2010版药典氨基酸检测方法(未施行)
简明氨基酸质量标准-CP2010

氨基酸类药 遮光,密封保存
氨基酸类药 遮光,密封保存
666 盐酸半胱氨酸
741 盐酸组氨酸
796 盐酸赖氨酸
801 盐酸精氨酸
851 胱氨酸
L-2-氨基-3-硫基丙 酸盐酸盐一水合物
C3H7NO2S.HCl.H2O 175.64
(L)2-氨基-3(1H-咪唑-4基)丙 酸盐酸盐一水合物 C6H9N3O2.HCl.H2O
含量 性状
色/状
气味 味道
98.0%-102.0%
≥98.5%
≥99.0%
≥98.5%
白色或类白色结晶性 白色或类白色结晶或 白色至类白色结晶性 白色或类白色结晶或
粉末
结晶性粉末
粉末
结晶性粉末
有类似蒜的臭气, 味酸 有引湿性
无臭 味微酸
无臭 味甜
有香气 味甜
溶解性
在水或乙醇中易溶
在热水中溶解,在水 中微溶,在乙醇中不 溶,在稀盐酸或氢氧 化钠中溶解
在水中易溶,在乙醇 、丙酮或乙醚中几乎 不溶
在水中微溶,在乙醇 中极微溶,在三氯甲 烷中不溶,在甲酸中 易溶,在稀盐酸或氢 氧化钠中溶解
在水中略溶,在乙醇 或乙醚中几乎不溶
在水中溶解,在乙醇 中几乎不溶
+21.0°~ +25.0° 化学方法一 色谱
+14.0°~ +15.6° 色谱
-30.0°~ -32.5° 色谱
5.5~ 6.5 ≥98.0%
5.0~ 6.5 ≥98.0%
≤0.02%
≤0.02% ≤0.02%
≤0.02%
≤0.02% ≤0.02%
≤0.02%
≤0.02% ≤0.02%
≤0.02%
氨基酸含量的测定

氨基酸含量的测定标准曲线绘制准确吸取200ug/ml的氨基酸标准溶液0.0,0.6,0.8,1.0,1.2,1.5,2.0 ml,分别置于25ml容量瓶或比色管中,各加水补充至溶剂为4.0ml,然后加入茚三酮和磷酸缓冲溶液各1ml,混合均匀,于水浴上加热15min,取出迅速冷至室温,再摇匀,加水至标线25ml,摇匀。
静置15min后,在570nm波长下,以试剂空白为参比液夨订其余各溶液的吸光度A。
以氨基酸的微克数为横坐标,吸光度A为纵坐标,绘制标准曲线。
样品的测定:将虾研磨冷却过滤后稀释10倍,吸取澄清的样品溶液1.5ml,平行三次,按标准曲线制作步骤,在相同条件下测定吸光度A 值,用测得的A值在标准曲线上即可查得氨基酸的微克数。
公式:氨基酸总量(ug/100g)=(c/m*1000)*100*10式中c是指从标准曲线上查得的氨基酸的ug数;M是指测定的样品溶液相当于样品的质量g;PH计酸度计测量ph的方法:(1)拿下笔帽(2)按on/off键,机器显示运作(3)将ph计放入待测液中(4)轻轻晃动ph计,保证内气泡逸出,使之于溶液充分接触,勿碰撞杯壁(5)ph计会立即显示数值,将笔置入待测液待数值稳定,30秒内将显示正确数值,(特:ph计数值上下浮动或不稳定是正常现象)(6)按hold键锁定数值,可在待测溶液外记录读取,继续按hold键解除锁定(7)按on/off键关闭ph计(8)轻甩PH计测试笔上多于的水,用蒸馏水或脱离子水冲洗,盖上笔帽测量温度方法在测试模式下,温度数值与ph数值同步显示在液晶面板上,但在校准模式下不显示,数值默认为摄氏温度。
(一)挥发性盐基氮(TVB-N)的测定半微量定氮法(1)原理:蛋白质在酶和细菌的作用下分解后产生碱性含氮物质,有氨、伯胺、仲胺等,此类物质具有挥发性,可在碱性溶液中被蒸馏出来,用标准酸滴定,计算含量。
(2)试剂①氧化镁混悬液(10g/L) 称取1.0g氧化镁,加100ml水,振摇成混悬液。
2010版药典氨基酸检测方法

附录XX 氨基酸分析法氨基酸分析法是指用于测定蛋白质、肽及其他药物制剂的氨基酸组成或含量的方法。
根据氨基酸组成分析可以对蛋白质及肽进行鉴别,氨基酸分析法可用于确定蛋白质、肽及氨基酸的含量,及测定可能存在于蛋白质及肽中的非典型氨基酸。
进行氨基酸分析前,必须将蛋白质及肽水解成单个氨基酸,具体水解方法由各品种项下规定。
蛋白质及肽水解后,其氨基酸分析过程与用于其他药物制剂中游离氨基酸的分析过程相同。
本法包括四种柱前衍生法,分别为异硫氰酸苯酯(PITC)法、6-氨基喹啉-N -羟基琥珀酰亚氨基氨基甲酸酯(AQC)法、邻苯二醛(OPA)和9-芴甲基氯甲酸甲酯(FMOC)法、2,4-二硝基氟苯(DNFB)法,以及一种茚三酮柱后衍生法。
不同的品种应针对自身所含的氨基酸种类及各氨基酸的含量选择适宜的氨基酸分析方法并做相应的方法学验证。
由于本法衍生过程中衍生溶液量较少,且容易挥发,外标法极易出现较大的误差,建议采用内标法进行测定,内标的确定由各品种项下规定。
在本法中,由于半胱氨酸或胱氨酸的衍生产物不稳定,因此对于含半胱氨酸或胱氨酸的样品衍生后应尽快测定,或者在衍生前对半胱氨酸或胱氨酸进行适当的处理,使其转化为稳定地产物(如磺基丙氨酸或半胱氨酸-硫代丙酸)后再衍生测定,具体方法由各品种项下规定。
在测定过程中,可根据所用的仪器、色谱柱品牌、色谱柱的长度及要分离的氨基酸种类,对流动相的有机溶剂和洗脱梯度作适当调整以获得较好的分离度。
第一法PITC柱前衍生氨基酸分析法本法系根据氨基酸与异硫氰酸苯酯(PITC)反应,生成有紫外响应的氨基酸衍生物苯氨基硫甲酰氨基酸(PTC-氨基酸),PTC-氨基酸经反相高效液相色谱分离后用紫外检测,在一定的范围内其吸光值与氨基酸浓度成正比。
本方法的线性浓度范围为0.025~1.25μmol/ml。
试剂(1)流动相A 0.1mol/L醋酸钠溶液(取无水醋酸钠8.2g,加水900ml溶解,用冰醋酸调pH至6.5,然后加水至1000 ml)-乙腈(93:7)。
02 2010年版《中国药典》微生物检查法

11
2010年版微生物限度检查法修订
9、附录109页,修订:供试品检查(法) • (取按验证的方法制备的均匀供试液,)按计数方法的验 证试验确认的程序进行供试液制备。用pH7.0无菌氯化钠蛋白胨缓冲液稀释成1∶10、1∶102、1∶103等稀释级。 • 1.平皿法 • (采用平皿法进行菌数测定时,应取适宜的连续2~3个稀 释级的供试液。)
10
2010年版微生物限度检查法修订
8、附录109页,增加: • 若没有适宜的方法消除供试品中的抑菌作用,那么验证试验中微生物 回收的失败可看成是因供试品的抗菌活性引起的,同时表明该供试品 不能被试验菌污染。然而,供试品也可能仅对试验用菌株具有抑制作 用,而对其他菌株没有抑制作用。因此,根据供试品须符合的微生物 限度标准和菌数报告规则,在不影响检验结果判断的前提下,应采用 能使微生物生长的更高稀释级的供试液进行方法验证试验。若验证试 验符合要求,应以该稀释级供试液作为最低稀释级的供试液进行供试 品检验。 • 计数方法验证时,若采用上述计数方法总存在一株或多株试验菌的回 收率达不到要求,那么选择回收情况最接近要求的方法和试验条件进 行供试品的检测。
16
二、培养基适用性实验的实施与操作
• 1.计数培养基的适用性检查
(一)应用范围;细菌、霉菌及酵母菌计数用的培养基应进行培养基的适 用性检查,包括成品培养基、由脱水培养基或按培养基处方配制的培 养基均应检查。 (二)菌种及菌液制备 验证试验所用的菌株传代次数不得超过5代(从 菌种保存中心获得的冷冻干燥菌种为第0代),并采用适宜的菌种保 藏技术,以保证试验菌株的生物学特性。 大肠埃希菌(Escherichia coli)〔CMCC(B) 44 102〕 金黄色葡萄球菌(Staphylococcus aureus)〔CMCC(B) 26 003〕 枯草芽孢杆菌(Bacillus subtilis)〔CMCC(B) 63 501〕 白色念珠菌(Candida albicans)〔CMCC(F) 98 001〕 黑曲霉(Aspergillus niger)〔CMCC(F) 98 003〕 菌液制备同微生物限度检查方法学验证
氨基酸检测方法药典

氨基酸检测方法药典
氨基酸的检测方法在药典中有多种,其中包括电位滴定法、高效液相色谱法等。
具体方法的选择取决于氨基酸的种类和检测要求。
对于某些氨基酸,如酪氨酸和组氨酸,药典中提供了详细的电位滴定法进行含量测定。
这种方法需要精密称取一定量的样品,溶解在特定的酸中,然后使用高氯酸滴定液进行滴定,并通过空白试验校正结果。
另外,高效液相色谱法也被广泛应用于氨基酸的检测。
例如,可以采用Waters C18色谱柱,以磷酸二氢钾溶液-甲醇为流动相,在280nm波长下进行检测。
这种方法可以高效、精准地检测酪氨酸的含量。
对于组氨酸的检测,除了电位滴定法外,还可以采用其他方法如茚三酮法、Pauly显色法、伏安法等,或者采用实验室组装的离子交换色谱分析仪和柱后衍生的高效液相色谱法等。
以上信息仅供参考,如需获取更准确的信息,建议查阅药典或咨询相关专家。
关于实施《中国药典》2010年版有关事宜的公告

关于实施《中国药典》2010年版有关事宜的公告国家食品药品监督管理局公告2010年第43号关于实施《中国药典》2010年版有关事宜的公告《中华人民共和国药典》2010年版(以下简称中国药典)已由卫生部2010年第5号公告颁布,自2010年10月1日起执行。
现就实施中国药典的有关事宜公告如下:一、中国药典包括凡例、正文及附录,是药品研制、生产、经营、使用和监督管理等均应遵循的法定依据。
所有国家药品标准应当符合中国药典凡例及附录的相关要求。
二、凡中国药典收载的品种,自执行之日起,原收载于历版药典、卫生部颁布药品标准、国家食品药品监督管理局颁布新药转正标准和地方标准上升国家标准的同品种药品标准同时废止。
药品注册标准不符合中国药典有关要求的,药品生产企业应按《药品注册管理办法》的有关规定提出补充申请。
对于药品注册标准中收载的检验项目多于中国药典规定的或质量指标高于中国药典要求的,在执行中国药典的基础上,应同时执行原标准的相应项目和指标。
中国药典品种项下未收载的制剂规格,其质量标准按中国药典同品种相关要求执行,规格项按原批准证明文件执行。
三、药品生产企业应根据中国药典的增修订内容,按照我局相关规定及程序变更药品说明书和标签。
2010年10月1日起生产的药品必须使用变更后的说明书和标签。
对于通用名称已作修订的药品,其原名称可作为曾用名过渡使用。
四、中国药典所收载的相同品种,如含有中国药典规定以外的杂质,应当增加杂质控制项目。
五、中国药典关于眼用制剂无菌要求的具体执行时间将根据《药品生产质量管理规范》实施的要求另行规定。
六、药品生产企业应积极做好执行中国药典有关准备工作,对在中国药典执行中发现的问题应及时报所在地省级食品药品监督管理局。
同时应不断加强质量标准研究,提高药品质量控制水平。
七、各级地方食品药品监督管理部门应配合做好中国药典的宣贯工作,加强中国药典执行中的监督与指导,及时收集和反馈相关问题和意见。
八、国家药典委员会负责中国药典执行中的具体指导等有关工作。
氨基酸测定方法
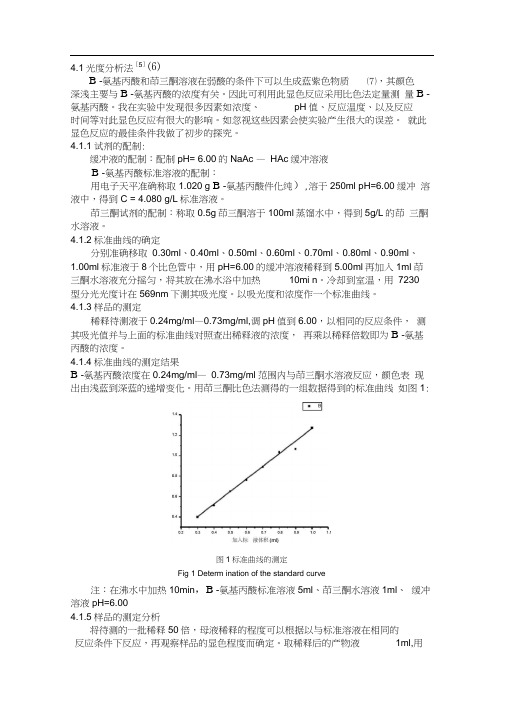
4.1光度分析法[5]⑹B -氨基丙酸和茚三酮溶液在弱酸的条件下可以生成蓝紫色物质⑺,其颜色深浅主要与B -氨基丙酸的浓度有关。
因此可利用此显色反应采用比色法定量测量B -氨基丙酸。
我在实验中发现很多因素如浓度、pH值、反应温度、以及反应时间等对此显色反应有很大的影响。
如忽视这些因素会使实验产生很大的误差。
就此显色反应的最佳条件我做了初步的探究。
4.1.1试剂的配制:缓冲液的配制:配制pH= 6.00的NaAc —HAc缓冲溶液B -氨基丙酸标准溶液的配制:用电子天平准确称取1.020 g B -氨基丙酸件化纯),溶于250ml pH=6.00缓冲溶液中,得到C = 4.080 g/L标准溶液。
茚三酮试剂的配制:称取0.5g茚三酮溶于100ml蒸馏水中,得到5g/L的茚三酮水溶液。
4.1.2标准曲线的确定分别准确移取0.30ml、0.40ml、0.50ml、0.60ml、0.70ml、0.80ml、0.90ml、1.00ml标准液于8个比色管中,用pH=6.00的缓冲溶液稀释到5.00ml再加入1ml 茚三酮水溶液充分摇匀,将其放在沸水浴中加热10mi n。
冷却到室温,用7230型分光光度计在569nm下测其吸光度。
以吸光度和浓度作一个标准曲线。
4.1.3样品的测定稀释待测液于0.24mg/ml—0.73mg/ml,调pH值到6.00,以相同的反应条件,测其吸光值并与上面的标准曲线对照查出稀释液的浓度,再乘以稀释倍数即为B -氨基丙酸的浓度。
4.1.4标准曲线的测定结果B -氨基丙酸浓度在0.24mg/ml—0.73mg/ml范围内与茚三酮水溶液反应,颜色表现出由浅蓝到深蓝的递增变化。
用茚三酮比色法测得的一组数据得到的标准曲线如图1:图1标准曲线的测定Fig 1 Determ ination of the standard curve注:在沸水中加热10min,B -氨基丙酸标准溶液5ml、茚三酮水溶液1ml、缓冲溶液pH=6.004.1.5样品的测定分析将待测的一批稀释50倍,母液稀释的程度可以根据以与标准溶液在相同的反应条件下反应,再观察样品的显色程度而确定。
《中国药典》2010年版(二部)

283
158 23 1871
261 / 15
144 / 24 0 1448
92.2%
91.1% 0 77.4%
化学药中由于未找到样品而未修订的品种有306个,占保留上版品种21.8%
2010年版与2005年版药典主要项目收载情况比对表
增修订项目 红外光谱鉴别 有关物质 残留溶剂 渗透压摩尔浓度 溶出度或释放度 含量均匀度 无菌检查方法 细菌内毒素 含量测定 HPLC法 原料 制剂 HPLC方法 2005年版 530 2 142 24 4 315 165 107 216 359 2010年版 580 73 707 97 45 414 219 132 372 694
依法进行该项检查外,其他未在“残留溶剂”项下明确列出的有机溶
剂与未在正文中列有此项检查的品种,如生产过程中引入或产品中残 留有机溶剂,均应按本版药典附录“残留溶剂测定法”检查并应符合
相应溶剂的限度规定。
主要内容
1 3 2 3 4 5 3 二部特点及品种收载情况
凡例的增修订情况
各论的增修订情况 现代分析技术的应用
脏器均应来自经检疫的健康动物,涉及牛源的应取自无牛海绵状脑病
地区的健康牛群;来源于人尿提取的药品,均应取自健康人群。上述 药品均应有明确的病毒灭活工艺要求以及质量管理要求。
凡例的增修订情况
项目与要求
• 十七、(第二段)对于生产过程中引入的有机溶剂,应在后续的生产 环节予以有效去除。除正文已明确列有“残留溶剂”检查的品种必须
Байду номын сангаас
应关注晶型和药效的关系。如确需利用熔点作为控制晶型的手段,则 标准中应收入。
各论的增修订情况(1):名称与性状
比旋度(原料药)
2010年版_中华人民共和国兽药典_三部增修订概况_杨京岚

2010年版《中华人民共和国兽药典》三部增修订概况杨京岚1,王占锋2,董义春1(1.中国兽医药品监察所,北京100081;2.北京中海生物科技有限公司,北京100081)[收稿日期]2011-07-11[文献标识码]C [文章编号]1002-1280(2011)08-0029-04[中图分类号]S859.2[摘要]针对农业部颁布实施的2010年版《中华人民共和国兽药典》三部的增修订总体情况进行了概述,除简单对新版兽药典收载原则和收载情况进行介绍外,重点对新版兽药典增修定的特点和要点等进行描述,以便广大使用者了解和掌握新版兽药典三部与2005年版兽药典三部之间的总体差异及技术标准变化等。
[关键词]中华人民共和国兽药典;兽用生物制品;标准;增修订作者简介:杨京岚(1972年-),女,副研究员,研究兽用生物制品注册管理及其国家标准。
E -mail :yangjinglan@ivdc.gov.cnRevisions of the Veterinary Pharmacopoeia ofthe People ’s Republic of China (2010Edition )Volume IIIYANG Jing -lan 1,WANG Zhan -feng 2,DONG Yi -chun 1(1.China Institute of Veterinary Drug Control ,Beijing 100081,China ;2.Beijing Zhonghai Biotech Co.,Ltd ,Beijing 100081,China )Abstract :This article described the revisions of the Veterinary Pharmacopoeia of PRC (2010Edition )Volume III ,which has been published by Ministry of Chinese Agriculture.Except for making a summary of the principle and the general situation of being recorded by the new Veterinary Pharmacopoeia ,this article introduced the revisions of the characteristics and key points importantly.These will help users to understand and get hold of the differences between the Veterinary Pharmacopoeia of 2010Edition and 2005Edition.Key words :the Veterinary Pharmacopoeia of PCR ;biological products ;standards ;revisions根据农业部公告1521号,2010年版《中华人民共和国兽药典》(以下简称《中国兽药典》)三部已于2010年12月颁布,并自2011年7月1日起施行。
中国药典2010年版中成药标准的介绍

药味改用饮片名表述 例如:香附(醋制) →醋香附 例如:香附(醋制) 麦芽( 麦芽(炒) →炒麦芽 百部(蜜炙) 百部(蜜炙) →蜜百部 山楂( 山楂(焦) →焦山楂 药典未收载的炮制品仍沿用原名称 例如:杜仲叶(盐炙) →杜仲叶(盐炙) 例如:杜仲叶(盐炙) 杜仲叶(盐炙) 处方中的炮制品如药典未收载的特殊炮制方法将 附在该品种项下
4
技术要求
中药成分复杂, 中药成分复杂,应根据所含成分的化学 性质选择适宜的专属性方法。 性质选择适宜的专属性方法。对于不宜达 到专属性要求的一般理化鉴别、 到专属性要求的一般理化鉴别、荧光鉴别 及光谱鉴别,一般不宜采用。 及光谱鉴别,一般不宜采用。 检查项主要包括安全性、有效性、 检查项主要包括安全性、有效性、均一 性与纯度要求四个方面, 性与纯度要求四个方面,应根据中药制剂 的具体情况,研究建立合理的检查项目。 的具体情况,研究建立合理ቤተ መጻሕፍቲ ባይዱ检查项目。 5
气相色谱法
中药指纹图谱分析方法的建立应 中药指纹图谱分析方法的建立应能体现中药的 整体特征
在满足表征中药化学成分群整体性质的前提下, 在满足表征中药化学成分群整体性质的前提下,要求 有较好的重现性。明确了指纹图谱认证、方法验证、 有较好的重现性。明确了指纹图谱认证、方法验证、 数据处理及计算分析的依据和方法。 数据处理及计算分析的依据和方法。
应选择专属性成分、 应选择专属性成分、活性成分作为含量测定的 指标
避免选择无专属性的指标成分、 避免选择无专属性的指标成分、低活性的微量成分或水 解产物作为测定指标。 解产物作为测定指标。当单一成分不能反映该药的整体 活性时,应采用多成分或多组分的检测方法。 活性时,应采用多成分或多组分的检测方法。
14
1、鉴别项 新增各项鉴别 、鉴别项--新增各项鉴别 新增各项鉴别2165项 项
2024版药典氨基酸检测方法

2024版药典氨基酸检测方法2024年版药典对氨基酸的检测方法尚未施行,因此在这篇文章中我们将着重介绍氨基酸的分析技术和方法。
氨基酸是构成蛋白质的基本单元,对于新药研发、蛋白质工程以及食品营养分析等领域来说,准确、快速、灵敏的氨基酸分析是非常重要的。
目前,氨基酸的分析方法主要包括色谱法、电泳法、质谱法和光谱法等。
色谱法是目前最常用的氨基酸分析方法之一、常见的色谱法包括高效液相色谱法(HPLC)和气相色谱法(GC)。
HPLC是一种基于液相色谱技术的分析方法,其优势在于可以同时测定多种氨基酸,并且具有准确度高、分析时间短等特点。
但是,HPLC的分离介质和流动相对氨基酸的分析结果有重要的影响,因此需要选用合适的HPLC柱和优化流动相的组成。
GC则是基于气相色谱技术的分析方法,其主要优势在于对样品的洗脱和检测具有较高的选择性。
但是,GC对于热敏性和易挥发性氨基酸的分析效果较好,而对于其他非极性或极性氨基酸的分析效果较差。
电泳法又被称为毛细管电泳和聚丙烯酰胺凝胶电泳,是基于电场作用下分离氨基酸的分析方法。
电泳法具有在线分离和检测、分离速度快、耗样量小等优点。
但是,由于电泳法对于氨基酸的分离度和灵敏度较低,因此电泳法常常与质谱法和光谱法结合使用,以提高分析的准确性和灵敏度。
光谱法是一种利用分子吸收、发射或散射光谱进行分析的方法。
氨基酸的紫外吸收光谱是其分析中最常见的光谱。
通过分析氨基酸在特定波长下的吸光度,可以定量测定其浓度。
另外,红外光谱和核磁共振光谱也可以用于氨基酸的结构和含量分析。
需要注意的是,以上介绍的氨基酸分析方法在样品前处理和仪器操作中都需要一定的专业知识和技能。
此外,针对特殊要求,也有一些新的技术正在不断发展和研究中,如基于表面增强拉曼光谱(SERS)的氨基酸检测方法和基于纳米孔技术的单分子氨基酸测定方法等。
综上所述,虽然2024年版药典对氨基酸的检测方法尚未施行,但是已有多种快速、准确、灵敏的氨基酸分析方法在实际应用中得到广泛的应用。
4氨基酸、多肽、蛋白质类药品检验

仪
碘量法
原理:半胱氨酸分子中含有-SH, 在酸性
条件下,与过量的碘作用,剩余的碘用硫代 硫酸钠溶液滴定。由硫代硫酸钠溶液所消耗 的量,间接求出硫化物的含量。
注意:滴定条件,指示剂,碘滴定液,
Na2S2O3 标液配制
碘量法的背景知识 (一) 方法简介 碘量法也是常用的氧化还原滴定方法之一。它 是以I2的氧化性和I-的还原性为基础的滴定分析法。 因此,碘量法的基本反应是 I 2 +2e - =2I - E IO/I- =0.535 V
定氮法
样品与浓硫酸共热,含 氮有机物即分解产生氨 (消化),氨又与硫酸 作用,变成硫酸氨。经 强碱碱化使之分解放出 氨,借蒸汽将氨蒸至酸 液中,根据此酸液被中 和的程度可计算得样品 之氮含量。
4.碘量法或溴量法 5.HPLC法
示例一:盐酸 示例一:三氨
基酸注射液-341 半胱氨酸水合物 的测定 示例二:六氨 示例一:L-胱氨 基酸注射液400 酸的测定 6.氨基酸自动分析
效价单位U
效价:指某一物质引起生物反应的功效单位,可用理 化方法检测,也可用生物检测方法测定,通常以重量 单位或效价单位来计量。不同的药物有各自的效价 的定义。
某些生化药物,其药物中含有一些杂质,不可能是 纯品。故不能以重量单位准确表示其含量,只能依 靠生物检定的方法与标准品进行比较来测定药物的 效价剂量。因此,采用特定的“单位”——“U”来 计量。产生相同效应药品的剂量比较时,所需剂量 越小,药物的效价就越高,反之效价就低。
第4章
氨基酸、多肽、蛋白质类 药品检验
易庆平
2010年5月6日
氨基酸类药物含量测定常用的方法 1.酸碱滴定法
示例1:谷氨酸的
2.非水溶液滴定法
2010版中国药典三部修改

欧洲药典
2、欧洲药典(EP)
《欧洲药典》由欧洲药品质量管理局(EDQM)下 属的职能机构欧洲药典委员会(European Pharmacopoeia Commission)出版。欧洲药典委员 会1964年成立。欧洲药典是欧洲药品质量控制标 准。已有多项法律使欧洲药典成为法定标准。 目前采用《欧洲药典》的国家已达28个,除 欧共体成员国和其他欧洲国家外,还有亚洲的土 耳其和塞浦路斯。
国际药典
《国际药典》的历史可以追溯到1874年, 由于当时需要将术语标准化以及明确药物 的剂量和成分,从而产生了制订一部国际 药典纲要的尝试。 《国际药典》1951年出第一版, 1967年出第二版, 1979年出第三版第一部,其第 二、三、四部分于1981年、1988年和1994 年出版,2003年出第五部。 2006年出第四版。
欧洲药典
《欧洲药典》最大的特点是其各论中只收载原 料药质量标准,不收载制剂质量标准。除此以外, 《欧洲药典》的附录也独具特色,《欧洲药典》 收载的附录,不仅包括各论中通用的检测方法, 而且凡是与药品质量密切相关的项目和内容在附 录中都有规定。另外,在收载的附录中,除了采 用通用的检测方法外,收载的先进技术也比较多, 如原子吸收光谱、原子发射光谱、质谱、核磁共 振谱和拉曼光谱测定法等,对色谱法还专门设立 一项色谱分离技术附录。从整体上看,《欧洲药 典》的附录是至今世界药典中最全面、最完善, 也是最先进的。
英国药典
《英国药典》与《欧洲药典》的关系
《英国药典》与《欧洲药典》有密切的关系。 按照惯例,《欧洲药典》中的全部各论与附录都收 录在相应版本的《英国药典》中。这些内容一般不 作任何编辑修改,只在必要的情况下,增加《英国 药典》相应的用法要求。
日本药典
《中国药典》2010年版一部凡例和中成药及相关标准增修订内容

1
1977版药典收载显微鉴别 1985版药典收载TLC鉴别 1990版药典收载对照药材的TLC鉴别和色谱 方法的含量测定 2000版药典建立了以色谱法含量测定为主 导的质控方法 2005版药典大幅度增加HPLC方法,重视特 征成分、活性成分的测定 2010版药典加强了新技术新方法的应用,加 强活性成分、多成分的测定,增加了指纹 图谱的测定,从整体上提高中药质量控制 水平
12
8、计量:规定了计量单位、符号及正文中各类温度、 浓度等的具体范围。 本版药典使用的滴定液和试液的浓度,以mol/L(摩 尔/升)表示者,其浓度要求精密标定的滴定液用 “XXX滴定液(YYYmol/L)”表示;作其他用途不需 精密标定其浓度时,用“YYYmol/LXXX溶液”表 示,以示区别。 溶液后记示的“(1→10)”等符号,系指固体溶质 1.0g或液体溶质1.0ml加溶剂使成10ml的溶液;未指 明用何种溶剂时,均系指水溶液;两种或两种以上液 体的混合物,名称间用半字线“-”隔开,其后括 号内所示的“:”符号,系指各液体混合时的体积 (重量)比例。
22
10
原料药的含量(%),除另有注明者外,均按重量计。 如规定上限为100%以上时,系指用本药典规定的分 析方法测定时可能达到的数值,它为药典规定的限 度或允许偏差,并非真实含有量;如未规定上限时, 系指不超过101.0%。 制剂的含量限度范围,系根据主药含量的多少、测 定方法、生产过程和贮存期间可能产生的偏差或变 化而制定的,生产中应按标示量100%投料。如已 知某一成分在生产或贮存期间含量会降低,生产时 可适当增加投料量,以保证在有效期(或使用期限) 内含量能符合规定。
14
《中国药典》2010年版培训试题汇总

2010年版药典培训考核试题一、填空题1、《中国药典》附录主要收载、、。
2、2010年版药典大幅度增加专属性鉴别,基本结束的历史。
3、片剂包衣必要的时候要检查。
4、新版药典全面禁用作溶剂。
工艺有使用有机溶剂的均检查。
5、新版药典明确了生产投料及入药形式均为。
6、新版药典从正式实施,其同品种的其他标准同时。
7、对于生产过程中引入的,应在后续的生产环节中有效去除。
8、对照品应按其使用说明书上规定的方法处理后按使用。
9、生物活性是以药物的为基础,以生物统计为工具,运用特定的实验设计,测定药物的一种方法。
10、HPLC法流动相宜选用流动相,尽量不加。
二、单选题1、一部药典中,片剂测崩解时限时()A、加挡板B、不加挡板C、可加可不加挡板2、下列关于2010版中国药典二部溶出度测定法(附录X C)描述不正确的是()A、溶出度测定法第一法(篮法)取样位置应在转篮顶端至液面中点,距溶出液内壁10mm处。
B、实际取样时间与规定时间的差异不得过±2%C、供试品溶液取样至滤过应在30分钟内完成D、结果判定:6片(粒、袋)中,如有1~2片(粒、袋)低于Q,但不低于Q-10%,且平均溶出量不低于Q,可判定为符合规定。
3、单一成分制剂含量如未规定上限时,应不超过()A、100.0%;B、101.0%;C102.0%;D105.0%4、下列对“重金属检查法”说法不正确的是()A、本法所指的重金属系指在规定试验条件下能与硫代乙酰胺或硫化钠作用显色的金属杂质B、制备1L贮备液称取硝酸铅的量为0.1599gC、对配置与贮存用的玻璃容器没有要求D、供试品若含高铁盐影响检查时,可在甲乙丙三管中分别加等量的维生素C三、判断题1、附录的指导原则系为执行药典、考察药品质量、起草与复核药品标准等所制定的指导性规定。
()2、比旋度与旋光度是一样的。
()3、制备纯化水应严格监测各生产环节,防止微生物污染,确保使用点的水质。
()4、影响因素用一批原料药或一批制剂进行试验。
生物药物分析与检验氨基酸、多肽和蛋白质类药品检验

• 掌握氨基酸定性鉴别的方法,熟悉氨基酸特殊杂质及 安全性检查,掌握氨基酸含量测定方法,掌握多肽因 子类药物的定性鉴别方法,熟悉多肽因子类药物的检 查方法,掌握多肽因子类药物生物效价的测定方法, 掌握蛋白质类药物的鉴别和检查方法,掌握蛋白质含 量测定和效价测定方法,了解几种氨基酸、多肽因子 和蛋白质药物的质量分析过程。
种天然氨基酸中,只有酪氨 酸、色氨酸和苯丙氨酸在紫 外区有最大吸收。 • 酪氨酸的max=275nm • 苯丙氨酸的max=257nm • 色氨酸的max=280nm
第7页,共42页。
第一节 氨基酸类药品检验
• 这三种氨基酸可以通过紫外 吸收光谱加以鉴别。
• 精密称取酪氨酸、色氨酸、 苯丙氢酸各适量。
第22页,共42页。
第二节 多肽因子类和蛋白质类药品 检验
一、多肽因子类药物的定性鉴别 • 基因重组多肽因子类药物的鉴别主要采用SDS-PAGE
法和免疫印迹法。 1、SDS-PAGE法 • 用SDS-PAGE鉴别生物样品必须是该品有极纯的标准
对照品,通过电泳结果的对比,确定所测样品是否与 标准品有相同的迁移率,从而鉴别该品。 • 缺点:必须有标准品;凝胶中多肽需保留生物活性, 或能够复性,或电泳前可将检测配基与待测多肽共价 交联。
• 所得的吸收图谱各主要吸收峰波长和各吸收峰间的相 互强度关系均应与对照的图谱一致。
第9页,共42页。
第一节 氨基酸类药品检验
3、薄层色谱鉴别法 • 在薄层板上点样,通过与标准氨基酸对照而鉴别氨基
酸。 (1)薄层板的制备
5g微晶纤 维 2 m 0 无 L 素 水 2 样 . 5 乙 m 水 L 醇 品 , 拌 成 糊状 均 匀 涂 溶 板 剂 挥 发 1 完 0 C 5 ,1 毕 . 5h 后 待用
氨基酸测定的方法_下_

%色氨酸= M色氨酸×186.1 毫克蛋白质/毫升溶液
%酪氨酸= M酪氨酸×163.1 毫克蛋白质/毫升溶液
如表 2 氨基酸的一些物理常数表如: 色氨酸 残基分子量=186.1, 酪氨酸残基分子量=163.1。
原理: 精氨酸除去胍乙酸后, 在碱性环境中可 与 α—萘酚及次溴酸钠产生颜色反应。 9.2.1 试剂
0.3% NaCl 液 ; α—萘 酚 及 尿 素 液 ( 在 无 水 CH3CH2OH 中 的 α—萘 酚 液 , 临 用 前 以 4 体 积 10%尿 素 液 冲 淡); 次 溴 酸 钠 液 (0.66mL 溴 溶 于 100mL5%NaOH 液中)。 9.2.2 操作
盐 酸 磷 酸 混 合 液 : 9 体 积 浓 HCl 与 1 体 积 H3PO4 相混合。 6.2 操作
1mLl4.3N NaOH 液, 取 1mL 含 1%蛋氨酸液, 0.3mL 新配制的 10%亚硝基铁氰化钠液, 混合后 加 入 5mL 待 测 液 中 , 将 此 试 管 置 于 已 热 至 35 ̄ 40℃的 水 浴 上 经 5 ̄10min, 然 后 在 冰 水 中 冷 却 2min, 一面混合一面加入 5mL 盐酸磷酸混合液, 再动 1min, 然后用水在室温下冷却 5~10min, 用 540mμ滤光片进行比色测定光密度。
将它放置于 4℃水浴中以供凝结醋酸蒸气之用。 5.2.5 粒状 NaOH
—48—
以收集最后残余的醋酸。 5.2.6 显色管
内 盛 15mL 浓 H2SO4 及 50~100mg 纯 对 二 羟 联苯, 将试管置于 0℃之冰水浴中, 全部时间均进 行空气抽滤。 5.3 操作
氨基酸的测定

酱油稀释液在加入甲醛后滴定至pH9.2所用NaOH标准溶液的体积V1=9.12mL
空白滴定在加入甲醛后滴定至pH9.2所用NaOH标准溶液的体积V2=1.36mL
NaOH标准溶液的浓度C=0.0498mol/L
根据电位滴定法,计算氨基酸态氮含量
V2---空白滴定在加入甲醛后滴定至pH9.2所用NaOH标准溶液的体积mL
C --- NaOH标准溶液的浓度mol/L
m ---吸取的酱油的质量g
0.014 ---氮的毫摩尔质量g/m mol
举例讲解:同时复习检验评价报告的写法。
现抽取南宁酱油厂生产的塑料袋装铁鸟牌酱油1袋(规格500mL)(生产日期20050520),要求测定氨基酸态氮含量。
②氨基酸的滴定
在上述滴定至pH8.2的溶液中加入10.00mL的中性甲醛溶液,再用NaOH标准溶液滴定至pH9.2,记下消耗的NaOH溶液体积。
③空白滴定
吸取80mL蒸馏水于250mL的烧杯中,用NaOH标准溶液滴定至pH8.2,然后加入10.00mL中性甲醛溶液,再用NaOH标准溶液滴定至pH9.2,记下加入甲醛后消耗的NaOH溶液体积。
可以看出0.36%与其它各次结果偏离太远,应为测定可疑值。
X2-X10.48%-0.36%
Q===0.86>0.64
Xn – X10.52%-0.36%
∵Q0.90=0.64∴0.36%测定结果值应舍去,应用其它四次测定值计算。
测定结果平均值X=(0.48%+0.49% +0.50%+0.52%)/4 = 0.50%(0.4975%)
(V1- V2)× C × 0.014
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
2010年版新增的“氨基酸检测方法”如下:新增附录附录XX 氨基酸分析法氨基酸分析法是指用于测定蛋白质、肽及其他药物制剂的氨基酸组成或含量的方法。
根据氨基酸组成分析可以对蛋白质及肽进行鉴别,氨基酸分析法可用于确定蛋白质、肽及氨基酸的含量,及测定可能存在于蛋白质及肽中的非典型氨基酸。
进行氨基酸分析前,必须将蛋白质及肽水解成单个氨基酸,具体水解方法由各品种项下规定。
蛋白质及肽水解后,其氨基酸分析过程与用于其他药物制剂中游离氨基酸的分析过程相同。
本法包括四种柱前衍生法,分别为异硫氰酸苯酯(PITC)法、6-氨基喹啉-N-羟基琥珀酰亚氨基氨基甲酸酯(AQC)法、邻苯二醛(OPA)和9-芴甲基氯甲酸甲酯(FMOC)法、2,4-二硝基氟苯(DNFB)法,以及一种茚三酮柱后衍生法。
不同的品种应针对自身所含的氨基酸种类及各氨基酸的含量选择适宜的氨基酸分析方法并做相应的方法学验证。
由于本法衍生过程中衍生溶液量较少,且容易挥发,外标法极易出现较大的误差,建议采用内标法进行测定,内标的确定由各品种项下规定。
在本法中,由于半胱氨酸或胱氨酸的衍生产物不稳定,因此对于含半胱氨酸或胱氨酸的样品衍生后应尽快测定,或者在衍生前对半胱氨酸或胱氨酸进行适当的处理,使其转化为稳定地产物(如磺基丙氨酸或半胱氨酸-硫代丙酸)后再衍生测定,具体方法由各品种项下规定。
在测定过程中,可根据所用的仪器、色谱柱品牌、色谱柱的长度及要分离的氨基酸种类,对流动相的有机溶剂和洗脱梯度作适当调整以获得较好的分离度。
第一法PITC柱前衍生氨基酸分析法本法系根据氨基酸与异硫氰酸苯酯(PITC)反应,生成有紫外响应的氨基酸衍生物苯氨基硫甲酰氨基酸(PTC-氨基酸),PTC-氨基酸经反相高效液相色谱分离后用紫外检测,在一定的范围内其吸光值与氨基酸浓度成正比。
本方法的线性浓度范围为0.025~1.25µmol/ml。
试剂(1)流动相A 0.1mol/L醋酸钠溶液(取无水醋酸钠8.2g,加水900ml溶解,用冰醋酸调pH至6.5,然后加水至1000 ml)-乙腈(93:7)。
(2)流动相B 乙腈-水(8:2)。
对照品溶液按各品种项下规定的方法制备。
供试品溶液按各品种项下规定的方法制备。
色谱条件与系统适用性试验用十八烷基硅烷键合硅胶为填充剂(4.6×250mm,5μm);流速为每分钟1.0ml;柱温为40℃;检测波长为254nm。
各氨基酸峰间的分离度均应大于1.0。
洗脱梯度如下:时间(min) 流动相A(%)流动相B(%)0 100 014 85 1529 66 3430 0 10037 0 10037.1 100 045 100 0测定法精密量取氨基酸对照品溶液200μl,置一2ml塑料离心管中,精密加入1mol/L三乙胺乙腈溶液100μl,混匀,精密加入0.1mol/L异硫氰酸苯酯乙腈溶液100μl,混匀,室温放置1小时,加0.8ml正己烷,剧烈振摇,放置10min,精密取下层溶液2μl,注入液相色谱仪,记录色谱图;另精密量取供试品溶液200μl,自“置一2ml塑料离心管中”起同法测定。
第二法AQC柱前衍生氨基酸分析法本法系根据氨基酸与6-氨基喹啉-N-羟基琥珀酰亚氨基氨基甲酸酯(AQC)反应,生成有紫外与荧光响应的不对称尿素衍生物(AQC-氨基酸),AQC-氨基酸经反相高效液相色谱后用紫外或荧光检测,在一定的范围内其吸光值与氨基酸浓度成正比。
本方法的线性浓度范围为 2.5~200nmol/ml。
试剂(1)流动相A 取醋酸铵10.8g或无水醋酸钠11.5g,加水900ml溶解,用磷酸调pH 至5.0,然后加水至1000 ml。
(2)流动相B 乙腈-水(3:2)。
(3)0.4 mol/L 硼酸盐缓冲液(pH 8.8) 取硼酸12.36g,加水400ml溶解,用40%氢氧化钠溶液调pH至8.8,然后加水稀释至500ml。
(4) AQC溶液取AQC适量,加乙腈溶解并稀释制成每1ml中含1mg的溶液。
对照品溶液按各品种项下规定的方法制备。
供试品溶液按各品种项下规定的方法制备。
色谱条件与系统适用新试验用十八烷基硅烷键合硅胶为填充剂(4.6×250mm,5μm);流速为每分钟1.4ml;柱温为37℃;检测波长为248nm。
各氨基酸峰间的分离度均应大于1.0。
洗脱梯度如下:时间(min) 流动相A(%)流动相B(%)0 88 1214 88 1229 80 2030 59 4137 59 4137.1 88 1245 88 12测定法精密量取对照品溶液10μl,置一直径为0.4cm、高度为5cm的小试管中,精密加入0. 4 mol/L 硼酸盐缓冲液(pH 8.8) 70μl,在涡旋混匀器上混匀,精密加入AQC溶液20μl,混匀,精密量取5μl,注入液相色谱仪,记录色谱图;另精密量取供试品溶液10μl,自“置一直径为0.4cm”起同法测定。
第三法OPA和FMOC柱前衍生氨基酸分析法本法系根据一级氨基酸,在巯基试剂存在下,首先与邻苯二醛(OPA)反应,生成OPA-氨基酸;反应完毕后,加入9-芴甲基氯甲酸甲酯(FMOC),剩余的二级氨基酸与FMOC继续反应,生成FMOC-氨基酸,两次反应生成的氨基酸衍生物经反相高效液相色谱分离后用紫外或荧光检测,在一定的范围内其吸光值与氨基酸浓度成正比。
本方法的线性浓度范围为0.025~2.5µmol/ml。
试剂(1)流动相A 称取醋酸钠7.5g,加水4000ml溶解,加三乙胺800μl,四氢呋喃24ml,混匀,用2%醋酸调pH至7.2。
(2)流动相B 称取醋酸钠10.88g,加水800ml溶解,用2%醋酸调pH至7.2,加乙腈1400ml,甲醇1800ml,混匀。
(3)0.4 mol/L 硼酸盐缓冲液(pH 10.4) 取硼酸24.73g,加水800ml溶解,用40%氢氧化钠溶液调pH至10.4,然后加水稀释至1000ml。
(4)OPA溶液取OPA 80mg,加0.4 mol/L 硼酸盐缓冲液(pH 10.4) 7ml,加乙腈1ml,3-巯基丙酸125μl ,混匀。
(5)FMOC溶液取FMOC 40mg , 加乙腈8ml溶解。
对照品溶液按各品种项下规定的方法制备。
供试品溶液按各品种项下规定的方法制备。
色谱条件与系统适用性试验用十八烷基硅烷键合硅胶为填充剂(4.6×150mm,5μm);柱温为40℃;检测波长为338nm(一级氨基酸),262nm(二级氨基酸)。
各氨基酸峰间的分离度均应大于1.0。
洗脱梯度及流速如下:时间(min) 流动相A(%)流动相B(%)流速(ml/min)0.0 100 0 1.017.0 50 50 1.045.0 0 100 1.045.1 0 100 1.550.0 0 100 1.550.1 100 0 1.053 100 0 1.0测定法精密量取对照品溶液50μl,置一1.5ml塑料离心管中,精密加入0. 4 mol/L 硼酸盐缓冲液(pH 10.2) 250μl,混匀,精密加OPA衍生剂50μl,混匀,放置30秒,精密加入FMOC 衍生剂50μl,混匀,精密量取4μl,注入液相色谱仪,记录色谱图;另精密量取供试品溶液50μl,自“置一1.5ml塑料离心管中”起同法测定。
附注:1、由于OPA-氨基酸不稳定,因此衍生后应立即进行分离测定。
2、本方法的衍生过程也可由自动进样器完成。
第四法DNFB柱前衍生氨基酸分析法本法系根据氨基酸与2,4-二硝基氟苯(DNFB)反应,生成有紫外响应的二硝基苯-氨基酸(DNP-氨基酸),DNP-氨基酸经反相高效液相色谱分离后采用紫外检测,在一定的范围内其吸光值与氨基酸浓度成正比。
本方法的线性响应范围为30~140 pmol。
本法所用的2,4-二硝基氟苯属易爆、剧毒物质,有强致癌性,且该法对色谱柱要求较高,易损坏色谱柱,衍生试剂水解生成的2,4-二硝基苯易干扰丝氨酸的测定。
除另有规定外,一般不宜采用本法。
试剂(1)流动相A)0.05mol/L醋酸钠溶液(取 4.1g无水醋酸钠,加水800ml溶解,加二甲基甲酰胺10ml,用稀醋酸调pH至6.4,用水稀释至1000 ml)。
(2)流动相B 流动相A-乙腈(1:1)。
对照品溶液按各品种项下规定的方法制备。
供试品溶液按各品种项下规定的方法制备。
色谱条件与系统适用新试验用十八烷基硅烷键合硅胶为填充剂(4.6×250mm,5μm);流速为每分钟1.0ml;柱温为40℃;检测波长为360nm。
各氨基酸峰间的分离度均应大于1.0。
洗脱梯度如下:时间(min) 流动相A(%)流动相B(%)0 75 256 75 256.1 65 3511 59 4114 59 4114.1 50 5022 45 5532 10 9037 10 9039 75 2550 75 25测定法精密量取氨基酸对照品溶液2ml,置一50ml量瓶中,加0.5mol/L碳酸氢钠溶液2ml,2,4-二硝基氟苯衍生化试剂(量取2,4-二硝基氟苯1ml ,用乙腈稀释至100ml)1ml,混匀,在60℃水浴中反应1小时,取20μl,注入液相色谱仪,记录色谱图;另精密量取供试品溶液2ml,自“置一50ml量瓶中”起同法测定。
第五法茚三酮柱后衍生氨基酸分析法本法系根据氨基酸经阳离子交换色谱柱分离后,与茚三酮反应,一级氨基酸生成在570nm处具有最大吸收的紫色化合物,二级氨基酸(如脯氨酸)生成在440nm具有最大吸收的黄色化合物,分别在570nm和440nm下检测上述反应产物,在一定的范围内其吸光值与氨基酸浓度成正比。
本方法的线性响应范围为20~500 pmol。
试剂(1)流动相A 取无水柠檬酸钠1.7g,盐酸1.5ml,加水溶解并稀释至100ml,用盐酸调pH至3.0。
(2)流动相B 取无水柠檬酸钠1.7g,盐酸0.7ml,加水溶解并稀释至100ml,用盐酸调pH 至4.3。
(3)流动相C 取氯化钠5g,无水柠檬酸钠1.9g,苯酚0.1g,加水溶解并稀释至100ml,用盐酸调pH至6.0。
(4)色谱柱再生溶液取氢氧化钠0.8g,加水溶解并稀释至100ml,用盐酸调pH至13。
(5)柱后衍生试剂取茚三酮18g,茚氮兰0.7g,加76.7%二甲基亚砜-0.7二水合醋酸锂-0.1%醋酸溶液900ml使溶解,在氮气下混合至少3小时。
(6)样品缓冲液2%无水柠檬酸钠-1%盐酸-0.5%硫代二乙醇-0.1%苯甲酸溶液。
对照品溶液按各品种项下规定的方法制备。
供试品溶液按各品种项下规定的方法制备。
色谱条件与系统适用新试验用磺化苯乙烯-二乙烯苯共聚物为填充剂(4.0×120mm,7.5μm);流动相流速为每小时14.0ml;柱后衍生试剂得流速为每分钟7ml,反应器温度为135℃;检测波长为440nm(一级氨基酸),570nm(二级氨基酸)。