(如定量时测量 谱峰强度时)。
最简单的本底去除方法是在用户感兴趣的谱 峰两端指定点间作直线—线性。
线性本底通常误差较大,是非物理的。 线性本底的改进涉及到的物理真实逼近—
Shirly本底。
线性本底
非线性本底 - Shirley Method
使用最普遍的非线性背景扣除方法
该方法认为能量损失是常数, 谱线上任 一点由非弹性散射电子引起的背景, 只 来源于更高动能电子的散射, 正比于更 高动能的积分光电子强度(面积)
原子百分数的计算
归一化面积(NA)由谱峰面积(IA)来计算
NA = IA /NF
因而样品中任一元素的原子浓度由下式算出:
CA = (At%)A = (NA/(NA + NB + NC..)) X 100 或
CA
N A 100 Ni
1
i
13
2.1、灵敏度因子
5光电子能谱分析

h Ek' Eb ' Eb h Ek' '
由于Φ’是谱仪的功函数,与样品无关,是固定值, 一的般X射仪线器能的量功,函也数是约已4e知V,的即,是于已是知只的要。在h实ν是验实中E验k测' 时得选光用电 子的动能,就可以计算Eb了。
3 XPS的仪器Instrumentation for XPS
9.2
1263.1
5.1
1271.0
0.8
1274.2
0.5
1302.0
2.0
Al 靶
能量(eV)
相对强度
1486.7
67.0
1486.3
33.0
1492.3
1.0
1496.3
7.8
1498.2
3.3
1506.5
0.42
1510.1
0.28
1557.0
2.0
❖X射线光电子谱仪
➢ 作为X射线光电子谱仪的激发源,希望其强度大、单色性好。
率高,可以作包括H元素的成分分析,而且还可以分析同位素。 (2)紫外光电子能谱(UPS)
激发源:紫外光 发射源(信号):价电子 (3)俄歇电子能谱(AES) 激发源:电子、X射(XPS)又称电子能 谱化学分析(ESCA: Electron Spectroscopy for Chemical Analysis ))
1 激发光源——X射线(软X射线;Mg Kα : hv = 1253.6 eV; Al Kα : hv = 1486.6 eV)或UV;
2 电子能量分析器-对应上述能量的分析器,只可能是表面 分析;
3 高真空系统:超高真空腔室super-high vacuum chamber ( UHV避免光电子与气体分子碰撞的干扰。
三、EDS定量分析方法与分析误差
考虑的物理过程比ZAF方法更符合实际。 适合于重、轻元素成分分析。
15
φ(z)函数
φ(z)是一个X射线深度分布函数,它表示试
样某一质量深度z 处一个薄层d(z)发射的X
射线强度,与在空间中孤立存在的同一厚度 的相同材料中发射的X射线强度之比值。
论计算结果导出。 1951年提出,可分析金属、氧化物等,对平的抛光
试样从Na-U元素的相对误差为~2% 。
8
基体校正
基体校正 :由于激发体积中其它元素 对电子散射、阻止本领、X射线产生与 传播以及二次荧光的影响,引起被测 元素的特征X射线强度的变化进行的 校正。包括:
1、吸收校正 2、原子序数校正 3、荧光校正。
kA=CA/C(A)·ZAFA/ZAF(A)
ZAFA和ZAF(A)分别为试样和标样的校正系数。
7
EDS定量校正方法
1. ZAF定量校正方法:
用原子序数校正因子Z、X射线吸收校正因子A, 及X射线荧光校正因子F,对试样与标样之间的差异 进行校正。 kA=CA/C(A)·ZAFA/ZAF(A) 注:Z、A、F因子是根据试验测量、经验拟合和理
标准物质系列来测定,或者通过ZAF或者Phi-Rho-Z模型理论测定。 是
一种经验方法,适合于 矿物、玻璃、氧化物、硅酸盐材料等分析。该方
法计算简便,准确度较高。式中
CA AB
是AO—BO二元氧化物中氧化物AO
相对于纯氧化物AO的浓度,K
A AB
是二元氧化物中A元素X射线强度相对于纯
氧化物AO中同名持征X射线强度之比,
9
1、原子序数(Z)校正
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1
1 3
电离截面计算值(Scofield)
U的相关结合能和电离截面
Scofield vs. Wagner?
两数据库都在用 – 但对同一套数据可能有不同结果!! 它们必须以稍有不同的方式来使用 Scofield – 理论灵敏度因子数据库, 基于 C1s = 1 为此我们需要增加一项来说明分析的深度 (即 λ 并通常取 KE0.6) Wagner -实验灵敏度因子数据库, 基于 F1s = 1 为此我们需要增加一项来修正不同(仪器)类型分析器 产生的因子 (A CMA rather than our HSA). 这可通过乘 以峰动能来实现。(λ项已包含在里面)
1、一级原理模型(First Principle Model)
l
l
从光电子发射的“三步模型”出发,将所观测到的谱线强 度和激发源,待测样品的性质以及谱仪的检测条件等统 一起来考虑,形成一定的物理模型。 由于模型涉及较多的因素,目前还缺乏必要精度的实验 数据,因此一级原理模型计算还未得到真正应用。
I ij = K ⋅ T ( E ) ⋅ Lij (γ ) ⋅ σ ij ⋅ ∫ ni ( z ) ⋅ e
−z λ ( E ) cos θ
dz ⋅
其中: Iij为i元素j峰的面积,K为仪器常数,T(E)为分析器的传输函数,Lij(γ)是i 元素j轨道的角不对称因子,σij为表面i元素j轨道的的光电离截面,ni(z)为表面i 元素在表面下距离z处的原子浓度,λ(E)为光电子的非弹性平均自由程,θ是测 量的光电子相对于表面法线的夹角。
l
深度剖析 l 离子束在样品表面扫描 l 样品表面物质被逐渐刻蚀掉 l 在刻蚀周期间采集XPS谱 l 建立起样品成分随深度变化的剖析图 l 剖析深度可达 1µm l 结构破坏技术
深度信息
l
角分辨XPS l 电子逃逸深度是有限的 l 掠射角方向的电子来自于近表面 l 以一系列的角度采集数据 l 计算膜厚可达5-10nm l 非结构破坏技术 l Theta Probe不必倾斜(tilting)样品即可达成 离子溅射深度剖析 (d< 1µm) l 离子束在样品表面扫描 l 样品表面物质被逐渐刻蚀掉 l 在刻蚀周期间采集XPS谱 l 建立起样品成分随深度变化的剖析图 l 结构破坏技术
谱峰 C 1s C 1s C 1s C 1s O 1s O 1s O 1s EB (eV) 285.0 286.6 289.0 291.6 532.1 533.7 538.7 面积 2000 700 700 100 1600 1685 85
*
*
〖一个定量分析例子〗
l
解:
nC SC 1 s = = nO I O SO1s
原子百分数的计算
归一化面积(NA)由谱峰面积(IA)来计算 NA = IA / Si 因而样品中任一元素的相对原子浓度由下式算出:
CA = NA × 100 ∑ Ni
i
1
1 3
2.1 灵敏度因子
灵敏度因子(归一化因子)包括下面几项:
X射线电离截面项 (特定跃迁将产生多少光电子) 分析深度项 (并入λ值中) 传输函数项 (谱仪对特定动能电子检测的能力) 不同仪器得出的灵敏度因子之间的归一化 (比如 CMA和HAS之间 )
第五章 定量分析和深度剖析
l 电子能谱的测量不仅可以给出从所含元素
简单的定性指认到复杂化学态分析,还可 以给出元素组成的定量分析及每个元素相 的分布状态信息(非均相样品)。
l l l
定量分析原理和方法 深度剖析方法(分层结构) 空间分布分析
一、定量分析方法
l
l
l l
在表面分析研究中我们不仅需要定性地确定试样的元素 种类及其化学状态,而且希望能测得它们的含量。对谱 线强度作出定量解释。 XPS定量分析的关键是要把所观测到的信号强度转变成 元素的含量,即将谱峰面积转变成相应元素的含量。这 里我们定义谱峰下所属面积为谱线强度。 实用XPS定量方法可以概括为标样法,元素灵敏度因子法 和一级原理模型。 标样法需制备一定数量的标准样品作为参考,且标样的 表面结构和组成难于长期稳定和重复使用,故一般实验 研究均不采用。目前XPS定量分析多采用元素灵敏度因子 法。该方法利用特定元素谱线强度作参考标准,测得其 它元素相对谱线强度,求得各元素的相对含量。
Elemental Symbol
2.1.1 X射线电离截面
X射线电离截面项 (特定跃迁将产生多少光电子) 一般使用下列两个数据库: (客户也可产生其自己的数据库, 大多数不必) Scofield – 理论灵敏度因子数据库, 基于 C1s = 1 (即一定量的光子作用到样品上后所产生光电子数目的 一个相对计算值) Wagner – 实验灵敏度因子数据库, 基于 F1s = 1 (即在某种谱仪上真实测量大量的已知化合物并计算出 相对灵敏度因子)
i
3
3、定量分析方法步骤
l
l
l
l
l
扣除背景 l Linear, Shirley, (Tougaard), Smart 测量峰面积 l 必要时进行峰拟合 应用传输函数 l 随不同的仪器而变 应用灵敏度因子 l 随不同元素(及厂商)而变 计算原子浓度
定义峰-本底类型及误差
l
l
l l
定义峰:为进行定量分析而计算峰面 积,就要确定峰的起点和终点,此两 点间的本底将被扣除。定义峰的起点 和终点位置对于定量计算的精确性是 重要的。 在实际加峰到谱峰表(Peak Table)中 之前,必须要考虑选取不同的本底类 型及其可能带来的误差。 有四种本底扣除的方法可选:Linear, Shirley,Tougaard和Smart。 应根据谱峰的实际峰型和情况来正确 选取本底类型
20
0 -1100
-900
-700
-500
-300
-100
Binபைடு நூலகம்ing Energy (eV)
〖一个定量分析例子〗
l
一材料的全谱扫描检测到只有碳和氧存在,高分辨C 1s和O 1s扫描 表明分别存在4个和3个子峰。用下面提供的数据计算C/O原子比和 每一组分在样品中存在的百分比。同时提出一个关于此样品的化 学结构,并给出对应每个子峰的自恰指认。激发源使用Al Kα X射 线。结合能值已对样品荷电进行过校正。C 1s和O 1s的原子灵敏度 因子分别为0.296和0.711。
C1s Sat C1s =OOH C1s -OH
峰面积 归一化峰面积
原子浓度
灵敏度因子
传输函数
原子百分数的精确度如何?
定量计算结果中存在几个不确定性的来源,它们来自于: l 峰强度的测定? 如何测量峰面积, 从什么位置到什么位置, 包括什么, 什么 形状背底等 l 灵敏度因子数据库的精确度? 不同的数据库给出不同的结果 – 哪个更好? l 传输函数的精确度? 对特定仪器传输函数定义的准确程度如何 l 其它因素包括λ的测定?
ni = I ij {K ⋅ T ( E ) ⋅ Lij (γ ) ⋅ σ ij ⋅ λ ( E ) cos θ } = I ij Sij
因此,
l
式中Sij=K⋅T(E)⋅Lij(γ)⋅σij⋅λ(E)cosθ ≈ T(E)⋅σij⋅λ(E) 定义为 原子灵敏度因子,它可用适当的方法加以计算,一般通 过实验测定。可取SF1s=1作为标准来确定其它元素的相 对灵敏度因子。 ni ∝ Iij / Sij =Ni
l
此外样品的不均匀性或表面有污染层存在以及元素化学 态不同
1
1 3
原子百分数的精确度如何?
l
使用原子灵敏度因子法准确度优于15%。在同一 仪器上使用标样测量准确度优于5%。在所有情形 下重复性(精密度)好于2%。 l 对于任一元素并非所有的XPS峰都有相同的强 度(面积比正比于其原子灵敏度因子)选 择具有最大原子灵敏度因子的峰以最大化灵 敏度 l 每一元素在复杂混合物中的灵敏度会变化。
1
1 3
元素的相对灵敏度因子
12
10
3d
Relative Sensitivity
8
4f
6
2p
4 2
1s
4d
0 Li B N F Na Al P Cl K Sc V M Co Cu G As Br Rb Y Nb Tc Rh Ag In Sb I Cs La Pr P Eu Tb Ho T Lu Ta Re Ir Au Tl Bi Be C O Ne M Si S Ar Ca Ti Cr Fe Ni Zn G Se Kr Sr Zr M Ru Pd Cd Sn Te Xe Ba Ce Nd S G Dy Er Yb Hf W Os Pt Hg Pb
l
厚度测量
l 某些情况下我们不仅需要知道表面的性质,而且想要得知样品内部( 体相)分布的信息。有两种方法来测定这些信息。
角分辨XPS (ARXPS) 用于小于XPS 分析深度的分析
Tungsten Titanium Alloy (W/Ti) 300 nm Silicon (Si) Titanium Silicide (TiSi) Silicon (Si)
元素灵敏度因子法
l
若某一固体试样中两个元素i和j,如已知它们的灵 敏度因子Si和Sj,并测出各自特定谱线强度Ii和Ij, 则它们的原子浓度之比为: 一般情况下:
ni I S = i i nj I j Sj
l
Ci =
I i Si ∑ Ij Sj
j
l
H和He的原子灵敏度因子非常小——在传统XPS中 不可测。
IC
(2000 + 700 + 700 + 100)
0.296 2.5 = 5 (1600 + 1685 + 85) 2 0.711
ð ð
C:C:C:O:O = 3:1:1:1:1 可能的分子结构: C5mHnO2m 或 C10HnO4