基因工程chapter 5 目的基因的分离与鉴定
第三章基因工程目的基因的获取分离

用氢氧化钠消化杂合双链 中的 mRNA 链
解离的第一链 cDNA 的 3‘末端就会形成一个发夹环 (发夹环的产生是第一链 cDNA 合成时的特性,原因 至今未知,据推测可能是 由帽子的特殊结构相关)
引导 DNA 聚合酶复制出第 二链,此时形成的双链之 间是连接在一起的
再利用 S1 核酸酶将连接处 (仅该位点处为单链结构) 切断形成平通过几种限制性内切酶切
割,所产生的基因组DNA片段与载体重组并克 隆,由此产N=ln(1DNA片段的出现概率。 f:是插入片段的大小与全基因组大小的比值。
第一:细胞总 RNA 的提取和 mRNA 分离;
mRNA
总RNA
tRNA 第二:第一链 cDNA 合成;
rRNA 第三:第二链 cDNA 合成;
2 合成cDNA第一条链 第四:双链 cDNA 克隆进质粒或噬菌体载体
(1)随机引物引导法
并导入宿主中繁殖。
(2)oligo(dT)引导法
(1)随机引物引导法 5´
链 cDNA 为模板合成一段段互补的序列。同时,大肠杆菌 DNA 聚合酶 Ⅰ 的 3'--5'
cDNA 片段。这些 cDNA 片段进而核酸外切酶的活性可将暴露出的第一链 cDNA 的
在 DNA 连接酶的作用下连接成一 3'-端部分消化掉,形成平端或差不多的平端。这
条链,即 cDNA 的第二链
种方法合成的 cDNA 在 5'-端存在几个核苷酸缺失, 但一般不影响编码区的完整。
查阅资料自学!
如何找回两膜或硝酸 纤维素滤膜之后,就可以同特异性
的核酸探针进行菌落或噬菌班杂交,
A 应用核酸探针分离克隆的目的基因 以便筛选出具有目的基因的阳性克 隆。这个过程叫做克隆基因的分离
分离目的基因实验报告(3篇)

第1篇一、实验目的本实验旨在通过分子生物学技术,学习并掌握目的基因的分离方法,包括基因组DNA提取、目的基因的克隆、扩增和鉴定等步骤。
通过实验,使学生熟悉实验原理、操作步骤和注意事项,提高学生的动手能力和实验技能。
二、实验原理目的基因分离是指从生物基因组中提取出特定的基因片段,并进行克隆、扩增和鉴定。
实验步骤主要包括以下几部分:1. 基因组DNA提取:利用各种方法从生物组织中提取出基因组DNA。
2. 目的基因的克隆:利用PCR技术扩增目的基因,并克隆到载体上。
3. 目的基因的鉴定:通过限制性内切酶酶切、DNA测序等方法对克隆的目的基因进行鉴定。
4. 目的基因的表达:将目的基因导入宿主细胞,进行表达和功能验证。
三、实验材料与试剂1. 实验材料:大肠杆菌、质粒载体、目的基因DNA模板等。
2. 试剂:DNA提取试剂盒、PCR试剂、限制性内切酶、DNA连接酶、DNA测序试剂盒等。
四、实验步骤1. 基因组DNA提取(1)取适量生物组织,按照DNA提取试剂盒说明书进行操作。
(2)提取的基因组DNA用琼脂糖凝胶电泳检测,确保DNA提取质量。
2. 目的基因的克隆(1)设计特异性引物,用于PCR扩增目的基因。
(2)按照PCR试剂盒说明书进行PCR扩增,获得目的基因。
(3)将PCR产物与载体连接,转化大肠杆菌。
(4)通过蓝白斑筛选,获得阳性克隆。
3. 目的基因的鉴定(1)对阳性克隆进行酶切鉴定,验证目的基因是否成功克隆。
(2)对阳性克隆进行DNA测序,确定目的基因序列。
4. 目的基因的表达(1)将目的基因克隆到表达载体上,构建表达系统。
(2)将表达载体导入宿主细胞,进行目的基因的表达。
(3)检测目的基因的表达产物,验证目的基因的功能。
五、实验结果与分析1. 基因组DNA提取:提取的基因组DNA在琼脂糖凝胶电泳中呈现清晰的主带,说明DNA提取成功。
2. 目的基因的克隆:通过PCR扩增,获得目的基因片段,大小与预期相符。
基因工程第五章-载体

cccDNA在较高高的温度和PH值条件下才会变性,(较线 性和开放环结构的DNA),如果变性,由于团在一起,条 件恢复后,会很快复性,形成cccDNA,线性和环状的变 性后,复性会形成杂乱的大分子团状结构,从而从体系中 沉淀下来。
3. 质粒DNA的分子量和拷贝数:
按质粒的分子量大小,可分为量大类: 3――7kb,拷贝数多; 70-150kb,含有与质粒功能有关的更多基因,可自我传 递(可编码一套控制质粒DNA转移的基因,所以可从一 个细胞转到另一个细胞),如果一个细胞含有这种质粒, 他可以带动小质粒进行传递转移。这种质粒的拷贝数低。
质粒的拷贝数:分子的多与寡是决定产量的重要因素。
pBR322 拷贝数大于25个 ColE1 15个 产量0.23ug/ml 0.11 ug/ml
基因克隆尽量选择松弛型质粒,如果是严紧型要进行质粒 复制子改造。
质粒的大小:通常质粒越大,拷贝数也越少,所以我们 在进行基因重组时,首先要选择分子量小的质粒,其次 质粒分子量大的要使外源基因的片断长度合适,不要过 大。
பைடு நூலகம் 2、pUC质粒载体:
pUC质粒载体:是由美国加利福尼亚大学的专家构建 的。是在pBR322质粒载体的基础上,组入了一个带 有多克隆位点的LacZ’基因,而发展成为具有双功能 检测特性的质粒载体。
最常用的克隆载体,优点: 具有更小的相对分子量和更高的拷贝数。 在构建pUC质粒载体时仅留下pBR322的复制起始起 始位点和AMP抗性基因,分子大大减小,他的拷贝数 可以达到500-700个/个细胞。 适合组织化学法检测重组子。有利于检测 具有多克隆位点,便于片断的插入。
沉淀出来。
第一步:(1)在5m1含相应抗生素的LB培养基中接种 一单菌落,于37℃剧烈振荡培养过夜。(2)将1.5m1 培养物倒入Eppendorf管中,用微量离心机于4℃以 12000rpm离心30秒。 (3)吸去培养液,使细菌沉淀 尽可能干燥。(4)将细菌沉淀重悬于100μl用冰预冷的 溶液I中(溶液Ⅰ:50mmol/L 葡萄糖, 25mmol/L Tris· (pH 8.0), 10mmol/L CI EDTA(pH 8.0,溶菌酶4-5mg,酶作用环境由缓冲 液提供)。剧烈振荡。须使细菌沉淀在溶液Ⅰ中完全分 散,将两个微量离心管的管底部互相接触,室温静止5 -30min,细胞壁降解。
高中生物选修基因工程目的基因的导入、检测与鉴定

高中生物选修基因工程目的基因的导入、检测与鉴定第8课时目的基因的导入、检测与鉴定[学习目标] 1.理解目的基因导入受体细胞的方法。
2.掌握检测与鉴定目的基因的方法。
[核心素养] 1.科学思维:结合生产实际,举例说出基因工程的基本程序。
2.社会责任:针对生产生活中的某一需求,尝试提出基因工程构想,完成初步设计。
一、将目的基因导入受体细胞1.转化的含义目的基因进入受体细胞内,并且在受体细胞内维持稳定和表达的过程。
2.转化的方法(1)导入植物细胞①农杆菌转化法:将目的基因导入双子叶植物和裸子植物时最常用的方法。
a.土壤农杆菌的特点:植物体受到损伤时,伤口处的细胞会分泌大量的酚类化合物,吸引农杆菌侵染,其Ti质粒上的T-DNA(可转移的DNA)可转移至受体细胞,并且整合到受体细胞的染色体的DNA上。
b.方法步骤:目的基因插入Ti质粒的T-DNA上→转入农杆菌→导入植物细胞→目的基因插入植物细胞中的染色体DNA上→目的基因表达。
②基因枪法:将目的基因导入单子叶植物常用的方法。
表达载体DNA包裹在微小的金粉粒子或钨粉粒子表面→用基因枪高速射入受体细胞或组织中→目的基因表达。
③花粉管通道法:在植物受粉后,花粉形成的花粉管还未愈合前,剪去柱头,滴加DNA(含目的基因),使目的基因借助花粉管通道进入受体细胞。
(2)导入动物细胞①方法:显微注射技术。
②常用受体细胞:受精卵。
③步骤:目的基因表达载体提纯→取卵(受精卵)→显微注射→胚胎早期培养→移植到雌性动物的输卵管或子宫内→获得具有新性状的转基因动物。
(3)导入微生物细胞①原核生物的特点:繁殖快、多为单细胞、遗传物质相对较少等。
②常用的方法a.用Ca2+处理细胞,使细胞处于一种能吸收周围环境中DNA分子的生理状态(这种细胞称为感受态细胞)。
b.感受态细胞和重组表达载体DNA分子在缓冲液中混合,在一定的温度下促进感受态细胞吸收DNA分子。
1.将胰岛素基因的表达载体导入大肠杆菌,从大肠杆菌中获得的“胰岛素”未能达到期待的生物活性,导致这种差异的原因可能是什么?提示大肠杆菌是原核生物,无内质网、高尔基体等细胞器,大肠杆菌中基因表达获得的蛋白质未经加工。
基因工程操作步骤

4.目的基因的检测与鉴定 在受体细胞中稳定遗传和正 确表达。
一、目的基因的获取
1 目的基因主要是编码蛋白质的基因: 如:与生物抗性相关的基因、与优良品质相 关的基因、与生物药物和保健品相关的基因、 与毒物降解相关的基因、与工业用酶相关的 基因、具调控作用的因子等。
合成
热稳定的DNA聚合酶
特点
半保留复制、 边解旋变复制
半保留复制、 全解旋再复制
结果
形成整个DNA分子
大量的DNA片段
随堂闯关 PCR技术扩增过程
a、DNA变性(90℃-95℃): 双链DNA模板 在热作用下, 氢断键裂,形成____单__链_
b、复性(55℃-60℃): 系统温度D降N低A ,引物 与DNA模板结合,形成局部__双__链____。
③例: 转基因抗虫棉
2.将目的基因导入动物细胞
①方法: 显微注射法
①程序
目的基因表达载体提纯
显微注射
受精卵
取卵(受精卵) 新性状动物
3.将目的基因导入微生物细胞
①微生物作受体细胞原因:
繁殖快、多为单细胞、遗传物质相对少
②常用菌: 大肠杆菌
③常用法: Ca2+处理获得感受态细胞
④过程:
Ca2+处理 大肠杆菌
1.2 基因工程的基本操作程序
知识回顾: 基因工程基本操作的四个步骤
有了目的基因, 我们才能赋予
1.目的基因的获取 一种生物以另一种生物的遗 传特性。
使目的基因在受体细胞中稳
2.基因表达载体的构建 定存在, 并可进行遗传、表达 和发挥作用
载体进入受体细胞稳定表达,
3.目的基因导入受体细胞 才能实现一种生物的基因在 另一种生物中的转化。
基因工程原理考试题库(精简版)

基因工程原理考试题库(精简版)一基因工程的基本概念1、基因工程的定义与特征。
定义:在体外把核酸分子(DNA的分离、合成)插入载体分子,构成遗传物质的新组合(重组DNA),引入原先没有这类分子的受体细胞内,稳定地复制表达繁殖,培育符合人们需要的新品种(品系),生产人类急需的药品、食品、工业品等。
特征:1、具跨越天然物种屏障的能力。
2、强调了确定的DNA片段在新寄主细胞中的扩增。
2、试述基因工程的主要研究内容。
(1)目的基因的分离(2)DNA的体外重组(载体、受体系统等)(3)重组DNA分子转移到受体细胞(4)重组DNA分子的筛选(5)基因在受体细胞内的扩增、表达、检测及其分析。
3、基因工程在食品工业上有何应用发展?主要是通过基因重组,使各种转基因生物提高生产谷氨酸、调味剂、酒类和油类等有机物的产率;或者改良这些有机物组成成分,提高利用价值。
4、转基因是一把双刃剑,请客观谈谈对转基因及转基因食品安全性的认识。
转基因技术所带来的好处是显而易见的,在人类历史进步和发展中起到了积极作用。
首先,通过该项技术可以提供人们所需要的特性,改良培育新品种;第二,延长食品保存时间或增加营养成分;第三,将抗虫防菌基因转入到作物中,使作物本身产生抵抗病虫害侵袭的能力,减少了农药的使用量,有利于环境保护;第四,转基因技术及基因食物在医学方面得到广泛研究和应用。
人们对转基因技术的主要担忧在于环境方面。
外源基因的导入可能会造就某种强势生物,产生新物种或超级杂草、损害非目标生物、破坏原有生物种群的动态平衡和生物多样性,也即转基因生物存在潜在的环境安全问题。
转基因作物的大面积种植已有数年,食用转基因食品的人群至少有10亿之多,但至今仍未有转基因食品对生命造成危害的实例;更何况目前每一种基因工程食品在上市前,都要经过国家法律认可,食品卫生部门和环境部门的严格检测。
只有测试合格了,才能投放市场。
因此公众完全可以安全地消费、大胆地食用转基因食品。
大学《基因工程学》教学大纲
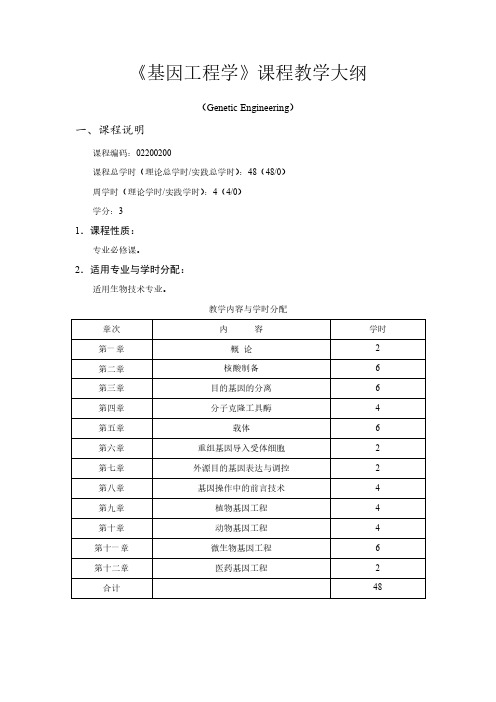
《基因工程学》课程教学大纲(Genetic Engineering)一、课程说明课程编码:02200200课程总学时(理论总学时/实践总学时):48(48/0)周学时(理论学时/实践学时):4(4/0)学分:31.课程性质:专业必修课。
2.适用专业与学时分配:适用生物技术专业。
教学内容与学时分配3.课程教学目的与要求:本课程的授课对象是生物技术专业的本科生。
课程简介:《基因工程》是生物技术专业的专业必修课程。
其以分子遗传学理论为基础,以分子生物学和微生物学的现代方法为手段而建立起来的一门技术学科。
基因工程兴起于20世纪70年代初,它的问世带动了生物技术的兴起和发展,是现代生物技术的核心内容。
基因工程课程的主要内容包括基因的分离、基因的克隆、基因的表达、植物基因工程、动物基因工程、药物基因工程和基因治疗等。
它是生命科学学院生物技术专业本科生的主干专业课程之一,它是生物工程(包括基因工程、细胞工程、酶工程、发酵工程)中最重要的课程,其它三大工程是建立在基因工程基础之上的,同时也为生物技术制药等后继学科奠定了重要的理论基础。
课程目标:设置本课程是为了让生物技术专业的学生理解和掌握基因工程的技术原理,通过本课程学习,掌握基因操作的工具酶,基因克隆常用载体,目的基因的分离与合成,重组体的构建,重组体向宿主细胞的导入,重组体克隆的筛选与鉴定以及克隆基因的表达,同时了解基因工程在生物学领域中的应用与发展前景。
对学生达到毕业要求贡献如下:1)了解基因工程学的历史、发展和前沿知识。
2)掌握基因工程学的基础理论、基本知识和基本技能;教学要求:学完基因工程学后,学生将具备以下能力:1)具有良好的自学能力;2)综合运用所掌握的基因工程学理论知识和技能、从事生物科学及其相关领域科学研究的能力。
4.本门课程与其它课程关系:先修课程为生物化学、微生物学、分子生物学、细胞学等,具备基础理论知识及实验能力是基因工程学课程的基础。
基因工程目的基因的分离和获得和重组基因的导入

基因工程目的基因的分离和获得和重组酵母 菌形及其周围限制酶切位目的基因的分离和获得和重组基因的导入
第15页
基因工程目的基因的分离和获得和重组基因的导入
第16页
基因工程目的基因的分离和获得和重组基因的导入
基因工程目的基因的分离和获得和重组增特定生物基因组 全部或个别片段,保留物种遗传种质。2.从基因中调出其中任何DNA片段或目标基 因。
3.构建基因是研究基因本身需要,如基因结构 分析、基目的基因的分离和获得和重组基因的导入
第20页
基因工程目的基因的分离和获得和重组基因的导入
第21页
(四)重组连接
制备出适当大小随机性基因组DNA与载体左臂右臂 进行连接,NA Sau3A I末端互补连接。
第2页
基因分离和取得常见方法
一、基因分离物理方法 二、鸟枪法(Sh因化学合成 六、聚合酶链反应技术(PCR)
基因工程目的基因的分离和获得和重组基因的导入
第3页
一、基因分离物理方法
依据DNA分子两条链存在着G≡C,A=T碱基配对。
如DNA分子中某段G≡C碱基对含量高,则其热稳定性就 高,其溶解温度(Tm)就高。这么,大家经过控制溶解 温度使富A=T区解链变性,而富G≡C区仍维持双链。 当利用单链核酸酶S,酶去除解开单链个别,得到富 G≡C区DNA片段 。
基因工程目的基因的分离和获得和重组基因的导入
第4页
二、鸟枪法(Shot gun) 又称霰弹法
基因工程目的基因的分离和获得和重组基因的导入
第5页
鸟枪法克隆目标基因基础战略
1、随机酶切成基因相当大小片段(约 1000bp);
2、重组入适当载体(质粒);
3、转化进宿主菌(E.coli),扩增;
基因工程目的基因的分离

基因组大小/bp
2×106(细菌)
2×107 (真菌)
2×109(动物)
a Ds
b
C座位 c-m突变
c Ac
Ac
C座位
····
转座子目前作为一种重要的标签(tag)在基因组图谱中 有重要位置 。
亲本杂交
筛选突变体 提取突变体基因,构建基因以转位子为探针筛选突变体克隆
亚克隆转位子两端基因芯片技术分离目的基因
二、基因的酶促合成(逆转录法)
mRNA
逆转录酶
cDNA
图1 基因的化学合成及克隆
图NA,通过几种限制性内切酶切 割,所产生的基因组DNA片段与载体重组并克隆,由此产生 的克隆群
5’
↓ EcoRⅠ
3’
3’
5’
↓
5’
3’ 5’
3’
3’
5’
5’
3’ ↓ 5’
3’
3’
5’
5’
3’ 5’
3’
3’
5’
5’
3’ ↓ 5’
3’
3’
5’
5’
3’ 5’
3’
3’
5’
图 DNA聚合酶Ⅰ的切口平移
四、mRNA差别显示分离目的基因
mRNA差别显示技术也叫DDRT-PCR(differential display RT-PCR),即差异显示RT-PCR。
基因芯片:是指将许多特定的DNA寡核苷酸或DNA片段(包 括cDNA)固定在芯片的每个预先设置的区域内,将待测样品 标记后同芯片进行杂交,通过杂交信息的分析来检测基因的功 能和基因组研究的分析系统。
1、制作方法
点接触法 喷墨法 光刻合成法
第5章基因工程基本流程-筛选与鉴定

分子量 Marker28
重组 克隆
载体
7. 限制性酶切图谱法 限制性酶切图谱:核酸经限制性内切酶消化成片段,用 电泳方法分离,不同的核酸形成的各自独特的条带图谱。
29
原理: 在外源片段大小及限制性酶切图谱已知的条件下,选择 一两种内切酶切割载体,电泳后比较电泳结果(DNA条 带数目和分子大小)差异进行区分。 或用合适的内切酶切下插入片断,再用其它酶切这个片 断,电泳后比较结果是否符合预计的结果。
6
无环丝氨酸培养基
7
(2)氨苄青霉素抗性插入失活选择过程 如果Ampr上插入外源DNA,导致氨苄青霉素 抗性基因失活,可用碘-青霉素指示液选择。
8
2. -半乳糖苷酶显色反应筛选法
乳糖
半乳糖 +
-半乳糖苷酶
X-gal
半乳糖 +
原理
葡萄糖
5-溴-4-氯靛蓝 深蓝色
载体上有一段-半乳糖苷酶基因(Lac Z)的片段(氨基 端),其上有外源DNA的插入位点。 受体菌基因组中有突变的-半乳糖苷酶基因(片段缺失)。 可以被IPTG诱导表达。 载体和受体菌基因组可以互补形成完 整有功能的-半乳糖苷酶。
30
A
B
A或B
31
不同克隆的酶切结果
32
选择过程
A
A
T T
TA AT
表型筛选加酶切筛选
33
8. DNA序列测定 原理:
通过对插入片段序列的测定确定是否是所需的重组子
34
选择过程 双脱氧末端终止法 化学降解法 焦磷酸降解法 高通量Solexa测序 ……
35
36
基于外源基因表达产物的筛选与鉴定
例:小鼠的二氢叶酸还原酶(DHFR)对三甲氧 苄二氨嘧啶有抗性。
基因工程原理讲义:目的基因的分离

第十章目的基因的分离中国科学院遗传与发育生物学研究所第十章目的基因的分离一、引言1.基因分离技术发展简介2.基因分离技术迅猛发展的客观要求3.目的基因的分离二、分离克隆基因的若干主要方法1.应用核酸探针分离克隆的目的基因2.应用差别杂交法或扣除杂交法分离克隆的目的基因3.低丰度mRNA之cDNA末端的快速扩增RACE法4.根据蛋白质家族的保守序列鉴定新的相关基因5.通过表达载体分离特异的cDNA三、应用mRNA差别显示技术分离目的基因1.引言2.mRNA差别显示原理3.mRNA差别显示实验过程及其局限性四、DNA标签法分离目的基因1.原理2.转位子标签法3.T-DNA标签法五、酵终双杂交与单杂交体系1.酵母双杂交体系2.酵母单杂交体系3.酵母三杂交体系第十章目的基因的分离一、引言1.基因分离技术发展简介*1.在上个世纪70年代中期已经具备了分离基因的能力这主要是取决于当时重组DNA技术的发展水平,特别是发展出了安全的寄主菌株和克隆载体:第一个安全的大肠杆菌寄主菌株——X1776品系的诞生第一批安全适用的克隆载体pM19和pBR322的问世在上述这两个条件的基础上,分子生物学家成功地克隆出第一批基因。
*2.在上个世纪70年代末期克隆基因分离的能力明显提高这主要应归功于基因克隆策略的改变。
由于发展出了改良的寄主菌株和克隆载体,人们提出了先克隆cDNA再分离目的基因的新策略。
*3.在上世纪80年代克隆基因分离技术得到了进一步的提高20世纪80年代之后,由于如下技术的进步,使人们分离克隆基因的能力得到进一步提高,这些技术主要有如下几项:a.寡核苷酸合成技术的改良;b.操作RNA和DNA的酶学方法的进步;c.PCR技术的发展与应用;*4.目前科学家们已经掌握了先进的分离克隆基因的能力就目前的能力而言,可以说几乎任何期望克隆和分离的目的基因都是有可能被分离出来的。
其主要原因是:a.技术的进一步发展;b.基因组全测序工作的迅速进展与成功;c.未知产物的发育调节基因分离技术的发明,例如mRNA差别显示技术基因定位克隆技术DNA标签技术等2.基因分离技术迅猛发展的客观要求近年来基因分离技术的迅猛发展,特别是产物未知的发育调节基因分离技术的发展尤为迅速,已发展出了基因定位克隆技术、mRNA差别显示技术、DNA标签法分离目的基因的技术(包括T-DNA标签法和转座子标签法)、以及酵母双杂交和单杂交技术。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
② 降解mRNA模板 用碱处理或用RNaseH降解mRNA-DNA杂交分子中的mRNA。
3’ 5’ 引物
mRNA
cDNA
反转录酶
5’
3’
3’
mRNA
5’
5’ 引物
cDNA第一链
3’
碱 或RNaseH
5’ 引物
cDNA第一链
3’
③ cDNA第二链合成
剩下的cDNA单链的3’末端一般形成一个弯回来的双链发卡结构(机理不明), 可成为合成第二条cDNA链的引物。用DNA聚合酶合成第二链DNA.。
5’
cDNA第一链
cDNA第二链合成
DNA聚合酶
5’ 3’
cDNA第一链 cDNA第二链
④去掉发卡结构
用核酸酶S1可以切掉发卡结构(但这会导致cDNA中有用的序列被切 掉!)。
5’ 3’
cDNA第一链 cDNA第二链
核酸酶S1
5’ 3’
cDNA第一链 cDNA第二链
3’ 5’
自 我 引 导 合 成 法
高浓度引物
3. 锚定PCR(anchored PCR, A-PCR) PCR技术需了解靶基因片段两侧的序列
对于未知序列和具有不同末端序列怎么办?
锚定PCR首先合成第一链cDNA,然后再添加一同聚物尾(polydG),与同 聚物尾配对的3’锚定引物(带有限制性内切位点polydC)一起作PCR扩增
1.反向PCR (reverse PCR)
目的:是用反向的互补引物来扩增两引物以外的DNA片段,对某个已知DNA 片段两侧的未知序列进行扩增。 用途:探索邻接已知DNA片段的序列;用于仅知部分序列的全长cDNA的克 隆;扩增基因的插入DNA;建立基因组步移。
未知序列
已知序列
未知序列
限制酶 未知序列
直接连接、同聚物加尾或人工接头。
① 粘性末端 直接连接
②同聚物加尾法
优点:能把任何片段连接起来 缺点:不能把插入片段再切下来
非酶切位点
③人工接头
人工接头:用化学合成法合成的一段10-12bp的特定限制性内切酶识别位点 序列的平端双链。
EcoRI linker: GGAATTCC CCTTAAGG
linker的作用:用T4 DNA连接酶连到平端DNA上,然后再用酶切,形成一 个人工粘性末端。
各种酶的接头细胞
重组噬菌体和cosmid都能在体外包装成噬菌体颗粒,以感染的 方式将重组DNA分子导入的局限性
✓ 并非所有的mRNA分子都具有polyA结构。 ✓ 细菌或原核生物的mRNA半衰期很短。 ✓ mRNA在细胞中含量少,对酶和碱极为敏感, 分离纯化困难。
✓ 仅限于克隆蛋白质.2 Pal RNA)提取 (2)mRNA的分离纯化
① 原理 利用mRNA都含有一段polyA尾巴,将mRNA从总RNA(rRNA、tRNA 等)中分离纯化。 mRNA只占总RNA的1%-5%。
宿主细胞DNA
基因组DNA 限制性内切酶
接入载体
进入细胞,培养 筛选单克隆基因组克隆2. 基因构建的一般步骤
1)基因组DNA大片段的制备
制备高分子质量的DNA:尽量避免机械切割, 避免其他DNA污染
断点完全随机,片断长度合适于载体连接。不能用一般的限制性内切酶消 化法。
物理切割法:
超声波(300bp)或机械搅拌(8kb)。
cDNA 末端核酸转移酶
3’GG…GG
5’
CC…CC 锚定引物
3’GG…GG
5’
CC…CC
3’GG…GG CC…CC
4. cDNA末端快速扩增技术RACE
原理:利用Oligo (dT) 对mRNA 进行反转录的同时在两头加上通 用接头(引物) ,从而可利用基因特异的引物,通过PCR 反应快速 获得目的序列的5′端和 3′端。
GGCCCTAGGGCC
接头自我连接
P-
-P
GATCCCGG GGCC
平末端DNA
BamHI adapter
GATCCCGGGGCC-
-CCGG -GGCCCTAG
酶切法:
内切酶Sau3A进行局部消化。可得到10-片段的制备 ①限制性内切酶法 用限制性内切酶把基因组DNA切成不同大小的片断。
酶切
优点:带有粘性末端,产物可以直接与载体连接。 缺点:目的基因内部也可能有该内切酶的切点。粒 λ-DNA Cosmid BAC YAC
15kb 25kb 45kb 连接
◆ PCR技术由于能从复杂DNA背景中将特定的DNA 区段扩增出来,使目标DNA在体系中大量合成,从 而很容易将其从体系中分离或克隆出来。
◆ 高效性、特异性
PCR原理
5.2.1 已知基因全长序列
要表达某种已知蛋白质
数据库中查到相应的序列 设计针对性的特异引物
RT-PCR扩增
……
/tools/种生物细胞的全部基因组DNA切割成大小合适的 片断,并将所有这些片断都与适当的载体连接,引入相 应的宿主细胞中保存和扩增。理论上讲,这些重组载体 片段重新组合起来可 以覆盖该生物的整个基因组。
人工接头2 DNA接头 (adapter)
一头平末端、另一头粘性末端(某种酶切位点序列)。
BamHI adapter 5‘p-GATCCCGG-OH3’ GGCC-p5’
用T4 DNA连接酶连到DNA分子的两端,直接成为人工粘性末端。
人工接头2 DNA接头 (adapter)
CCGGGATCCCGG
例如:人的基因组是 3×109 bp,插入DNA片断的平均长度如果是2×104 bp
G
N= 4.61× F
= 4.61×
3×109 2×104
= 6RNA为模板逆转录出的DNA称cDNA。
3’
mRNA
5’
5’ 引物 cDNA
反转录酶
3’
2. cDNA library
BamH I
BamH I
BamH I
策略:随机片断化
目的基因
限制性内切酶局部消化法:控制内切酶的用量和消化时间,使基因组 中的酶切位点只有一部分被随机切开。
② 机械切割法
a.超声波
超声波强烈作用于DNA,可使其断裂成约300bp的随机片断。
b.高速搅拌
1500转/分下搅拌30min,可产生约8kb的随机片断。
利用某种生物的总mRNA合成cDNA,再将这些cDNA与载体连接,转入细菌 细胞中进行保存和扩增,称cDNA。基因组 cDNA
3. cDNA的特点(1)不含内含子序列。 (2)可以在细菌中直接表达。 (3)包含料,适用于某些RNA病毒基因组结构研究。
② mRNA的分离纯化
分离mRNA用商业化的Oligo dT纤维柱。
(3)cDNA的合成 ① cDNA第一链合成
逆转录酶能以RNA为模板合成DNA。 用Oligo dT(或随机引物)作引物,合成cDNA的第一链。
3’AAAAAAA 5’ TTTTTTT
cDNA
mRNA
反转录酶
5’
Oligo dT引物
已知序列
限制酶 未知序列
连接酶
2. 不对称PCR
目的:扩增产生特异长度的单链DNA。 方法:采用两种不同浓度的引物。分别称为限制性引物和非限制性引物,其 最佳比例一般是0.01:0.5 μM,关键是限制性引物的绝对量。 用途:制备核酸序列测定的模板
制备杂交探针 基因组DNA结构功能的研究
低浓度引物
目的基因
载体 真核生物
连接 电穿孔或显微注射
重组载体
转化或转导
因的连接
• 直接连接 • 同聚物加尾 • 人工接头
直接连 接-1
直接连接1 同一粘性末端
缺点: 自我环化 无方向性 武汉工商学院《基因工程》
直接连接2 不同粘性末端
优点: 不会自我环化 定向性
存在的问题: 目的基因两端不一定 有酶切位点
同聚物加尾法
优点: 能把任何片段连接起来 缺点: 插入片段不能再切下来
非酶切位点
人工接头1
衔接物:用化学合成法合成的一段10-12bp的特定限制性内 切酶识别位点序列的平端双链。
GGAATTCC CCTTAAGG
武汉工商学院《基因工程》
优点:可酶切,还可连接Polylinker 缺点:如果插入片段内部也有该酶位点,不能得到完整的插入片段
妙用RNaseH
这种酶能识别mRNA-cDNA杂交分子中的mRNA,并将其降解成许多小片断。 小片断正好成为DNA聚合酶的引物,用来合成冈崎片断。 DNA Pol I除去引物并修补后再使用DNA连接酶连成一整条DNA链。
3’ 5’ 引物
mRNA
cDNA
反转录酶
3’ 5’ 引物
mRNA cDNA第一链p: 包含了整个基因组DNA的概率(99%) f: 插入载体的DNA片段的平均长度占整个基因组DNA的百分数 N:所需的重组载体数(克隆数)
N=
ln (1-p) ln (1-f)
=
ln (1-99%)
ln (1-f)
= 4.61×
G F
N=
4.61×
G F
G:Genome大小;F:Fragment大小
(1)对载体的要求
载体容量越大,所要求的DNA片断数目越少,所需的重组子越少。
(2)目前常用的载体 载体系列: 容量为 24 kp
Cosmid: 容量为 50 kb
YAC:
容量为 500kb
BAC:
容量为 300 kb基因组的大小一个要包含99%的基因组DNA时所需要的克隆数目。
N=
ln (1-p) ln (1-f)
RACE流程
RACE流程
SMART RACE cDNA synthesis Kit原理
cDNA第一链的合成机构建的步骤。 4. 简述RT-PCR法获得目的基因的原理及步骤。