药理学辅导资料:药动学-基本参数及概念
药理学—— 药动学知识点归纳

药理学——药动学知识点归纳一、药物的体内过程药物从进入机体至离开机体,可分为四个过程:简称ADME系统→与膜的转运有关。
(一)药物的跨膜转运:※药物在体内的主要转运方式是:被动转运中的简单扩散!Ⅰ、被动转运——简单扩散1.概念:指药物由浓度高的一侧向浓度低的一侧扩散,以浓度梯度为动力。
2.特点:(1)不消耗能量。
(2)不需要载体。
(3)转运时无饱和现象。
(4)不同药物同时转运时无竞争性抑制现象。
(5)当膜两侧浓度达到平衡时转运即停止。
3.影响简单扩散的药物理化性质(影响跨膜转运的因素)(1)分子量分子量小的药物易扩散。
(2)溶解性脂溶性大,极性小的物质易扩散。
(3)解离性非离子型药物可以自由穿透。
离子障是指离子型药物被限制在膜的一侧的现象。
4.体液pH值对弱酸或弱碱药物的解离的影响:从公式可见,体液pH算数级的变化,会导致解离与不解离药物浓度差的指数级的变化,所以,pH值微小的变动将显著影响药物的解离和转运。
例题:一个pK a=8.4的弱酸性药物在血浆中的解离度为A.10%B.40%C.50%D.60%E.90%『正确答案』A『答案解析』pH对弱酸性药物解离影响的公式为:10 pH-pKa=[解离型]/[非解离型],即解离度为10 7.4-8.4=10-1=0.1。
※总结:体液pH值对药物解离度的影响规律:◇酸性药物在酸性环境中解离少,容易跨膜转运。
达到扩散平衡时,主要分布在碱侧。
◇碱性药物在碱性环境中解离少,容易跨膜转运。
达到扩散平衡时,主要分布在酸侧。
同性相斥、异性相吸或“酸酸碱碱促吸收;酸碱碱酸促排泄”例题:某弱酸性药物pK a=3.4,若已知胃液、血液和碱性尿液的pH 值分别是1.4、7.4和8.4。
问该药物在理论上达到平衡时,哪里的浓度高?A.碱性尿液>血液>胃液B.胃液>血液>碱性尿液C.血液>胃液>碱性尿液D.碱性尿液>胃液>血液E.血液>碱性尿液>胃液『正确答案』A『答案解析』同性相斥、异性相吸。
药代动力学及其参数基本概念

正常受试者药代动力学研究
——单剂量给药的临床药代动力学研究
二、试验设计
一般应选用高、中、低3个剂量组,根据人体 耐受性试验的结果 高剂量组的剂量一般应高于临床试验的治疗 剂量,但不应超过人体的最大耐受剂量 受试人数:每组8~12例
正常受试者药代动力学研究
——单剂量给药的临床药代动力学研究
三、 试验操作步骤
三种单剂量的药代动力学试验结果反映不同药物 剂量(小、中、大剂量)的吸收和消除动力学的 规律是线性或非线性动力学
正常受试者药代动力学研究
——单剂量给药的临床药代动力学研究
五、药代动力学参数的估算
线性或非线性动力学的判断标准举例:依立雄胺 (epristeride)的9名健康男性受试者单剂量口服 5 mg、10 mg、20 mg爱普列特片剂进行药代动 力学研究结果如下(表8-2、表8-3)
或因与血浆蛋白结合力高,不易进入组织,其Vd 值常较小,约为0.15~0.3L/kg;与此相反,碱性 有机药物如苯丙胺、山莨菪碱等易被组织所摄取, 血中浓度较低,Vd值常超过体液总量(60kg的正 常人,体液约36L,即0.6L/kg)。例如,地高辛 的Vd达600L(10 L/kg),说明该药在深部组织大 量储存。
物效的 浓最 度临低 。床中最毒佳浓效度果,是(维C持SS)药min物大的于(药CS物S)m的ax最小低于有药
(六)负荷剂量(Loading dose,DL)
概念:临床上为了使药物尽快到达稳态 从而尽早发挥疗效,常常先给予一个较维持 剂量大的剂量使药物迅速达到稳态水平,然 后在预定的给药间隔时间给予维持剂量维持 稳态水平,这个在第一次使用的剂量称为负 荷剂量。
应用
3. 根据表观分布容积调整剂量 通常药物的表观分布容积与体表面积成正
生物药剂学与药动学——药动学概述

生物药剂学与药动学——药动学概述一、药动学定义药动学是应用动力学的原理和数学处理方法,研究药物在体内的吸收、分布、代谢和排泄过程(即ADME 过程)的量变规律的学科,即药动学是研究药物体内过程动态变化规律的一门学科。
二、血药浓度与药物效应(一)治疗浓度范围治疗浓度范围即治疗窗,是指给药后产生药效的最低有效浓度和产生毒性的最低中毒浓度之间的浓度范围。
治疗窗窄的药物,其治疗浓度相对较难控制,易发生治疗失败或不良反应,常需进行治疗药物监测。
(二)血药浓度与药物效应的关系对于大多数药物及其制剂,药物进入体内后,血中的药物浓度与药物作用靶位的实际浓度呈正相关,从而间接反映药物的临床效应,包括治疗效果及不良反应。
药动学中常以血液中的药物总浓度作为观察指标。
三、药动学的基本概念和主要参数(一)血药浓度-时间曲线药动学的研究中,将药物制剂通过适当的方式给予受试者,然后按照适当的时间间隔抽取血样,检测血样中的药物浓度,每一个取血时间点有一个对应的药物浓度,由此就得到一系列的血药浓度相对于时间的实验数据,简称为药-时数据。
将其用坐标图表示,称为血药浓度-时间曲线,简称药-时曲线。
血管内给药的药-时曲线通常为曲线,而血管外给药的药-时曲线一般为拋物线。
根据研究的需要,常将药-时曲线的不同时间段用吸收相、平衡相和消除相来表示,表明该时间段(时相)体内过程的主要影响。
(二)血药浓度-时间曲线下面积血药浓度-时间曲线图中,药-时曲线与时间轴共同围成的面积称为血药浓度-时间曲线下面积,简称药-时曲线下面积,用AUC表示。
其与药物吸收的总量成正比,能够反映药物吸收的程度。
AUC越大,表明制剂中的药物被生物体吸收越完全。
血药浓度-时间曲线下面积是评价制剂生物利用度和生物等效性的重要参数。
(三)峰浓度和达峰时间血管外给药的药-时曲线一般为拋物线,其中有两项特征性参数,即血药峰浓度和达峰时间。
血药峰浓度即药-时数据中的最大浓度,用C max表示,C max的大小能够反映药物的疗效情况和毒性水平。
药理学02药动学

药动学与药效学的研究方法
实验研究
通过动物实验和人体实验,观察药物在不同个体内的药动学和药 效学表现。
临床研究
通过临床试验,评估药物在患者中的疗效和安全性,同时监测其药 动学参数。
数学建模
建立药动学和药效学的数学模型,用于预测药物在不同个体内的表 现,以及优化给药方案。
THANKS
感谢观看
联合用药
同时使用其他药物可能会影响药物的排泄速 度。
药物排泄的研究方法
尿液检测
通过检测尿液中药物的浓度,了解药物排泄的情况。
血液检测
通过检测血液中药物的浓度,了解药物在体内的代谢和排泄情况。
动物实验
利用动物模型研究药物的排泄机制和过程。
体外实验
利用离体器官或组织进行研究,了解药物排泄的机制。
06
人体试验
通过在人体上进行药物吸 收试验,研究药物的吸收 特性,如药代动力学研究。
体外实验
利用离体组织或细胞进行 药物吸收研究,如细胞培 养、组织切片等实验方法。
03
药物分布
药物分布的机制
被动扩散
药物通过细胞膜的被动转运,由高浓度区域向低浓度区域扩散,与 细胞膜的通透性有关。
主动转运
药物通过细胞膜的主动转运,需要消耗能量,如Na+依赖性转运、 Ca2+依赖性转运等。
主动转运
药物通过细胞膜由低浓度一侧向高浓度一侧的逆 浓度梯度转运,需要载体并消耗能量。
3
胞饮
大分子和脂溶性药物通过细胞膜的特殊转运方式, 即细胞膜内陷形成小囊泡将药物包裹在内,从而 完成药物的转运。
影响药物吸收的因素
药物的理化性质
药物的脂溶性、解离度、分子大小等都会影响其吸收速率 和程度。
药理学3、药动学

二、药物的分布
分布 (distribution)
是指药物随血液循环转运到组织器官的过程。
多数药物在体内的分布是不均匀的 影响药物在体内分布的因素有:
(一) (二) (三) (四)
药物与血浆蛋白的结合; 药物理化性质和体液pH; 细胞膜屏障; 其他因素: 组织器官的血流量、药物与组织的亲和力等
三、药物的代谢
肝药酶的诱导剂和抑制剂
药酶诱导剂:凡能增强药酶活性或促进药酶生成的药物。
① 常见诱导剂:巴比妥类、水合氯醛、苯妥英钠、利福 平等。 ② 意义:可加速自身代谢和其他药物代谢。药酶诱导作 用可解释连续用药产生的耐受性、交叉耐受性、停药 敏化现象、使其他药效力下降等。 ③ 例如:苯巴比妥,合并用香豆素时,则因肝药酶活性 增强,加速了对双香豆素的灭活,从而使双香豆素疗 效降低。
药 理 学 Pharmacology
第三章 药物代谢动力学
概 述
药物代谢动力学 (药动学)(pharmacokinetics):
是研究机体对药物处置过程及体内血药浓度随
时间变化规律的一门科学。
药物的体内过程:
包括药物的吸收、分布、代谢和排泄等过程。
第一节 药物的跨膜转运
药物的跨膜转运: 药物在体内被吸收、分布、代谢和排
二、主动转运
主动转运 (active transport): 是药物从生物膜低浓度一侧向高浓度
一侧转运(又称上山转运)的过程。
主动转运的特点:
⒈低浓度到高浓度 ⒊需要消耗能量 ⒌存在竞争性抑制现象
⒉需要载体 ⒋有饱和性
三、膜动转运
膜动转运 : 大分子物质的转运伴有膜的运动,膜的表面张
排泄 (excretion) 是指药物原形及其代谢产物通过排泄器官或
第三章 第二节 药动学基本参数及概念
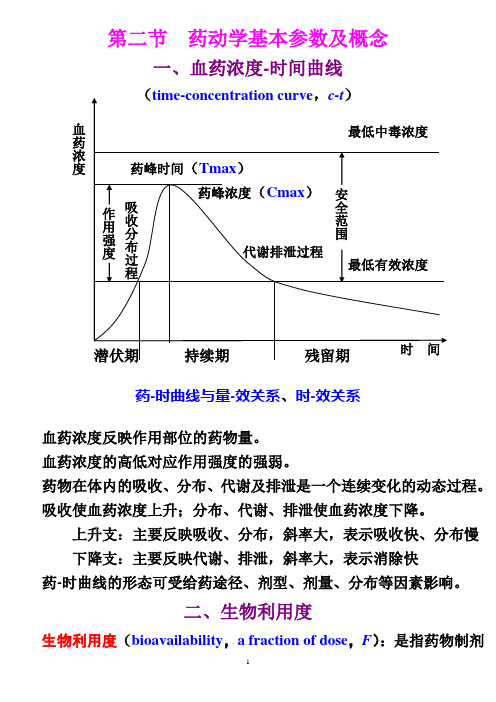
第二节 药动学基本参数及概念一、血药浓度-时间曲线药-时曲线与量-效关系、时-效关系血药浓度反映作用部位的药物量。
血药浓度的高低对应作用强度的强弱。
药物在体内的吸收、分布、代谢及排泄是一个连续变化的动态过程。
吸收使血药浓度上升;分布、代谢、排泄使血药浓度下降。
上升支:主要反映吸收、分布,斜率大,表示吸收快、分布慢 下降支:主要反映代谢、排泄,斜率大,表示消除快药-时曲线的形态可受给药途径、剂型、剂量、分布等因素影响。
二、生物利用度生物利用度(bioavailability ,a fraction of dose ,F ):是指药物制剂血药浓度被机体吸收的速率和吸收程度的一种量度。
即:实际被吸收利用的量(A)占服用总量(D)的百分比。
F=A/D×100%药物的吸收量可通过测定给药后的药-时曲线下面积(area under the time concentration curve,AUC)来估算。
绝对生物利用度F=AUC po×D iv/AUC iv×D po×100%相对生物利用度F=AUC t×D r/AUC r×D t×100%AUC:曲线下面积D:剂量po:口服iv:静注t:试验制剂r:参比制剂影响因素:可因制剂质量、剂型、给药途径、患者具体情况等不同,同一药物的生物利用度会有差异。
意义:评价各种药物制剂的生物等效性;评价药物的首过消除与作用强度;指导临床合理用药;查明药物无效或中毒的原因。
三、表观分布容积药物在体内的分布是不均的。
当药物在体内分布达到动态平衡时,体内药量与血药浓度的比值称表观分布容积(apparent volume of distribution,V d)。
V d=A/C。
V d:表观分布容积A:给药量(mg/kg)C:血药浓度(mg/L)意义:表示药物在组织中分布范围的广窄,结合程度的高低。
V d ≈0.045 L/kg,主要分布在血浆;V d = 0.14-0.29 L/kg,主要分布在细胞外液;V d = 0.3-0.4 L/kg,主要分布在细胞内液;V d ≈0.6L/kg,表示分布在细胞内外液;V d >>0.6L/kg,表示药物在某组织中集中分布。
药动学的参数

药动学的参数药物动力学是研究药物在体内吸收、分布、代谢和排泄过程的科学。
正确认识和掌握药物动力学的参数对于合理用药、临床治疗以及新药开发都具有重要意义。
本文将从吸收、分布、代谢和排泄四个方面分别介绍药物动力学的主要参数。
一、吸收参数1. 生物利用度(bioavailability):药物经口给药后进入体内的总吸收量与直接注射的总吸收量的比值。
一般通过体内外药物浓度随时间的变化曲线来计算。
2. 最大血浓度(Cmax):药物在给药之后血浆中达到的最高浓度。
该参数反映了药物吸收的速度和程度。
3. 时间到达峰浓度(Tmax):最大血浓度出现所需要的时间。
4. 血药浓度-时间曲线(AUC):药物在体内的总曝露量,通过血浆药物浓度随时间的变化曲线下的面积来计算。
AUC值越大,表示总体内曝露量越大。
二、分布参数1. 分布容积(Vd):反映药物在体内分布的广泛程度。
Vd值越大,说明药物在体内的分布范围越广。
2. 血浆蛋白结合率(protein binding):部分药物在体内主要以结合蛋白的形式存在,该参数反映了药物与蛋白质结合的程度。
3. 大脑-血浆分布系数(BBB):反映了药物能否越过血脑屏障进入脑组织,对于中枢神经系统药物很重要。
三、代谢参数1. 代谢速率(metabolic clearance):单位时间内从血浆中清除药物的速率。
代谢速率越大,药物代谢越快。
2. 代谢半衰期(t1/2):药物浓度下降到初始浓度的一半所需要的时间。
代谢半衰期可间接反映药物在体内停留的时间。
四、排泄参数1. 肾脏清除率(renal clearance):药物在单位时间内通过肾脏清除的速率。
2. 肝脏清除率(hepatic clearance):药物在单位时间内通过肝脏清除的速率。
药物动力学参数反映了药物在体内的代谢、分布、排泄等过程。
了解这些参数有助于合理用药,指导临床用药,也有助于评价新药的临床前药代动力学特性。
对于药物研究、开发和用药安全有着重要的意义。
药物动力学参数

• Vd不具有直接的生理意义,绝大多数情况下不涉及真正的容积。
Vd
=
D0 C0
=
D静注 C0
=
������ ������������������ ∙ ������
D-体内药量 C-血药浓度 AUC-血药浓度-时间曲线下的面积 K-消除速率常数
ห้องสมุดไป่ตู้0.693
K
������1/2 = ������
Vd
������1/2;
0.693 ������1/2 = ������
三、单室模型静脉给药
(一)静脉注射给药 • 例题:某单室模型药物作快速静脉注射,剂量为500mg,并立即测
得其血药浓度为32µg/ml,已知该药的t1/2=8h,试求Vd值及静注12h 的血药浓度,并计算出何时血药浓度达8µg/ml?
– 多数药物以肝的生物转化和肾的排泄两种途径从体内消除
ClTBCL=Cl肝+Cl肾+……
一、常用术语
(五)体内总清除率 • 单位时间内从体内清除的表观分布容积数。 • 不同的人对同一药物的半衰期有差异,但TBCL基本是相同的。因
此,当同一药物使用在不同个体时,只要他们的TBCL相同,则单 位时间内从体内清除的表观分布容积数也就相同。
• 单位时间内机体消除药物的量即为消除速率常数K,具有加和性,
反应体内药物总清除的情况。
k =ke+kb+kbi+klu+…....
ke-肾排泄速率常数 kb-代谢速率常数 kbi-胆汁排泄速率常数 klu-肺消除速率常数
k是衡量药物在体内消除快慢、表示药物在体内停留时间的重要参数之一。
药动学的参数

药动学的参数药动学是研究药物在体内吸收、分布、代谢和排泄过程的学科,是药物治疗学的重要理论基础之一。
药动学参数是评价药物在体内行为的重要工具,它们可以帮助我们了解药物的药效、药代动力学和安全性,从而指导临床用药。
下面我们就药动学参数进行详细的介绍。
一、吸收1. 生物利用度(Bioavailability)生物利用度是指口服给药后药物在体内被吸收并发挥药效的比例,通常用AUC(曲线下的药物浓度时间曲线下面积)来评价。
生物利用度是衡量口服给药的药物吸收情况的重要参数。
2. 吸收速率常数(Ka)吸收速率常数描述的是药物被吸收到体内的速度,它是影响给药后血药浓度上升的一个重要参数。
吸收速率常数的大小决定了药物的吸收速度,对于快释放的药物比较重要。
3. 最大血浆浓度(Cmax)最大血浆浓度是指在给药后的一段时间内,血药浓度达到最高值。
Cmax是药物吸收速度和程度的重要指标,通常也与药物的毒性和疗效密切相关。
二、分布1. 分布容积(Vd)分布容积描述了药物在体内的分布情况,它反映了药物在水溶液中的分布情况,是需要在临床用药中考虑的重要参数。
2. 蛋白结合率(Plasma protein binding)蛋白结合率是指药物在血浆中与蛋白质结合的比例。
蛋白结合率对药物在体内的分布、代谢和排泄有重要影响,也是影响药物在体内的有效浓度的重要因素。
三、代谢1. 代谢常数(Km)代谢常数描述了药物在体内的代谢速率,它是由药物代谢酶介导的,是影响药物代谢速度的重要因素。
2. 代谢清除率(Clm)代谢清除率是指药物在体内通过代谢途径被清除的速率,它是评价药物代谢速度的重要参数之一。
四、排泄1. 生物半衰期(t1/2)生物半衰期是指体内药物浓度下降到初始浓度一半所需的时间,它是评价药物在体内清除速度的重要参数。
2. 肾脏清除率(ClR)肾脏清除率描述了药物在体内通过肾脏被清除的速率,它是评价药物排泄速度的重要参数。
通过以上介绍,我们了解了药动学参数在评价药物在体内行为中的重要作用,它们可以帮助我们更好地理解药物的药效、药代动力学和安全性,为临床用药提供重要的参考依据。
药理学第3章药动学

孕妇应避免使用
四、药物的代谢
药物的代谢:药物在体内发生化学变化
药物生物转化的意义:使药理活性改变 灭活: 活化:
无活性变为有活性药物:
SL • • • • • • • • • • • • • • • • 1-3 min
Transdermal • • • • • • • • • 40-60 min
三、药物的分布和影响因素
影响因素: 1. 与血浆蛋白结合 2. 局部器官的血流量 3. 药物与组织的亲和力 4. 体液的pH值和药物的理化性质 5. 体内屏障
Absorption
Drug Administration
Drug in Tissues of Distribution
Drug Concentration in
Systemic Circulation
Drug Metabolism or
Excreted
Distribution
Elimination
药理作用 解热止痛 抗焦虑 抗风湿 抗抑郁 抗心律失常
(一)药物代谢反应 1、第一相反应:氧化、还原、水解。 2、第二相反应:结合(与葡萄糖醛酸、 硫酸、乙酰基、氨基酸、谷胱甘肽、 甲基等)
I 相反应 & II 相反应的比较
I相 反应类型 氧化
还原 水解 增加亲水性 小
II 相 结合
大
机制
暴露功能基团
(1) 一般来说, 药物可迅速分布到血流量大的 组织器官,达到平衡。
(2) 重分布: 药物首先分布到血流丰富的组织 器官, 然后再向分布容积大的组织转移, 称之为重分布。 例:硫喷妥钠 脑 脂肪 分布 分布 效应 效应消失
药动学的参数

药动学的参数药物动力学(pharmacokinetics)指的是描述药物在体内的吸收、分布、代谢和排泄的过程。
它是药理学的一个重要分支,对于理解药物在体内的行为和对治疗效果的影响至关重要。
在这篇文章中,我们将介绍药物动力学的一些重要参数,以帮助读者更好地理解药物在体内的表现。
一、吸收(absorption)1. 生物利用度(bioavailability)生物利用度是指服用药物后在体内的有效利用程度,通常以百分比表示。
它受到很多因素的影响,包括药物的溶解性、吸收速度、首过效应等。
了解药物的生物利用度对于确定适当的给药途径和剂量至关重要。
2. 最大血药浓度(Cmax)最大血药浓度是指药物在体内吸收后在血液中达到的最高浓度,通常在给药后数小时内测得。
它反映了药物的吸收速度和强度,对于评估药物的起效时间和药效有着重要的意义。
二、分布(distribution)1. 分布容积(volume of distribution,Vd)分布容积是一个描述药物在体内分布的参数。
它反映了药物在血液和组织间的分布程度,通常以体积单位表示。
了解分布容积可以帮助我们理解药物在体内的分布情况,为合理用药提供重要信息。
2. 蛋白结合率(protein binding)蛋白结合率是指药物在血浆中与蛋白结合的比例,通常以百分比表示。
这个参数直接影响着药物在体内的有效浓度,因为只有游离态的药物才能发挥药效。
了解药物的蛋白结合率有助于我们评估药物与蛋白的亲和力,以及对药物在体内的影响。
三、代谢(metabolism)1. 清除率(clearance)清除率是指单位时间内身体清除给定药物的能力,通常以血浆中单位时间内药物减少的速率表示。
清除率受到肝脏和肾脏等器官功能的影响,是评估药物在体内代谢与排泄的重要参数。
2. 体内半衰期(half-life)半衰期是指体内一半药物被清除的时间,通常表示药物的代谢和排泄速度。
了解药物的半衰期有助于我们确定用药频率,以及评估药物在体内的停留时间。
主管药师专业知识讲义-药理学——第三节 药动学
药理学——第三节药动学考纲一、药物的体内过程药物从进入机体至离开机体,可分为四个过程:简称ADME系统→与膜的转运有关。
(一)药物的跨膜转运:※药物在体内的主要转运方式是:简单扩散!Ⅰ、被动转运——简单扩散(3)转运时无饱和现象。
(4)不同药物同时转运时无竞争性抑制现象。
(5)当膜两侧浓度达到平衡时转运即停止。
3.影响简单扩散的药物理化性质(影响跨膜转运的因素)(1)分子量分子量小的药物易扩散。
(2)溶解性脂溶性大,极性小的物质易扩散。
(3)解离性非离子型药物可以自由穿透。
离子障是指离子型药物被限制在膜的一侧的现象。
※药物的解离程度受体液pH值的影响离子型非离子型4.体液pH值对弱酸或弱碱药物的解离的影响:※pK a(解离常数)的含义:>>是指解离和不解离的药物相等时,即药物解离一半时,溶液的pH值。
>>每一种药物都有自己的pK a。
若为弱酸性药物,则:若为弱碱性药物,则:从公式可见,体液pH算数级的变化,会导致解离与不解离药物浓度差的指数级的变化,所以,pH值微小的变动将显著影响药物的解离和转运。
一个pK a=8.4的弱酸性药物在血浆中的解离度为A.10%B.40%C.50%D.60%E.90%『正确答案』A『正确解析』pH对弱酸性药物解离影响的公式为:即:解离度为107.4-8.4=10-1=0.1。
※四两拨千斤:体液pH值对药物解离度的影响规律:◇酸性药物——在酸性环境中解离少,容易跨膜转运。
达到扩散平衡时,主要分布在碱侧。
◇碱性药物——在碱性环境中解离少,容易跨膜转运。
达到扩散平衡时,主要分布在酸侧。
同性相斥、异性相吸或“酸酸碱碱促吸收;酸碱碱酸促排泄”A.碱性尿液>血液>胃液B.胃液>血液>碱性尿液C.血液>胃液>碱性尿液D.碱性尿液>胃液>血液E.血液>碱性尿液>胃液『正确答案』A『正确解析』同性相斥、异性相吸。
在碱性尿液中弱碱性药物A.解离多,重吸收少,排泄快B.解离少,重吸收多,排泄快C.解离多,重吸收多,排泄快D.解离少,重吸收多,排泄慢E.解离多,重吸收少,排泄慢『正确答案』D『正确解析』酸酸碱碱促吸收;酸碱碱酸促排泄。
临床药理学药动学概述文档

药物动力学(pharmacokinetics)
应用动力学原理与数学处理方法, 定量描述药物在体内动态变化规律的学 科。
应用
药物的体内过程
吸收 分布 代谢 排泄
组织分布 吸收 血液循环 排泄
代谢
吸收(absorption)
吸收指药物从用药部位进入体循环的过程。 静脉注射和静脉滴注药物,直接进入体循环不存在
➢水和食物 餐后pH显著 ➢疾病 十二指肠溃疡 pH ➢药物 组胺 pH
阿托品类、阿司匹林抑制胃酸分泌
➢胃排空速率
药物从胃幽门排至小肠上部的速度
胃排空速率决定了药物到达肠道的速度,对
药物的起效快慢,药效强弱及持续时间有显
著影响
主要受胃内容物影响
胃
食物的组成
排
空
速
粘度 渗透压
度 减
主动吸收的药物,如VB2
➢ 食物的影响 食物不仅能改变胃排空速率而影响吸收,而且 由于其他多种因素对药物的吸收产生不同程度 、不同性质的影响.
• 延缓或减少药物吸收 食物消耗胃肠内水分,使胃肠黏液减少,使药物的崩 解、溶出减慢。 如: 空腹服用对乙酰氨基酚,tmax为20 min; 早饭后服用对乙酰氨基酚,tmax为2 h; 食物可减慢苯巴比妥的吸收而使其不能起到 催眠作用
拟为不同隔室的组合
▪ 一室模型 ▪ 二室模型 ▪ 多室模型
周边室1
一室模型
药物进入体内后,能迅速向各组织器官分布,很 快在血液和各组织脏器间达到动态平衡
BODY
二室模型
药物进入体内后,能很快进入机体的某些部位,
口服给药形成的曲线是由迅速上升的以吸收为主的吸收相和缓慢下降的以消除为主的消除相两部分组成。
药理学辅导资料:药动学-基本参数及概念

药理学辅导资料:药动学-基本参数及概念一、时量关系和时量曲线血浆药物浓度随时间的变化过程称时量关系。
以浓度或对数浓度为纵坐标和以时间为横坐标作图为时量曲线。
二、房室概念与房室模型1、一室模型:假定身体由一个房室组成,给药后药物立即均匀地分别于整个房室,并以一定的速率从该室消除。
单次静注给药时,时量(对数浓度)曲线呈单指数消除。
2、二室模型:假定身体由两个房室组成,即中央室(血流丰富的器官如心、肝、肾)和周边室(血流量少的器官如骨、脂肪)。
给药后药物立即分布到周边室。
单次静注给药时,时量(对数浓度)曲线呈双指数衰减即分为分布相和消除相。
三、生物利用度:不同剂型的药物能吸收并经首过消除后进入体循环的相对份量及速度。
四、血药浓度——时间曲线下面积:表示在服用某一剂量后的一定时间内吸收入血的药物相对量。
五、表观分布容积(Vd)药在体内达到平衡时,按血药浓度(C)推算体内药物总量(A)在理论上应占有的体液容积。
六、药物消除动力学1、恒量消除(零级动力学消除):每单位时间内消除恒定数量的药物,这是由于血药浓度过高,超出了机体消除能力的极限。
当血药浓度下降至消除能力以下时,则按一级动力学消除。
特点:(1)药物血浆半衰期(t1/2)随血浆浓度高低而变化;(2)时量曲线用普通坐标时为直线。
2、恒比消除(一级动力学):每单位时间内消除恒定比例的药量,由于血药浓度较低,未超出机体消除能力的极限。
特点:(1)药物血浆t1/2 恒定,不随血药浓度高低而变化;(2)时量曲线用普通坐标时为曲线,血药浓度改为对数尺度时呈直线。
七、清除率每单位时间内能将多少升血中的某药全部消除(L/min或h)。
八、消除速率常数(K)某单位时间内药物被消除的百分速率数。
九、半衰期(t1/2)血浆血药浓度下降一半所需的时间,t1/2=0.693/K.t1/2是决定给药间隔时间的重要依据。
十、连续多次给药或单次给药血浆药物浓度的变化和稳态血药浓度1、恒速静脉滴注药物时,血药浓度没有波动地逐渐上升,经5个t1/2达到稳态浓度(Css,坪值)。
药代动力学基本概念及参数

(二).组织器官 器官血流量、 器官亲和力 均可影响再分布
(三).屏障(barrier) 血脑屏障 (blood-brain barrier) 血眼屏障(blood-eye barrier) 病灶纤维组织 胎盘屏障(blood-eye barrier)
(四).组织pH
四.生物转化(biotransformation)
(二).理化性质 (三).制剂 (四).吸收环境 (五).生物利用度(bioavilability)
F = 体内药量/给药量×100%
三、 分布(distribution) 因素:
(一)药物与血浆蛋白结合
药物与血浆蛋白结合: 疏松、按比例结合、可逆的、影响分布
和疗效、无药理活性、是一种暂时的贮存形 式、饱合性;药物过量,游离增加,肝硬化, 营养不良病人,竞争性。
弱酸性药物在碱性环境中,易解离,主要呈离 子状态,不易通过膜。
弱碱性药物在碱性环境中,难解离,主要呈分 子状态,易通过膜。
弱碱性药物在酸性环境中,易解离,主要呈离 子状态,不易通过膜。
3.转运平衡时 弱酸性药物主要堆集在碱侧, 弱碱性药物主要堆集在酸侧.
例如:药物吸收;巴比妥中毒;药物的细胞 内外分布;(吗啡中毒)
Cl=Vd.Ke
AUC = C / Ke C = Ke. AUC Vd = A / C = A / Ke. AUC
Cl A/AUC =Vd.Ke = (A / Ke. AUC ). Ke =
(8)峰浓度 Cmax
(9)达峰时间 Tmax
END!
前一章
下一章
机体对药物的化学处理
(一).方式:
(1).第一阶断 氧化 还原 分解. 结构改 变.作用改变
作用消失
药物动力学的参数是什么_什么叫药物动力学的参数

药物动力学的参数是什么_什么叫药物动力学的参数药物动力学是近20年迅速发展的新领域,所以很多人都对这个新领域表示一无所知,更不知道药物动力学的参数,那么药物动力学的参数是什么呢?下面是店铺为你整理的药物动力学的参数是什么的相关内容,希望对你有用!药物动力学的参数消除速度常数消除是指体内药物不可逆失去的过程,它主要包括代谢和排泄。
其速度与药量之间的比便常数K称为表观一级消除速度常数,简称消除速度常数,其单位为时间的倒数,K值大小可衡量药物从体内消除的快与慢。
药物从体内消除途径有:肝脏代谢、肾脏排泄、胆汁排泄及肺部呼吸排泄等,所以药物消除速度常数K等于各代谢和排泄过程的速度常数之和,即:K=Kb+Ke+Kbi+Klu+……消除速度常数具有加和性,所以可根据各个途径的速度常数与K 的比值,求得各个途径消除药物的分数。
生物半衰期生物半衰期(Half-life time)简称半衰期,即体内药量或血药浓度下降一半所需要的时间,以t1/2表示,单位为时间。
药物的生物半衰期与消除速度常数之间的关系为:因此,t1/2也是衡量药物消除速度快慢的重要参数之一。
药物的生物半衰期长,表示它在体内消除慢、滞留时间长。
一般地说,正常人的药物半衰期基本上相似,如果药物的生物半衰期有改变,表明该个体的消除器官功能有变化。
例如肾功能、肝功能低下的患者,其药物的生物半衰期会明显延长。
测定药物的生物半衰期,特别是确定多剂量给药间隔以及肝肾器官病变时给药方案调整都有较高的应用价值。
根据半衰期的长短,一般可将药物分为:t1/2<1小时,称为极短半衰期药物;t1/2在1~4小时,称为短半衰期药物;t1/2在4~8小时,称为中等半衰期药物;t1/2在8~24小时,称为长半衰期药物;t1/2>24小时,称为极长半衰期药物。
清除率整个机体(或机体内某些消除器官、组织)的药物消除率,是指机体(或机体内某些消除器官、组织)在单位时间内消除掉相当于多少体积的流经血液中的药物。
第三章 第二节 药动学基本参数及概念
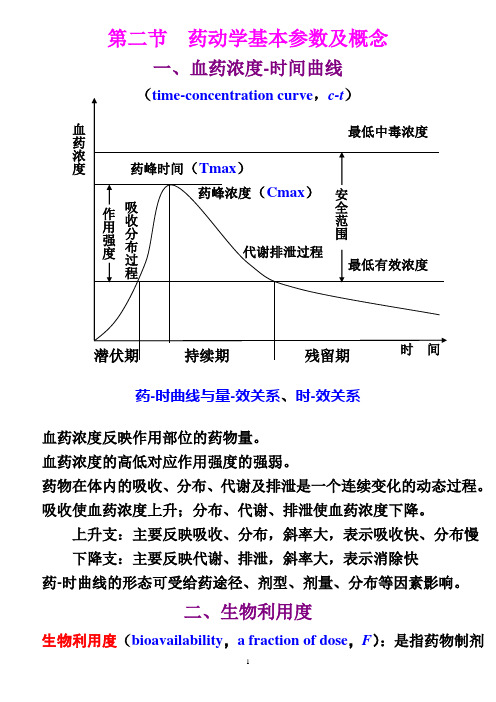
第二节 药动学基本参数及概念一、血药浓度-时间曲线药-时曲线与量-效关系、时-效关系血药浓度反映作用部位的药物量。
血药浓度的高低对应作用强度的强弱。
药物在体内的吸收、分布、代谢及排泄是一个连续变化的动态过程。
吸收使血药浓度上升;分布、代谢、排泄使血药浓度下降。
上升支:主要反映吸收、分布,斜率大,表示吸收快、分布慢 下降支:主要反映代谢、排泄,斜率大,表示消除快药-时曲线的形态可受给药途径、剂型、剂量、分布等因素影响。
二、生物利用度生物利用度(bioavailability ,a fraction of dose ,F ):是指药物制剂血药浓度被机体吸收的速率和吸收程度的一种量度。
即:实际被吸收利用的量(A)占服用总量(D)的百分比。
F=A/D×100%药物的吸收量可通过测定给药后的药-时曲线下面积(area under the time concentration curve,AUC)来估算。
绝对生物利用度F=AUC po×D iv/AUC iv×D po×100%相对生物利用度F=AUC t×D r/AUC r×D t×100%AUC:曲线下面积D:剂量po:口服iv:静注t:试验制剂r:参比制剂影响因素:可因制剂质量、剂型、给药途径、患者具体情况等不同,同一药物的生物利用度会有差异。
意义:评价各种药物制剂的生物等效性;评价药物的首过消除与作用强度;指导临床合理用药;查明药物无效或中毒的原因。
三、表观分布容积药物在体内的分布是不均的。
当药物在体内分布达到动态平衡时,体内药量与血药浓度的比值称表观分布容积(apparent volume of distribution,V d)。
V d=A/C。
V d:表观分布容积A:给药量(mg/kg)C:血药浓度(mg/L)意义:表示药物在组织中分布范围的广窄,结合程度的高低。
V d≈0.045 L/kg,主要分布在血浆;V d= 0.14-0.29 L/kg,主要分布在细胞外液;V d= 0.3-0.4 L/kg,主要分布在细胞内液;V d≈0.6L/kg,表示分布在细胞内外液;V d>>0.6L/kg,表示药物在某组织中集中分布。
1-10药动学参数

药动学参数u 生物利用度1.概念非血管给药时,吸收进入血循环量占给药总量的百分比。
F = ×100%A (吸收药量)D (给药总量)2. 简单计算公式:1.概念:表现分布容积(Vd)是假设药物在血浆和组织内分布达到平衡时,按照血药浓度(C)推算体内药物总量(A)在理论上应占有的体液容积。
u 表观分布容积V d =A/C2.计算公式u表观分布容积3.意义:(1)仅反映所测药物在组织中分布的范围、结合程度的高低。
(2)根据Vd可推测药物分布范围。
(3)根据Vd还可推算体内药物总量、血药浓度、达到某血药浓度所需药物剂量,以及排泄速度。
u 消除(2)恒量消除:(1)恒比消除:(3)非线性消除:2.类型:消除是指进入血液循环的药物由于分布、代谢和排泄,血药浓度不断衰减的过程。
1.概念:单位时间内按恒定比例消除药物。
单位时间内按恒定的量消除药物。
恒比与恒量混合型消除。
u清除率清除率(CL)指单位时间内有多少容积血将中药物被清除。
计算公式为:CL=k·Vd其中k为消除速度常数,Vd 为表观分布容积它反应肝肾的功能。
肝肾功能不全时CL值会降低,药物易蓄积。
u半衰期( t)1/21.概念:血浆药物浓度下降一半所需要的时间。
2.意义:(1)药物分类的依据,超短效,短效,中效,长效,超长效。
给药一次 。
(2)确定给药时间,通常一个t1/2u 半衰期2.意义:(4)估计药物基本消除时间。
停药后, 大约经过5个t 1/2药物基本消除。
(3)估计药物到达稳态血药浓度的时间。
(每隔一个t 1/2用药, 约经过5个t 1/2达稳态血药浓度。
)u稳态血药浓度1.概念恒比或恒量消除的药物,连续恒速或分次恒量给药,当给药速度等于消除速度时,血药浓度维持在一个相对稳定的水平,称稳态血药浓度(Css)。
其波动的峰值为峰浓度(Cmax),谷值为谷浓度(Cmin),二者之间相对距离为波动幅度。
u稳态血药浓度2.意义(1) Css的高低与给药总量成正比。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
药理学辅导资料:药动学-基本参数及概念
一、时量关系和时量曲线
血浆药物浓度随时间的变化过程称时量关系。
以浓度或对数浓度为纵坐标和以时间为横坐标作图为时量曲线。
二、房室概念与房室模型
1、一室模型:
假定身体由一个房室组成,给药后药物立即均匀地分别于整个房室,并以一定的速率从该室消除。
单次静注给药时,时量(对数浓度)曲线呈单指数消除。
2、二室模型:
假定身体由两个房室组成,即中央室(血流丰富的器官如心、肝、肾)和周边室(血流量少的器官如骨、脂肪)。
给药后药物立即分布到周边室。
单次静注给药时,时量(对数浓度)曲线呈双指数衰减即分为分布相和消除相。
三、生物利用度:
不同剂型的药物能吸收并经首过消除后进入体循环的相对份量及速度。
四、血药浓度——时间曲线下面积:表示在服用某一剂量后的一定时间内吸收入血的药物相对量。
五、表观分布容积(Vd)
药在体内达到平衡时,按血药浓度(C)推算体内药物总量(A)在理论上应占有的体液容积。
六、药物消除动力学
1、恒量消除(零级动力学消除):每单位时间内消除恒定数量的药物,这是由于血药浓度过高,超出了机体消除能力的极限。
当血药浓度下降至消除能力以下时,则按一级动力学消除。
特点:
(1)药物血浆半衰期(t1/2)随血浆浓度高低而变化;
(2)时量曲线用普通坐标时为直线。
2、恒比消除(一级动力学):每单位时间内消除恒定比例的药量,由于血药浓度较低,未超出机体消除能力的极限。
特点:
(1)药物血浆t1/2 恒定,不随血药浓度高低而变化;
(2)时量曲线用普通坐标时为曲线,血药浓度改为对数尺度时呈直线。
七、清除率
每单位时间内能将多少升血中的某药全部消除(L/min或h)。
八、消除速率常数(K)
某单位时间内药物被消除的百分速率数。
九、半衰期(t1/2)
血浆血药浓度下降一半所需的时间,t1/2=0.693/K.t1/2是
决定给药间隔时间的重要依据。
十、连续多次给药或单次给药血浆药物浓度的变化和稳态血药浓度
1、恒速静脉滴注药物时,血药浓度没有波动地逐渐上升,经5个t1/2达到稳态浓度(Css,坪值)。
2、连续分次给药,即每隔一定时间(如一个t1/2)给予等量的药物时,血药浓度波动地上升,经5个t1/2达Css.当首次剂量加倍(负荷剂量)时,血药浓度迅速达到Css.
3、单次给药时,经5个t1/2体内药量基本消除(>96%)。
【。