设计验证和生产确认ppt课件
确认与验证PPT课件

三 确认与验证的主要内容
设备确认V-model模式
变更控制
系统使用与维护
新项目需求
验证
用户/系统需求 风险评估
项目确认计划 功能文件
性能确认 运行确认
设计文件
安装确认
设计确认
接受测试
设备确认的时序性
• 用户需求 • 风险分析 • 项目与确认计划 • 设计确认 • 工厂测试 • 安装确认
• 运行确认 • 性能确认
确认VS验证
验
证
确认
验证总计划---验证方针
所有上市销售的产品都应经过一系列相关验证, 以确保工艺的重现性和可靠性,且按照相应的 法规进行生产
对产品质量、安全、纯度或效力有重要影响 的生产工艺、清洁程序、生产中使用的分析 检测方法、储存和销售必须经过验证,与上 述相关使用的设备、厂房、设施及公用系统 必须经过确认
确认 就是要用数据证明我们是不是制造 了正确的产品。注意这里强调的是结果 的正确性
验证 就是要用数据证明我们是不是在正 确的制造产品。注意这里强调的是过程 的正确性
再确认与验证、持续工艺确认
验 证
再确认/再验证
生产工艺、设备、系统经过确认/验证并使用 一个阶段后,采用质量风险管理方法评估变 更对产品质量、质量管理体系、文件、验证、 法规符合性、校准、维护和其他系统的潜在 影响,必要时进行的活动,旨在证实其确认/ 验证状态没有发生变化
验证总计划---验证分类
检验方法的确认 运输确认 设备及公用工程系统确认 工艺验证 清洁验证
验证总计划---验证常用方法
前验证 同步验证 回顾性验证 再验证
验证总计划---验证实施时间
按照管理要求首次进行的 发生变更、偏差等事件,经评估需再验证的 验证周期到期后或经回顾发现验证状态发生 漂移的实施再验证
设计和开发验证和确认的区别
设计开发评审
策划时作出安排(一般是对输入和设计过程)
评审点 评审 选择 方式
评审 人员
评审 准备
评审 要求
评审 内容
评审 结果
意见 处理
改进设计
新设计
评审
设计更改
评价结果
满足要求的
能 力
适宜性 充分性 有效性
识别不足
解决问题的
措 施
保持记录
设计和开发验证
策 划 时 作 出 安 排(一般是对输出)
通常是最终产品 (样品、图纸)
使用者代表或权威 专家
一般在批量生产前, 也可分阶段
新产品鉴定会 确认签字
设计开发更改的控制
评
审
➢根据更改范围的大小、
后
重要性决定是否评审、
验证、确认
验
更
➢评价更改后对产品部
更改及措施的
证
分或整体功能、性能、 批准
后
改
结构等方面的影响 ➢评价对已交付产品的
记录 应保持
验证点
验证内容
验证方式
设计 输入
几种验证方式
❖变换方法计算 ❖与已证实的类
似设计比较 ❖试验和演示 ❖对设计文件评审
验证
设计 输出
输出是否满足 输入要求?
不足部分
验证结果
采取有效 措施解决
保持记录
设计和开发确认
设计开发完成后,批量正式生产/服务正式提供前,策 划时作出安排
确认点
确认内容
确认条件
确认方式
要
职责和权限
开发中的职责权限
求
规定
接口关系
组织结构与 技术上的接口
可行性报告
设计开发 计划
设计验证培训PPT

样品制备
根据方案中“样品信息”的样品规格、样品数量、 样品状态的要求制备样品
样品制备过程中的记录满足可追溯性要求,包含 原材料信息、制作过程信息、过程检验信息及灭 菌信息(要求灭菌的样品)。具体见 MP/AS4.2.4.-03 《验证和确认的原始记录管理 规定》。
制备完成的样品入品质部仓库。由使用样品的人 员从品质库领取出库。
8.1 General
8.2 Sampling
8 Design Evaluation
8.3 Conditioning of test samples 8.4 Reporting
8.5 Delivery system and stent sys
8.6 stent
8.7 Preclinical in vivo evaluation
否则,需重新制定方案 。
设计验证方案
设计验证方案包含的内容
目的 范围 人员和职责 原材料信息 测试项目和接受标准 测试方法 目标 样品信息(样品规格、数量、状态、灭菌信息) 数据分析方法 原始记录收集方法 参考文件
注:动物实验方案、生物学性能验证方案见相关程序文件 的要求
职责
生物学工程师
负责生物学性能验证; 负责安排验证样品的制作和测试; 负责验证的原始记录的可追溯性。
职责
研发工程师
协助制定各设计验证方案; 对验证报告的编制提供帮助。
职责
TC测试中心
负责样品测试,出具测试记录; 负责保存测试原始记录。
职责
研发支持部档案室
保存OA系统中设计验证方案和报告打印件; 保存样品制作原始记录、测试原始记录(适用
其它要求
发生设计更改
对于设计评审I之后发生的设计更改,见MP/AS7.304《设计更改控制》的规定;
QA必备--验证与确认 ppt课件

5.验证的生命周期
6.验证的文件管理
•URS •FS/DS/FDS •DQ •FAT/SAT •IQ/OQ/PQ •状态维护
•方案 •记录和报告 •文件管理
PPT课件
2
1验证概述
验证的定义:
中国GMP 2010版:证明任何操作规程(或方法) 、生产工艺或系统能够达到预期结果的一系列活 动。
3新版GMP附录确认与验证
第一章 范围 “药品生产质量管理过程中”的所有确认与验证活动。ICHQ10 制药
验证与确认培训PPT课件

验证与风险管理
• 设施、设备和公用系统的确认 :有助于基于使用的关键性确定试机及确认的范围与程度。
第20页/共89页
验证与风险管理
• 工艺验证 :通过工艺关键性分析,识别验证过程中需注意的高风险区域。这些高风险区域应该是和产品安 全性、有效性的关键质量属性相关联的。
第21页/共89页
验证与风险管理
• 验证方案应在审核并得到批准后实施。
第35页/共89页
3.8验证方案
• 封面 • 硫酸庆大霉素注射液工艺验证方案审批 • 方案起草人:日期: 年 月 日 • 方案审核人:日期: 年 月 日 • 方案批准人:日期: 年 月 日
第36页/共89页
3.8验证方案
• 第2页:验证小组名单 • 小组职务 姓名 工作部门 职 务
第27页/共89页
3.6验证文件的管理
• 验证总计划( validation Master Plan,VMP) • 是项目工程整个验证计划的概述,是指导企业进行验证的纲领性文件。是为公司的整个验证工作的实施提
供政策、导向以及公司生产、设施、系统和质量计划的总体情况。
第28页/共89页
3.6验证文件的管理
第11页/共89页
3.验证与再验证
• 再验证—一项生产工艺,一个系统或设备经过验证并在使用了一个阶段以后,旨在证实其验证状态没有发 生变化而进行的验证活动
第12页/共89页
3.1.工艺验证与工艺优选
• 工艺验证—证明工艺在预期参数范围内运行时,能有效地、重复地生产出符合预定质量标准和质量属性的 产品的有文件记录的一系列活动。
第40页/共89页
3.11.验证实施的结果
• — 各种验证试验的主要结果。 • 偏差及措施 —阐述验证实施过程中所发现的偏差情况以及所采取的措施。
确认与验证课件

确认与验证
主要内容
第六章 工艺验证
第一节 一般要求
第十九条 工艺验证应当证明一个生产工艺按照规定的工艺参数能 够持续生产出符合预定用途和注册要求的产品。工艺验证应当包 括首次验证、影响产品质量的重大变更后的验证、必要的再验证 以及在产品生命周期中的持续工艺确认,以确保工艺始终处于验 证状态。
第二十条 企业应当有书面文件确定产品的关键质量属性、关键工 艺参数、常规生产和工艺控制中的关键工艺参数范围,并根据对 产品和工艺知识的理解进行更新。
确认与验证
主要内容
第五章 确 认
第三节 运行确认
第十五条 企业应当证明厂房、设施、设备的运行 符合设计标准。运行确认至少包括以下方面:
(一) 根据设施、设备的设计标准制定运行测试项 目。
(二) 试验/测试应在一种或一组运行条件之下进行 ,包括设备运行的上下限,必要时选择“最差条 件”。
第十六条 运行确认完成后,应当建立必要的操作 、清洁、校准和预防性维护保养的操作规程,并 对相关人员培训。
确认与验证
主要内容
第四章 文 件
第九条 当确认或验证分阶段进行时,只有当上一阶 段的确认或验证报告得到批准,或者确认或验证活 动符合预定目标并经批准后,方可进行下一阶段的 确认或验证活动。
上一阶段的确认或验证活动中不能满足某项预先设 定标准或偏差处理未完成,经评估对下一阶段的确 认或验证活动无重大影响,企业可对上一阶段的确 认或验证活动进行有条件的批准。
确认与验证
主要内容
第三章 验证总计划
第三条 所有的确认与验证活动都应当事先计划。确认与验证 的关键要素都应在验证总计划或同类文件中详细说明。
第四条 验证总计划应当至少包含以下信息:
新版GMP之确认与验证PPT培训课件

验证概念迅速被各国法规所接受,并扩展到各个剂型,同时提出了对于
计算机系统等新技术的验证要求
2011年1月25日 FDA 颁布新的工艺验证指南,贯彻产品生命周期的工
艺验证,从产品研发到商业化生产
欧盟附录15, WHO GMP 中国2010版GMP
中国GMP确认与验证附录
要时选择“最差条件”,
性能确认—确认设备符合预期的性能 使用生产物料、适当的替代品或者模拟产品来进行试验/测试;应当评估测
试过程中所需的取样频率。
确认与其他体系之间关系
工艺验证
生命周期内保持验证状态
工艺验证应当包括首次验证、影响产品质量的重大变更后的验证、必要的
再验证以及在产品生命周期中的持续工艺确认,以确保工艺始终处于验证 状态。不再提回顾性验证。
测试项目与测试方法 测试结果 结论与签名
第三方提供的确认验证必须得到企业的审核、批准,确认方案、数据或
验证报告,偏差调查
确认
通常发生在FAT/SAT之后 包括设计确认/安装确认/运行确认/性能确认 清晰的用户需求标准至关重要 更加清晰描述设计、安装、运行和性能之间的关系,需要连贯性
新版GMP之确认 与验证
目录
验证的发展 中国GMP附录
产品生命周期和验证与确认 验证主计划 验证文件 确认 工艺验证 运输确认 清洁验证 再确认和再验证
工艺验证解读
验证的发展
1965年-1975年 美国市场大容量注射液市场召回超过600起,54人死亡 败血症案例引起FDA特别工作组调查 1976年6月1日 FDA 大容量注射剂GMP规程草案, 首次将验证以文件
验证主计划
确认与验证版gmp培训PPT课件

在完成影响评估以后,关键功能应被评估以确定直接 影响系统的组分的关键程度,以确保验证行动关注在 对产品质量造成风险的部分。
如果一个间接影响或非影响系统包含一个或更多关键 成分,则是对系统进行了错误的分类,或者是对成分 进行了错误的评估。
◦ 组分用于创建或者保护关键系统的状态 ◦ -Volume 5 commissioning and qualification ISPE
.
17
.
18
参见GAMP-5
.
19
工厂验证总计划 (Site Validation Master Plan)
◦ 目的 ◦ 公司简介(产品,剂型) ◦ 验证的指导方针与规程(工厂SOP) ◦ 责任(验证工作的分工,批准职责,批准人至少有生产负责人,质量负责人) ◦ 验证行为 ◦ 时间表与进程 ◦ 版本历史 ◦ 附录 ◦ 参考
◦ 参考与附录 ◦ 版本历史
-FS/DS应在设计确认开始之前得到用户和质量部门的批准
.
24
安装确认应当证明厂房、设施、设备的建造和安装 符合设计标准;
◦ 安装确认将确保
关键部件的安装符合被记录的设计标准。
直接影响系统及其部件的主要特征,如材质证明应被记录。
具有足够的信息以确保可以对关键项目进行安全、有效以及 一致性地操作与维护。
◦ 批准签字页 ◦ 介绍(项目简介,是什么引出的验证活动) ◦ 目的 ◦ 范围(风险评估的结果,什么系统,什么设备需要验证) ◦ 验证活动方法-活动内容,执行顺序(DQ,IQ,OQ,
PQ,PV) ◦ 接受标准 ◦ 支持性工作 ◦ 时间进程表 ◦ 角色与职责 ◦ 参考与附录 ◦ 回顾历史
.
确认与验证-药品生产质量管理课件

FDA在工艺验证总则指南(1987年5月)中描述:“……验证是为确保一个 专门的过程……可以持续地生产满足一个产品的预设规格与质量特征而反复 建立的书面依据。
”ICH关于确认的定义:“证明并记载设备或辅助系统安装适当、使用正确 并实际上产生期望的结果。
中国GMP(2010年修订)中引入了通过风险评估确定确认验证范围和程度 的要求。
验证应该通过风险分析确定哪些步骤和具体操作是决定产品质量的关键工艺 参数。
验证过程中应注意这些关键的步骤和操作,通过进一步分析识别关键工艺参 数。
将系统分为直接影响系统、间接影响系统和无影响系统。
直接或间接影响药品质量的与制药工艺过程、质量控制、清洁、消毒或灭菌 等方面相关的制药机械设备属于必须验证的范围,其他起辅助作用或不对药 品质量产生影响的系统可不列为验证的范围。
变更已批准的确认与验证方案,应当进行评估并采取相应的控制措施。 确认或验证报告应当经过书面审核、批准。
当确认或验证分阶段进行时,只有当上一阶段的确认或验证报告得到批准, 或者确认或验证活动符合预定目标并经批准后,方可进行下一阶段的确认或 验证活动。
上一阶段的确认或验证活动中不能满足某项预先设定标准或偏差处理未完成, 经评估对下一阶段的确认或验证活动无重大影响,企业可对上一阶段的确认 或验证活动进行有条件的批准。
确认与验证
本章学习要求
1.掌握确认与验证的定义、分类、范围及 文件要求。 2.了解厂房设施验证的流程和主要内容。 3.了解分析方法验证的内容和方法。 4.掌握工艺验证和清洁验证的内容和方法。 5.掌握GMP对运输确认的要求。
尽管98版GMP对验证提出了基本要求,但在实施过程中,验证技术在我国 药品生产企业中的推进仍有很大的发展空间。
PPT课件-确认与验证

PPT课件-确认与验证⽬录为什么要开展确认或验证如何进⾏确认或验证值得关注的⼏个问题如何进⾏计算机化系统验证验证案例验证时容易出现的问题为了验证⽽验证不清楚什么时候该验证,为什么要验证?验证⽅案不是基于风险评估⽽设计的验证⽅案是抄来的模板,没有对范围和程度进⾏风险分析参加验证的⼈员没有进⾏培训操作⼈员不清楚验证⽬的,对关健参数不了解没有对验证所获得的数据进⾏科学分析验证得到的数据(完整性,⼀致性等)不归纳总结,出现偏差没有处理验证结果评价不全⾯验证报告与验证⽅案不⼀致验证与实际⽣产脱节如没有及时更新相关的SOP为什么要开展确认或验证验证⾏为在⽇常⽣活⾥⽆处不在,验证技术存在于各⾏各业当中对于药品和医疗器械⽣产企业⽽⾔,验证⾏为是企业定标及达标运⾏的基础,验证⽂件则是有效实施GMP的重要证据。
企业可以通过贯穿于产品⽣命周期全过程的确认或验证⼯作来证明影响质量的关键要素能够得到有效控制,为持续⽣产出合格药品提供保证。
对于监管部门来讲,验证是保证药品和医疗器械质量的根本,必须从法规上强制性加以规定和要求。
质量管理系统的⼦系统数据完整性是指数据的准确性和可靠性,⽤于描述存储的所有数据值均处于正确的状态。
并不是计算机化系统实施后才出现的。
适⽤于电⼦数据和⼿⼯(纸质)数据。
企业应当处于⼀种基于数据完整性风险的可接受控制状态。
数据的属性A(attributable)-可追踪⾄产⽣数据的⼈L(legible)-清晰的,能永久保存C(contemporaneous)-同步O(original(or‘true copy’)-原始(或真实复制)A(accurate)-准确数据完整性的检查基于风险,判断重点深⼊调查,不蜻蜓点⽔有疑问的数据⼀定要证实客观真实性追踪最原始的数据QC实验室,尤其是稳定性试验的数据物料发放流转的数据各项记录的发放和填写企业质量管理体系对数据完整性的覆盖数据完整性直接表现企业的质量管理⽔平设计控制总结确认与验证的定义确认 Validation是对本⾝性能的认定,内涵归属于性能检验。
确认与验证培训课件

无忧PPT整理发布
“确认与验证〞的定义
只有了解何为确认与验证,才能更好地开展确认与验证工作
确认
•证明厂房、设 施、设备和检 验仪器能正确 运行并可到达 预期结果的一 系列活动。
确认常用于厂房、设施、设备 及检验仪器
验证
证明任何操作规 程〔或方法〕、 检验方法、生产 工艺或系统能到 达预期结果的一
验证的生命周期
确认的阶段
1
设计确认〔DQ〕
2
安装确认〔IQ〕
3
运行确认〔OQ〕
4
性能确认〔PQ〕
确认
❖确认应在生产工艺验证实施前完成。确认的过程应有逻 辑性、系统性,应起始于厂房、设备、公用设施和设备 的设计阶段。
❖根据设备、公用设施或系统的功能和操作,可能仅要求 安装确认(IQ)和运行确认(OQ),因为设备、公用设 施或系统的正确运行足以证明其性能。设备、公用设施 和系统随后应按照常规方案进行维护、监控和校准。
❖ 主要设备以及关键公用设施和系统需要进行IQ、OQ和 PQ
❖ 确认期间,应准备操作、维护和校验的所有SOP。 ❖应对操作人员进行培训并保存培训记录。
设计确认
❖ 应提供文件证据证明符合设计标准。
安装确认
❖ 应提供文件证据证明安装已完成且结果符合要求。 ❖ 在安装确认过程中,应核对采购标准、图纸、手册、备
预防性维护 保养
校验
变更控制
生产过程控 产品年度回 再验证管理
制控制
忆
验证状态保持的主要手段
GMP条例
❖第一百四十条 应当建立确认与验证的文 件和记录,并能以文件和记录证明到达以 下预定的目标。
❖第一百四十五条 企业应当制定验证总方 案,以文件形式说明确认与验证工作的关 键信息。
设计和开发验证和确认的区别
使设规用定计要的求 输已知入的
预期用途
实际条件下/ 模拟状态下的
试验
确认
设计 开发的 产品
产品是否满足 使用要求?
不足部分
确认结果
采取有效 措施解决
保持记录
设计开发过程控制图
市场 调研
设计 开发 策划
可行 性 报告
任 务 书
输 入
(产品
要求)
评审
设计和 开发
硬件
软件
输 出
试 制
样品 流程性
材料
验证
要
职责和权限
开发中的职责权限
求
规定
接口关系
组织结构与 技术上的接口
可行性报告
设计开发 计划
策划 形成 文件
变 更
修改后 文件
日程进度 工作计划
设计和开发的输入和输出
法律法规要求
标准 特别是强制性
• • •
功
保 密 性
安 全 性
使 用 性
能 要 求
• • • •
化 学
电 气
机 械
物 理
性 能
性性性性要
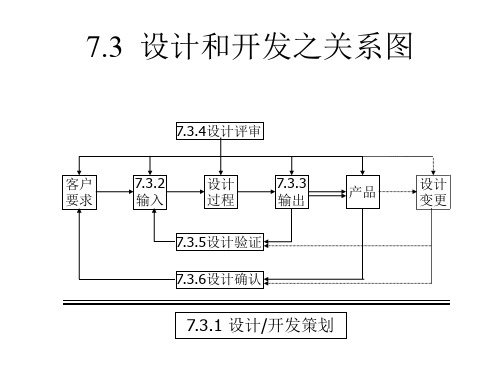
7.3 设计和开发之关系图
7.3.4设计评审
客户 要求
7.3.2 输入
设计 过程
7.3.3 输出
产品
设计 变更
7.3.5设计验证 7.3.6设计确认
7.3.1 设计/开发策划
设计开发的策划
划分
设计开发阶段
规定每阶段的 工作内容和要求
策
划 的
确定
评审验证确认
活动时机、参与 人员和活动要求
内
容
与
明确
有关部门在设计
服务
第六章-确认与验证 PPT课件

提出必要时经过药品监督管理部门批准的要求。
33
2020/3/29
33
五、验证
(一)工艺验证 类型
1.前验证:前验证是指新工艺、新产品、新设备等在正式
投入生产使用前,必须完成并达到设定要求的验证。
是正式投产前的质量活动。 前验证常用于:产品要求高或有特殊质量要求的产
②文件:设备规格标准与使用说明书;设备安装图及质量验 收标准;设备部件与备件清单;设备相应的公用工程及建 筑设施资料;安装、操作、清洁的SOP草案;有关记录表 格等。
2020/3/29
18
⑶运行确认
①内容:按SOP草案对设备的单机或系统进行空载试车;考 察设备运行参数的波动性;对仪表在确认前后各进行一次 校验,以确定其稳定、准确;设备运行的稳定性;SOP草 案的适应性;人员的培训。
协调、验证计划的制定和监督实施、验证文件的管理等。
3.提出验证项目 4.制定确认和验证的计划
2020/3/29
10
5.制定确认与验证方案 6.审批验证方案 7.组织实施 8.验证报告,进行偏差处理 9.审批验证报告 10.发放验证证书
2020/3/29
11
四、确认
包括
设计确认 (DQ)
安装确认 (IQ)
25
⑷初始清洁
2020/3/29
26
⑸校准
2020/3/29
27
⑹文件
2020/3/29
28
⑺偏差
2020/3/29
29
验证
常见的有
工艺验证 清洁验证 分析方法验证 计算机化系统验证
2020/3/29
新版GMP设计确认84页PPT

一、新版GMP法规要求
第九条 无菌药品生产所需的洁净区可分为以下4个级 别:
A级:高风险操作区,如灌装区、放置胶塞桶和与无菌 制剂直接接触的敞口包装容器的区域及无菌装配或连 接操作的区域,应当用单向流操作台(罩)维持该区 的环境状态。单向流系统在其工作区域必须均匀送风, 风速为0.36~0.54m/s(指导值)。应当有数据证明单 向流的状态并经过验证。
(二)生产特殊性质的药品,如高致敏性药品(如青 霉素类)或生物制品(如卡介苗或其他用活性微生物 制备而成的药品),必须采用专用和独立的厂房、生 产设施和设备。青霉素类药品产尘量大的操作区域应 当保持相对负压,排至室外的废气应当经过净化处理 并符合要求,排风口应当远离其他空气净化系统的进 风口;
一、新版GMP法规要求
在密闭的隔离操作器或手套箱内,可使用较低的风速。
一、新版GMP法规要求
B级:指无菌配制和灌装等高风险操作A级洁净区所处 的背景区域。
C级和D级:指无菌药品生产过程中重要程度较低操作 步骤的洁净区。
以上各级别空气悬浮粒子的标准规定如下表:
一、新版GMP法规要求
洁净度 悬浮粒子最大允许数/立方米
一、新版GMP法规要求
洁净区微生物监测的动态标 准如下:
洁净度级别 浮游菌 cfu/m3
沉降菌 (φ90mm)
Cfu/4小时
表面微生物
接触( φ55mm ) 5指手套
cfu/碟
Cfu/手套
A <1
<1
<1
<1
B 10
5
5
5
C 100
50
பைடு நூலகம்
25
-
D 200
100
50
-
98版GMP对洁净区的级别要求:
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Do, Study, Act
Sub-System子系统
Plan
Do, Study, Act
Component零部件
Plan计划 Do, Study, Act 做,研究,行动
.
Lower Level Interactions 较低级别的相互作用
.
Systems Approach to Manufacturing
• DV和PV在贯穿Ve系hic统le V模型的系统工程中得到最好的
实施。
System
Sub-System
.
Component
Defining Requirements 定义需求
• The requirements cascade down the left side.
• 需求的逐级传递如左侧
• The cI ascade is an iterative process.
进ign Verification 设计验证
.
Engineering Targets
工程目标
Requirements Cascade 需求逐级传递
• Working in a Systems Engineering context supports
systemic thinking.
• 系统工程中的工作支持系统的考虑 • Opposite of Traditional approach to engineering. • 与传统的工程方法相反
• 根据制造状态,一个产品的功能要符合顾客所期望的方
式,而. 且能以所要求的产量进行生产。
DV&PV Within Systems Engineering
系统工程内的DV&PV
.
Verification and Systemic Thinking
验证和系统思想
Customer Wants 顾客的需要
• 根据设计状态,一个产品的功能要符合顾客所期望的方
式。
• Production Validation:
生产确认
• as manufactured, a product functions in the manner that
the customer expects and can be manufactured at required volumes.
• 验证RA可能包括分析试验和物理试验的组合。
T
反I V
System
复E
计P
划
L A
N
N
I
N
.
G
Sub-System Component
S E Q U
E工
N
T作
I
A顺
L
序
D O I N G
DV & PV and PDSA
Vehicle整车
Plan
Do, Study, Act
System系统
Plan
Applied Consumer Focus
应用顾客的关注
FTEP
福特技术培训项目
DV & PV
设计验证&生产确认
.
Tolerance Design
公差设计
Systems Engineering Fundamentals
系统工程基本原理
FMEA
失效模式后果与分析
Experimental Design
实验设计
Parameter Design 参数设计
Course Structure 课程结构 • Introduction to DV & PV
• 对设计验证及生产确认的介绍 • Tools for DV & PV • 设计验证及生产确认的工具 • Vehicle Level DV • 整车级别的设计验证 • System/Sub-System Level DV • 系统/子系统级别的设计验证 • Component Level DV • 零部件级别的设计验证 • System/Sub-System Level PV • 系统/子系统级别的生产确认 • Component Level PV • 零部件级.别的生产确认 • Summary
.
Design Verification and Production Validation
• Design Verifica设tio计n: 验证和生产确认
设计验证
• as designed, a product will function in the manner that the
customer expects.
Design Verification and
Production Validation 设计验证和 生产确认
.
Design Verification and Production Validation
设计验证和生产确认
.
Benefit to Ford Motor Company 对福特汽车公司的益处
sequential process.
• 验证试验是按照自下至上的顺序过程执行的
• Verification may include a combination of Analytical and
Top Down 自上至下 Bottom Up 自下至上
physTIical tests.
E
Vehicle
.
Systems “V” Model 系统V模型
• DV&PV is a requirements driven process • DV和PV 是一个需求驱动的过程 • DV&PV is best conducted in a systems engineering
context through the systems V.
T
•E
Vehicle
该逐RA级传递是一反复的过程
T
I V
System
E
Top Down 由上到下
P L A N N I N G.
Sub-System Component
Verification Bottom-up 验证自下至上
• Verification testing is implemented as a bottom-up
Welcome
Design Verification and Production Validation . 设计验证和生产确认
Ford Technical Education Program福特技 术培训项目
Global 8D
全球8D
Statistical Engineering
统计工程学
Reliability 可靠性