Gaussian软件的基本原理与应用
《Gaussian培训》课件

结果分析
对计算结果进行分析,如 能量分解、轨道分析、电 荷分布等。
结果导出
将计算结果导出为其他软 件格式,如Xyz、Pdb等, 以便在其他软件中进行进 一步分析。
03
Gaussian软件进阶使用
高级计算方法
1 2 3
MP2方法
MP2方法是基于Hartree-Fock自洽场方法的二 阶Møller-Plesset微扰理论,适用于计算相对精 确的基态能量和电子相关效应。
反应机理研究案例
总结词
利用Gaussian软件进行反应机理研究,涉及反应路径计算、过渡态寻找和动力学模拟等。
详细描述
反应机理研究对于理解化学反应的本质和过程至关重要。Gaussian软件提供了反应路径计算和过渡态 寻找的方法,可以确定反应的最低能量路径和关键中间体。动力学模拟可以帮助研究反应速率和反应 机制,为实验设计和改进提供理论支持。
计算材料性质
02
材料模拟可以计算材料的电子结构、光学性质、力学性质、热
学性质等。
模拟软件
03
常用的材料模拟软件包括VASP、Quantum ESPRESSO和
CASTEP等。
04
Gaussian软件常件计算中最常见的问题之一,通常表 现为计算无法完成或结果不准确。
1990年代
Gaussian软件不断更新和 完善,逐渐成为广泛使用 的量子化学计算软件。
2000年代至今
Gaussian软件持续发展, 不断推出新版本,支持更 多的理论方法和基组,应 用领域也不断扩大。
02
Gaussian软件基本操作
文件操作
打开文件
使用“File”菜单中的 “Open”选项打开Gaussian 输入文件(.gjf或.gjfx)或输出文
量子化学软件Gaussian应用

Gaussian是做半经验计算和从头计算使用最广泛的量子化学软件,可以研究:分子能量和结构,过渡态的能量和结构化学键以及反应能量,分子轨道,偶极矩和多极矩,原子电荷和电势,振动频率,红外和拉曼光谱,NMR,极化率和超极化率,热力学性质,反应路径。
计算可以模拟在气相和溶液中的体系,模拟基态和激发态。
Gaussian 03还可以对周期边界体系进行计算。
Gaussian是研究诸如取代效应,反应机理,势能面和激发态能量的有力工具。
功能①基本算法②能量③分子特性④溶剂模型Gaussian03新增加的内容①新的量子化学方法②新的分子特性③新增加的基本算法④新增功能(1)基本算法可对任何一般的收缩gaussian函数进行单电子和双电子积分。
这些基函数可以是笛卡尔高斯函数或纯角动量函数多种基组存储于程序中,通过名称调用。
积分可储存在内存,外接存储器上,或用到时重新计算对于某些类型的计算,计算的花费可以使用快速多极方法(FMM)和稀疏矩阵技术线性化。
将原子轨(AO)积分转换成分子轨道基的计算,可用的方法有in-core(将AO积分全部存在内存里),直接(不需储存积分),半直接(储存部分积分),和传统方法(所有AO积分储存在硬盘上)。
(2)能量使用AMBER,DREIDING和UFF力场的分子力学计算。
使用CNDO, INDO, MINDO/3, MNDO, AM1,和PM3模型哈密顿量的半经验方法计算。
使用闭壳层(RHF),自旋非限制开壳层(UHF),自旋限制开壳层(ROHF) Hartree-Fock波函数的自洽场SCF)计算。
使用二级,三级,四级和五级Moller-Plesset微扰理论计算相关能。
MP2计算可用直接和半直接方法,有效地使用可用的内存和硬盘空间用组态相互作用(CI)计算相关能,使用全部双激发(CID)或全部单激发和双激发(CISD)。
双取代的耦合簇理论(CCD),单双取代耦合簇理论(CCSD),单双取代的二次组态相互作用(QCISD), 和Brueckner Doubles理论。
《Gaussian培训》课件

Gaussian软件将继续拓展应用领域,涉及到更多的化学领域和材料科学领域,为科研和应 用提供更广泛的服务。
THANKS
谢谢您的观看
在计算参数设置中,可以指定计 算方法、基组、优化算法等参数 ,以便进行后续计算。
在完成分子结构和计算参数设置 后,可以点击计算按钮执行计算 ,得出结果。
在结果窗口中,可以查看计算得 到的能量、构型、振动频率等结 果,并可以通过图形界面分析结 果。
03
Gaussian软件高级功能
计算多重态
多重态计算是指根据分子的电子组态计算分子 的能量或其他物理性质。
02
Gaussian软件安装及使用
软件安装步骤
准备安装文件
从官方网站下载Gaussian软件的安 装文件。
安装向导
运行安装文件,进入安装向导,选 择安装路径,点击“下一步”。
组件选择
选择需要安装的组件,如Gaussian 主程序、Lynx等,点击“下一步” 。
完成安装
等待安装程序完成安装过程,关闭 向导。
Gaussian软件基于量子力学原理,采用从头算和密度泛函理 论等方法,能够模拟分子的结构和性质,为化学科研工作者 提供重要的理论依据和工具。
软件背景
01
Gaussian软件是由美国Gaussian公司开发的一款商业软件,最初由John Gaussian在20世纪70年代创立。
02
自1970年以来,Gaussian软件已经经历了多个版本的不断更新和发展,成为了 国际上最为流行的计算化学软件之一。
和使用。 • 可定制性强:Gaussian软件支持自定义模块和功能,可根据用户的需求进行定制和扩展。 • 软件缺点 • 学习曲线较陡峭:对于初学者来说,Gaussian软件的学习曲线比较陡峭,需要一定的时间和精力来熟悉和
gaussian频率计算

gaussian频率计算Gaussian频率计算是一种常用的计算化学方法,用于研究分子的振动和光谱性质。
在化学和材料科学领域中,频率计算是理解和解释分子结构、反应机理和光谱谱线的重要工具。
本文将详细介绍Gaussian频率计算的原理和应用,并探讨其在科学研究中的重要性。
我们来了解一下Gaussian频率计算的基本原理。
频率计算是基于量子力学的原理,通过求解分子的力常数矩阵来获得分子的振动频率。
在Gaussian软件中,通过输入分子的几何构型和相关参数,利用量子力学的原子核运动方程求解,得到分子的振动频率和振动模式。
这些振动频率可以用来计算分子的红外光谱、拉曼光谱以及其他光谱性质,从而进一步研究分子的结构和性质。
Gaussian频率计算的应用非常广泛。
首先,它可以用于确定分子的构型和几何参数。
通过计算不同构型下的振动频率,可以确定分子的平衡几何构型和键长、键角等几何参数。
这对于理解分子的稳定性、反应机理以及与其他分子的相互作用非常重要。
Gaussian频率计算可以用于预测和解释分子的光谱性质。
振动频率与分子的红外光谱和拉曼光谱密切相关。
通过计算分子的振动频率和振动模式,可以预测和解释实验观测到的红外光谱和拉曼光谱谱线。
这对于确定分子的化学键、官能团和分子结构非常有帮助。
Gaussian频率计算还可以用于研究分子的振动性质和能量。
通过计算不同振动模式的振动能量和振动强度,可以了解分子的振动能级和振动强度分布,从而进一步研究分子的能量分布和振动态。
Gaussian频率计算在材料科学中也有重要应用。
振动频率对于材料的力学性质、热学性质和光学性质具有重要影响。
通过计算材料的振动频率和振动模式,可以研究材料的弹性、热膨胀、热导率、光学吸收等性质,为材料设计和性能优化提供指导。
Gaussian频率计算是一种重要的计算化学方法,可以用于研究分子的振动和光谱性质。
它在理论研究、实验解释和材料设计等方面都具有广泛的应用。
Gaussian的使用(一)

主要计算项目包括: • 分子的能量与几何结构 • 化学反应过渡态的能量与几何结构 • 振动频率分析 • 红外与拉曼光谱 • 分子的热化学性质 • 键能与化学反应能 • 化学反应途径 • 分子轨道的能量与性质 • 原子电荷分布(电子布居分析)和自旋密度分布
• 分子的多极矩(永久偶极矩和四极至十六极矩) • NMR 屏蔽常数、化学位移及分子的磁化率
用默认的外部浏览器显示分子结构的 3D 图形
用外部文本编辑器打开输出文件,阅读、分析和编辑计算结果
2.在“作业编辑对话框”内进行在线编辑 打开软件,进入 Gaussian03 主窗口。在主窗口中打开下拉菜单“File”, 点击“New”选项后将跳出“作业编辑对话框” (如图 1 所示) , 框内工具 条上各按钮的功能为
图 1 中已标出每一段的主要作用和功能。
图 2 乙炔分子编辑对话框
其中 OPT 表示优化分子结构,HF 表示采用从头算 Hartree-Fock 方法,6 31G * 表示采用的基函数,0 和 1 分别表示乙烯分子的电荷和 自旋多重度。 分子说明部分采用的是分子的内坐标, 它主要是通过分子中原子 的空间构型进行设置,具体格式为: 元素标记,参考原子,键长,参考原子,键角,参考原子,二面角
包含多个输入行,每行的行首均用#号开始。段后需加一空白行。 题目段 作业计算内容与目的的简要描述,以便于输出文件的阅读。
可包含多个输入行,段后需加一空白行。 电荷与自旋行 给定分子所带的净电荷数(正负整数)及自旋多重度
(正整数) 。限一行,行后不加空白行。 分子说明段 给定分子中各原子的坐标。 笛卡儿坐标或分子内坐标均
Water Energy
0 O1 H2 0.
1 0. 0.
0.ห้องสมุดไป่ตู้84 -0.554 0.
gaussian计算相互作用能

gaussian计算相互作用能【原创实用版】目录1.引言2.Gaussian 计算相互作用能的原理3.Gaussian 计算相互作用能的具体步骤4.Gaussian 计算相互作用能的优点与局限性5.总结正文一、引言在量子化学中,计算分子间的相互作用能是一项重要的研究任务。
Gaussian 是一款广泛应用于量子化学计算的软件,能够有效地计算分子间的相互作用能。
本文将介绍 Gaussian 计算相互作用能的原理、具体步骤以及优点与局限性。
二、Gaussian 计算相互作用能的原理Gaussian 基于密度泛函理论(DFT)计算分子间的相互作用能。
DFT 将电子密度作为基本变量,通过引入交换关联作用,可以有效地描述电子相关性。
Gaussian 使用分组方法将分子划分为不同的组,然后计算每组之间的相互作用能,最后将各组间的相互作用能相加得到总相互作用能。
三、Gaussian 计算相互作用能的具体步骤1.准备输入文件:在计算相互作用能之前,需要首先准备输入文件,包括分子的几何结构、元素种类、轨道类型等参数。
2.运行 Gaussian 软件:将输入文件提交给 Gaussian 软件,软件将自动进行计算。
3.分析输出结果:Gaussian 计算完成后,会生成输出文件,其中包括相互作用能的结果。
需要对输出结果进行分析,提取相互作用能的数值。
四、Gaussian 计算相互作用能的优点与局限性1.优点:Gaussian 计算相互作用能具有较高的准确性和可靠性,可以广泛应用于各种分子体系。
此外,Gaussian 软件操作简便,计算效率较高,适用于大规模计算。
2.局限性:尽管 Gaussian 在计算相互作用能方面具有很多优点,但它仍然有一定的局限性。
例如,对于某些具有较大空间伸展性的分子,Gaussian 计算结果可能存在误差。
此外,Gaussian 计算相互作用能的计算成本较高,对于计算资源有限的研究者而言可能存在一定的限制。
2._Gaussian_09_软件基本介绍

V Vij
2013-7-2
量子力学三大基本理论
分子轨道理论
• R. S. Mulliken和F. Hund
价键理论
• L. C. pauling和slater
密度泛函理论
• Hohenberg和Kohn
2013-7-2 7
密度泛函理论 Density Functional Theory
2013-7-2 24
研究激发态反应
基于卟啉结构的敏化太阳能电池设计
电子的激发态性质 是研究者普遍关心的一 个研究领域,所涉及的 到领域有光化学、光谱 学、生物化学、材料学 等。Gaussian 09 提供多 种激发态计算方法,可 以很好的预测分子或材 料的吸收和发射光谱。
Santhanamoorthi, N. et. al. JPC Lett. 2013, 4, 524
历史悠久
GAUSSIAN 70, The Nobel Prize in Chemistry 1998 J. A. Pople (1925-2004) Gaussian-系列程序 GAUSSIAN 76, GAUSSIAN 80,
GAUSSIAN 82,
GAUSSIAN 86, GAUSSIAN 88,
17
2
3
4
5
2013-7-2
GAUSSIAN 的功能
研究范围 • 各种物质形态,如气相、 液相、固相、多相 • 各种物质形式,如无机 物、有机物、生物分子、 晶体等 • 各种尺度,如分子尺度、 纳米尺度、介观尺度 • 周期性与非周期性体系 • 基态与激发态
研究内容 • 分子的平衡结构及过 渡态结构 • 化学反应机理 • 分子间相互作用 • 分子性质 • 光谱
Gaussian计算软件的使用

可执行各类不同精度和理论档次的MO 计算,包括Hartree-Fock 水平从 头算(HF)、Post-HF 从头算(各级CI 和MP)、MC-SCF法、密度泛 函理论(DFT),以及多种半经验量子化学方法,进行分子和化学反应 性质的理论预测。
2
Gaussian 的启动
3
4
工作环境初始化设置
G98W 安装后的首次启动时需进行工作环境初始化设置,规定主程序、检 查点文件、输入输出文件等默认子目录和路径。其内容记录在G98W 根目 录下的初始化文件g98w.ini 上。用户可以在启动G98W 前用Notepad 对 该文件直接编辑修改,也可点击“File”下拉菜单中的“Preference”选项 在程序界面上进行设定。下面介绍后一种初始化方式。在主窗口上打开下 拉菜单File
练习
35
36
37
38
39
40
41
42
优化时常用的关键词
• %chk=td-fang-2-2 • #P B3LYP/6-31G* POPT(tight,
maxcycle=200,restart) SCF(direct,maxcycle=200) GFINPUT IOP(6/7=3) TEST • • td at 12.26
• 或者 • # CIS/3-21g* POPT=restart TEST
48
TD光谱计算
• #P B3LYP/6-31G* TD(singlets,Nstates=30, root=1) TEST
• 如果中间断电可以用下面来接, • #P B3lyp/3-21g* TD=restart guess=read
HF,GVB,MCSCF,CASSCF,MPn等。改变模型方法通常 也会收敛性质。通常,精度更高的方法更难收敛。精度 比较低的方法产生的计算结果可以作为高精度计算的初 始猜测。 • 7)增加最大循环步数 • Gaussian默认的最大循环步数为64 (SCF=DM或 SCF=QC方法则为512),
第9章 量子化学程序Gaussian的原理和功能

31
可以使用的完备基组
y(r,q,f)=Rnl(r)· lm(q,f) Y 径向部分 原子轨道(STO) 角度部分(s, p, d, f …) 由轨道符号确定 N·n-1e-z r ( z = 0-) r Gaussian函数(GTF) N· exp(-ar2) (a = 0-) 平面波函数(PW) 用于固体计算 exp(-ip· (p = 0-) r)
5
II. 模型化学
计算机迅速发展 大型量子化学软件包
模型化学
对化学体系或过程
便于计算和预测
简化或理想化, 化学结构,性质和功能 化学分子:0K气体状态 溶剂效应:连续可极化模型 液体和固体:大晶胞的周期结构
6
模型化学
量子化学方法 分子力学方法
<500
准确, 化学键
~1万—100万
粗略, 只有结构 二者杂化方法 (内层用量子化学方法,外 层用分子力学方法)
10
III. 波函数的确定
准确的分子波函数无法得到 只能采取近似的方法,得到分子的近似的
波函数 各种近似在Gaussian中的体现 近似波函数的合理性判定
11
理论研究中的近似概念
绝大部分理论都是近似的。 近似的目的是抓住主要因素,发现规律,
解决问题。 近似必须是可以被不断修正的。 化学和生物学理论需要近似研究方法
23
Hamilton量的高级校正(相关能)
来源:单粒子算符Fi不能完全代替多粒子算
符H H=H(0)+H’=H(0)+(H-SFi) H’作为微扰,把相关能逐步考虑进来 HF已经是MP方法的一级能量了,因此从二 级考虑(MP2)以及(MP3, MP4)等
gaussian 16 的引用

gaussian 16 的引用Gaussian 16是一款广泛应用于计算化学领域的软件包,它提供了一系列功能强大的计算化学工具和方法。
本文将介绍Gaussian 16的一些主要功能和应用领域。
Gaussian 16是Gaussian系列软件的最新版本,它能够进行从简单到复杂的分子结构计算,包括基态和激发态的计算、分子动力学模拟、能量表面扫描等。
Gaussian 16采用了量子力学的基本原理,基于密度泛函理论(DFT)和哈特里-福克(HF)方法,结合了多种基组和方法,可以模拟各种化学反应和性质。
Gaussian 16的主要功能之一是计算分子的几何结构和能量。
通过输入分子的初始几何结构和基组信息,Gaussian 16可以计算出分子的最稳定几何结构和对应的能量。
这对于研究分子的稳定性、反应机理以及分子间相互作用等具有重要意义。
此外,Gaussian 16还可以计算分子的振动频率和红外光谱,从而进一步了解分子的结构和性质。
另一个重要的功能是计算分子的电子结构和性质。
Gaussian 16可以计算分子的电子轨道和轨道能级,以及相应的电子密度分布。
这对于研究分子的电子云分布、化学键的性质以及分子的光谱性质具有重要意义。
此外,Gaussian 16还可以计算分子的电荷分布和电荷转移,从而揭示分子的化学反应机理和反应活性。
Gaussian 16还可以模拟分子间相互作用和化学反应。
通过分子动力学模拟方法,可以模拟分子的运动和相互作用过程,从而研究分子的结构转变、反应速率和反应路径等。
此外,Gaussian 16还可以计算分子的反应能垒和反应活化能,从而评估化学反应的难易程度和速率。
除了上述功能,Gaussian 16还具有一些特殊的应用领域。
例如,Gaussian 16可以用于计算催化剂的活性位点和反应机理,从而指导催化剂的设计和优化。
此外,Gaussian 16还可以用于计算生物分子的结构和性质,例如蛋白质和核酸,从而揭示生物分子的功能和相互作用机制。
gaussian反应过程能量计算

Gaussian反应过程能量计算一、概述Gaussian计算是一种常用的计算化学方法,通过计算分子结构和反应能量,可以帮助化学研究人员理解分子的结构和性质,以及预测反应的速率和产物。
其中,Gaussian反应过程能量计算是指利用Gaussian软件来计算反应的过渡态能量、反应能垒以及反应热等参数。
本文将介绍Gaussian反应过程能量计算的基本原理和方法。
二、原理1. 反应过渡态在化学反应中,反应过渡态是反应物转变为产物的临时结构,其能量高于反应物和产物。
Gaussian软件可以通过计算反应物、产物和过渡态的能量,来判断反应的稳定性和速率。
2. 能量计算方法Gaussian软件使用量子化学方法来计算分子和反应的能量。
常用的方法包括DFT(密度泛函理论)、HF(Hartree-Fock方法)和MP2(二阶摄动方法)等。
这些方法可以通过数值计算,求得分子的电子结构和能量。
三、计算步骤1. 绘制反应势能面在进行反应过程能量计算之前,需要首先绘制反应势能面图。
通过对反应物、产物和过渡态的几何结构进行优化,可以得到它们的稳定构型和能量。
2. 选择计算方法根据反应的性质和分子的大小,选择合适的计算方法。
一般情况下,DFT方法适用于大分子和复杂体系的能量计算,而HF方法则适用于小分子和简单体系。
3. 进行能量计算利用Gaussian软件进行能量计算,得到反应物、产物和反应过渡态的能量值。
通过对比能量值的大小,可以确定反应的稳定性和能垒大小。
四、实例分析以SN2反应为例,假设反应物为溴甲烷(CH3Br),产物为溴乙烷(CH3CH2Br),过渡态为CH3-Br-C2H5。
通过Gaussian软件优化得到这三种分子的几何结构和能量分布。
选择合适的计算方法,进行能量计算。
最终得到反应过渡态的能量高于反应物和产物,证明该反应是一个SN2反应。
五、结论通过Gaussian反应过程能量计算,可以得到反应的能量参数,帮助化学研究人员理解反应的机理和动力学。
Gaussi an 量子化学计算技术与应用

Gaussi an 量子化学计算技术与应用Gaussian 是做半经验计算和从头计算使用最广泛的量子化学软件,可研究诸如分子轨道,结构优化,过渡态搜索,热力学性质,偶极矩和多极矩,电子密度和电势,极化率和超极化率,红外和拉曼光谱,NMR,垂直电离能和电子亲合能,化学反应机理,势能曲面和激发能 QM/MM 计算等化学领域的许多课题。
应用非常广泛,而且易于上手。
一、理论计算化学理论及相关程序入门1 理论计算化学简介1.1 理论计算化学概述1.2 HF理论及后HF方法(高精度量化方法)1.3 密度泛函理论和方法1.4 不同理论计算方法的优缺点及初步选择1.5 基组及如何初步选择基组2 Gaussian安装及GaussView安装及基本操作2.1 Gaussian安装及设置(Win版和Linux版)2.2 GaussView安装及设置2.3 GaussView使用及结构构建3 Linux、Vi编辑器等及Gaussian基本介绍3.1 学习Linux基本命令及Vi编辑器3.2 详细认识输入文件和输出文件(Win和Linux)3.3 构建Gaussian输入文件并提交任务二、Gaussian专题操作及计算实例4 Gaussian专题操作Ⅰ:(均含操作实例)4.1 结构几何优化及稳定性初判4.2 单点能(能量)的计算及如何取值4.3 开壳层与闭壳层计算4.4 频率计算及振动分析(Freq)4.5 原子受力计算及分析(Force)4.6 溶剂模型设置及计算(Solvent)5 Gaussian专题操作Ⅱ:(均含操作实例)5.1 分子轨道、轨道能级计算及查看5.2 HOMO/LUMO图的绘制5.3 布居数分析、偶极矩等计算及查看5.4 电子密度、静电势计算及绘制(SCF、ESP)5.5 自然键轨道分析(NBO)三、 Gaussian进阶操作及计算实例6 Gaussian进阶操作I:势能面相关(均含操作实例)6.1 势能面扫描 (PES)6.2 过渡态搜索(TS和QTS)6.3 反应路径IRC等6.4 反应能垒:熵,焓,自由能等7 Gaussian进阶操作II:——各类光谱计算及绘制(均含操作实例)7.1 紫外吸收,荧光和磷光7.2 红外光谱IR7.3 拉曼光谱RAMAN7.4 核磁共振谱NMR7.5 电子/振动圆二色谱(ECD/VCD)7.6 外加电场与磁场(Field)8 Gaussian进阶操作III:——激发态专题8.1 垂直激发能与绝热激发能8.2 垂直电离能与电子亲和能8.3 重整化能(重组能)8.4 激发态势能面8.5 激发态能量转移(EET)8.6 自然跃迁轨道(NTO)8.7 激发态计算方法讨论9 Gaussian进阶操作IV:——高精度和多尺度计算方法9.1 多参考态(CASSCF)方法及操作9.2 背景电荷法9.3 ONIOM方法与QM/MM方法及操作9.4 结合能( Binding Energy )和相互作用能(包含BSSE 修正,色散修正等)9.5 非平衡溶剂效应及其修正四、 Gaussian计算专题与实践应用10 Gaussian综合专题I:Gaussian报错及其解决方案10.1 如何查看报错及解决Gaussian常见报错10.2 专项:SCF不收敛解决方案10.3 专项:几何优化不收敛(势能面扫描不收敛)解决方案10.4 专项:消除虚频等解决方案10.5 专项:波函数稳定性解决方案11 Gaussian综合专题II:常用密度泛函和基组分类、特点及选择问题11.1 Jacobi之梯下的交换相关能量泛函11.2 常见交换相关泛函优缺点及用法11.3 长程修正泛函、色散修正泛函等11.4 常见基组特点及用法选择(自定义基组等,基组重叠误差等)12 Gaussian文献I: 聚集诱导荧光(AIE)和激发态分子内质子转移(ESIPT)12.1 聚集诱导荧光(AIE)与聚集诱导猝灭(ACQ)12.2 激发态质子转移ESIPT12.3 晶体结构及分子建模12.4 QM/MM与ONIOM计算12.5 重整化能,圆锥交叉及质子转移(文献:Dyes and Pigments Volume 204, August 2022, 110396 )13 Gaussian文献专题II: 热激活延迟荧光(TADF)13.1 热激活延迟荧光TADF机理13.2 分子内能量转移Jablonski图13.3 旋轨耦合与各类激发能13.4 辐射速率、非辐射速率、(反)系间穿越等13.5 评估荧光效率(文献: ACS Materials Lett. 2022, 4, 3, 487–496 )14 其他量化软件简介及总结Molcas/Molpro, Q-chem, lammps, Momap, ADF, Gromacs等详情:【腾讯文档】Gaussian量子化学、LAMMPS分子动力学课程。
Gaussian软件的基本原理与应用

利用变分原理,单粒子薛定谔方程中的单电子哈密顿算符可以被明确地 给出,这样得到的单电子薛定谔方程就称为哈特利—福克方程,或简称 为H-F方程,它是在总能量准确到一级近似的情况下得到的,具有如下形 式:
Hˆ (r1 ) K (q1 ) K K (q1 )
其中,Ĥ(r1)可称为H-F哈密顿算符。
计算简单,并可预测一些简单的分子,有时也可作为其它 近似方法进行迭代的基础
因此,HMO法仍为一种重要的分子轨道方法。
后来,Hoffmann把HMO法推广到包括σ电子的体系中去, 称为推广的休克尔分子轨道法(简称EHMO法)
虽然EHMO法的某些计算结果说明它是有前途的,但由于 它忽略了电子之间的排斥作用而受到激烈批评
缺点:HF方法忽略了电子相关,在一些特殊体系的应用 上,就显得不足。
(1)RHF 方程: 闭壳层体系,是指体系中所有的电子均按 自旋相反的方式配对充满某些壳层(壳层指一个分子能级或 能量相同的即简并的两个分子能级)。
体系的特点:是可用单斯莱特行列式表示多电子波函数(分 子的状态),描述这种体系的HFR方程称为限制性的HFR方 程,所谓限制性,是要求每一对自旋相反的电子具有相同的 空间函数。限制性的HFR方程简称RHF方程
计算资源 CPU、内存、硬盘…
计算精度: eV、kcal/mol、kJ/mol…
量子化学方法 从头算法、密度泛函、半经验…
计算化学程序的功能 能量以及能量梯度
活化能、离解能、电离能以及不同异构体之间的能量差…
指定电子态的分子性质
平衡结构、偶极矩 、极化率、振动频率、NMR位移…
不同电子态之间的跃迁特性
系;分子与分子之间的相互作用;分子与分子之间的相互碰撞和反应 等
gauc计算

gauc计算Gaussian计算是一种常用的计算化学方法,它可以用来计算分子的结构、能量、振动频率、光谱等性质。
Gaussian是一款商业软件,由Gaussian Inc.开发,目前已经发展到了Gaussian 16版本。
本文将介绍Gaussian计算的基本原理、使用方法以及一些常见的应用。
一、Gaussian计算的基本原理Gaussian计算的基本原理是量子力学。
在量子力学中,分子的运动状态可以用波函数来描述。
Gaussian计算就是通过求解分子的波函数来计算分子的性质。
具体来说,Gaussian计算可以分为以下几个步骤:1.构建分子模型。
在Gaussian中,可以通过手动输入分子的坐标或者从文件中读取分子的坐标来构建分子模型。
2.选择计算方法。
Gaussian支持多种计算方法,包括Hartree-Fock方法、密度泛函理论方法、半经验方法等。
不同的计算方法适用于不同的分子体系和性质计算。
3.设置计算参数。
在进行计算之前,需要设置一些计算参数,如基组、收敛标准、计算级别等。
这些参数的选择会影响计算结果的精度和计算时间。
4.进行计算。
在设置好计算参数之后,就可以进行计算了。
Gaussian会自动求解分子的波函数,并计算分子的性质。
5.分析计算结果。
计算完成后,需要对计算结果进行分析。
Gaussian可以输出分子的结构、能量、振动频率、光谱等性质,可以通过这些性质来判断计算结果的可靠性和分子的性质。
二、Gaussian计算的使用方法Gaussian计算是一款非常强大的计算化学软件,但是对于初学者来说,使用起来可能会比较困难。
下面介绍一些Gaussian计算的使用方法,帮助初学者更好地使用这款软件。
1.学习基本原理。
在使用Gaussian之前,需要先学习一些基本的量子化学原理,如分子轨道理论、Hartree-Fock方法、密度泛函理论等。
只有掌握了这些基本原理,才能更好地理解Gaussian计算的原理和方法。
gaussian在电催化中的应用

gaussian在电催化中的应用
Gaussian是在电催化中常用的量子化学计算软件,用于模拟分子的电子结构和性质。
在电催化反应中,反应物分子在电极表面吸附并发生电子转移和化学转化,生成产物分子。
这个过程中涉及到的吸附能、电子转移能、化学转化能等性质都可以通过Gaussian进行计算和预测。
以下是一些Gaussian在电催化中应用的具体例子:
1. 计算反应能垒:通过Gaussian计算反应物和产物分子的能量,可以预测反应的能垒,从而推断出反应速率常数和反应条件下的反应方向。
2. 预测吸附能和吸附模式:电催化反应中,反应物分子需要在电极表面吸附才能进行反应。
Gaussian可以计算分子在电极表面的吸附能,从而预测分子的吸附模式和稳定性。
3. 模拟电子转移过程:在电催化反应中,电子转移是关键步骤之一。
Gaussian可以模拟电子转移过程中分子的电子结构和电荷分布变化,从而深入理解电子转移机制。
4. 优化催化剂结构和性能:电催化反应中,催化剂的性能对反应速率和选择性有很大影响。
Gaussian可以通过计算催化剂的电子结构和性质,预测其活性和选择性,为催化剂的设计和优化提供理论指导。
总的来说,Gaussian在电催化中的应用可以帮助我们深入理解电催化反应的机制和过程,预测反应的能垒、反应速率、产物选择性等性质,优化催化剂的结构和性能,为电催化技术的发展提供理论支持。
gaussian 单个原子能量计算

gaussian 单个原子能量计算Gaussian单个原子能量计算引言:在化学和物理学领域,原子能量计算是一项重要的研究内容。
而Gaussian是一款被广泛使用的计算化学软件,可以用于模拟和计算分子系统的能量和性质。
本文将探讨Gaussian在单个原子能量计算中的应用。
一、Gaussian软件简介Gaussian是一款基于量子力学原理开发的计算化学软件。
它采用了密度泛函理论(DFT)和分子力场(MM)等方法,能够对分子和原子进行精确的能量计算和结构优化。
Gaussian具有丰富的功能和灵活的参数设置,可以模拟和计算各种化学反应和性质。
二、Gaussian在单个原子能量计算中的原理Gaussian使用基于量子力学的方法来计算原子的能量。
在计算中,原子的电子结构被建模为一组波函数,通过求解薛定谔方程来获得这些波函数。
Gaussian采用了Hartree-Fock方法和密度泛函理论等不同的计算方法,可以得到不同精度的能量计算结果。
三、Gaussian在单个原子能量计算中的步骤1. 原子结构优化:首先,需要通过Gaussian进行原子结构的优化。
通过调整原子的几何构型,使得原子的能量达到最低点。
这一步需要设置优化的算法和收敛准则,以确保得到准确的原子结构。
2. 波函数计算:在得到优化的原子结构后,Gaussian将计算出原子的波函数。
波函数描述了原子的电子分布和能级,是计算原子能量的基础。
3. 能量计算:基于得到的波函数,Gaussian可以计算出原子的能量。
能量计算是通过对波函数进行积分和求和得到的,包括动能、势能和相互作用能等各项贡献。
四、Gaussian在单个原子能量计算中的应用举例以氢原子为例,使用Gaussian进行能量计算的步骤如下:1. 设置计算参数:选择合适的方法和基组,如Hartree-Fock方法和6-31G基组。
2. 原子结构优化:通过优化算法,调整氢原子的几何构型,使得能量达到最低点。
Gaussian软件应用

计算采用的基组,如 6-31G, Lanl2DZ; 布局分析方法,如 Pop=Reg; 波函数自恰方法,如 SCF=Tight. Pop=Reg 只在输出文件中打印出最高的 5 条 HOMO 轨道和最低的 5 条 LOMU 轨道,而采用 Pop=Full 则打印出全部的分子轨道. SCF 设置是指波函数的收敛计算时的设定,一般不用写,SCF=Tight 设置表示采用比 一般方法较严格的收敛计算. 计算的名称 一般含有一行,如果是多行,中间不能有空行.在这里描述所进行的计算. 分子结构 首先是电荷和自旋多重度 电荷就是分子体系的电荷了,没有就是 0, 自旋多重度就是 2S+1,其中 S 是体系的总自旋量子数,其实用单电子数加 1 就是了. 没有单电子,自旋多重度就是 1. 然后是分子几何构性,一般可以用迪卡尔坐标,也可以用 Z-矩阵(Z-Matrix) 多步计算 Gaussian 支持多步计算,就是在一个输入文件中进行多个计算步骤. 2.3 输出文件中的信息 例 2.1 文件 e2_01 甲醛的单点能 标准几何坐标. 找到输出文件中 Standard Orientation 一行,下面的坐标值就是输入分子的标准 几何坐标. 能量 找到 SCF Done: E(RHF)= -113.863697598 A. U. after 6 cycles 这里的树脂就是能量,单位是 hartree.在一些高等级计算中,往往有不止一个能量 值,比如下一行 E2=-0.3029540001D+00 EUMP2=-0.11416665769315D+03
Gaussian 包含多种化学模型,比如 计算方法 Gaussian 关键词 方法 HF Hartree-Fock 自恰场模型 B3LYP Becke 型 3 参数密度泛函模型,采用 Lee-Yang-Parr 泛函 MP2 二级 Moller-Plesset 微扰理论 MP4 四级 Moller-Plesset 微扰理论 QCISD(T) 二次 CI 具体在第六章讨论 基组 基组是分子轨道的数学表达,具体见第五章 开壳层,闭壳层 指电子的自旋状态,对于闭壳层,采用限制性计算方法,在方法关键词前面加 R 对于开壳层,采用非限制性计算方法,在方法关键词前面加 U.比如开壳层的 HF 就是 UHF.对于不加的,程序默认为是闭壳层. 一般采用开壳层的可能性是 1. 存在奇数个电子,如自由基,一些离子 2. 激发态 3. 有多个单电子的体系 4. 描述键的分裂过程 模型的组合 高精度的计算往往要几种模型进行组合,比如用中等算法进行结构优化,然后用高 精度算法计算能量. Gaussian 软件应用 第二章 单点能计算
gaussian基本概念和用法
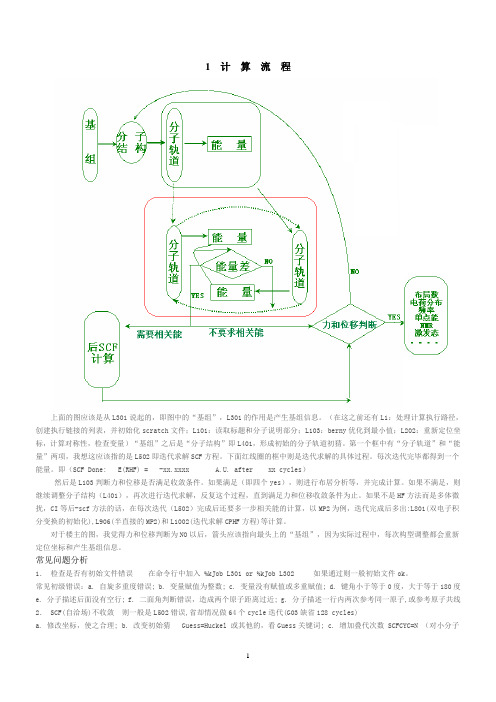
1 计算流程上面的图应该是从L301说起的,即图中的“基组”,L301的作用是产生基组信息。
(在这之前还有L1:处理计算执行路径,创建执行链接的列表,并初始化scratch文件;L101:读取标题和分子说明部分;L103:berny优化到最小值;L202:重新定位坐标,计算对称性,检查变量)“基组”之后是“分子结构”即L401,形成初始的分子轨道初猜。
第一个框中有“分子轨道”和“能量”两项,我想这应该指的是L502即迭代求解SCF方程。
下面红线圈的框中则是迭代求解的具体过程。
每次迭代完毕都得到一个能量。
即(SCF Done: E(RHF) = -xx.xxxx A.U. after xx cycles)然后是L103判断力和位移是否满足收敛条件。
如果满足(即四个yes),则进行布居分析等,并完成计算。
如果不满足,则继续调整分子结构(L401),再次进行迭代求解,反复这个过程,直到满足力和位移收敛条件为止。
如果不是HF方法而是多体微扰,CI等后-scf方法的话,在每次迭代(L502)完成后还要多一步相关能的计算,以MP2为例,迭代完成后多出:L801(双电子积分变换的初始化),L906(半直接的MP2)和L1002(迭代求解CPHF方程)等计算。
对于楼主的图,我觉得力和位移判断为NO以后,箭头应该指向最头上的“基组”,因为实际过程中,每次构型调整都会重新定位坐标和产生基组信息。
常见问题分析1.检查是否有初始文件错误在命令行中加入 %kJob L301 or %kJob L302 如果通过则一般初始文件ok。
常见初级错误:a. 自旋多重度错误; b. 变量赋值为整数; c. 变量没有赋值或多重赋值; d. 键角小于等于0度,大于等于180度e. 分子描述后面没有空行; f. 二面角判断错误,造成两个原子距离过近; g. 分子描述一行内两次参考同一原子,或参考原子共线2. SCF(自洽场)不收敛则一般是L502错误,省却情况做64个cycle迭代(G03缺省128 cycles)a. 修改坐标,使之合理;b. 改变初始猜 Guess=Huckel 或其他的,看Guess关键词;c. 增加叠代次数 SCFCYC=N (对小分子作计算时最好不要增加,很可能结构不合理); d. iop(5/13=1)这样忽略不收敛,继续往下做。
gaussian 磷光光谱

gaussian 磷光光谱摘要:1.概述2.磷光光谱的基本原理3.gaussian 磷光光谱的应用4.总结正文:1.概述磷光光谱是一种测量物质发光性质的光谱技术,通过记录样品在激发光源照射下产生的发光强度与波长关系,可以获得磷光光谱。
在众多磷光光谱软件中,Gaussian 是一个广泛使用的软件,它不仅可以模拟各种光谱,还可以进行分子轨道计算和其他相关计算。
本文将介绍Gaussian 磷光光谱的原理及其应用。
2.磷光光谱的基本原理磷光光谱的基本原理是:当分子或原子受到能量激发后,会从基态跃迁到激发态,然后再返回到基态。
在返回基态的过程中,分子或原子会释放出能量,这个过程就是发光。
磷光光谱通过测量样品在不同波长下的发光强度,可以获得样品的磷光光谱。
3.gaussian 磷光光谱的应用Gaussian 磷光光谱在许多领域都有广泛应用,例如材料科学、生物化学、环境监测等。
以下是Gaussian 磷光光谱在几个领域的应用实例:(1) 材料科学:通过研究有机材料的磷光光谱,可以了解材料的发光性能、能量传递和激发态寿命等性质,从而优化材料的发光性能。
(2) 生物化学:在生物化学领域,Gaussian 磷光光谱可用于研究生物大分子(如蛋白质、核酸等)的结构和功能。
此外,通过对生物分子的磷光光谱研究,还可以了解其在生物体内的作用机制。
(3) 环境监测:Gaussian 磷光光谱可以用于监测环境中的有害物质,如重金属离子、有机污染物等。
通过检测这些物质在特定波长下的磷光信号,可以实现对环境污染的快速、灵敏检测。
4.总结Gaussian 磷光光谱作为一种重要的光谱分析方法,在材料科学、生物化学和环境监测等领域具有广泛的应用。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
(1)RHF 方程: 闭壳层体系,是指体系中所有的电子均按 自旋相反的方式配对充满某些壳层(壳层指一个分子能级或 能量相同的即简并的两个分子能级)。
体系的特点:是可用单斯莱特行列式表示多电子波函数(分 子的状态),描述这种体系的HFR方程称为限制性的HFR方 程,所谓限制性,是要求每一对自旋相反的电子具有相同的 空间函数。限制性的HFR方程简称RHF方程
将LCAO法用于H-F方程中,并经一系列数学处理,便可 得到下列代数方程组:
(F K S )aK 0
式中:μ=1,2,···,n
(1-5)
式(1-5)即是罗汤方程。
求解罗汤方程的困难之处,还在于: (1) 计算矩阵元时要计算大量的积分,积分的数量与方程阶数n的4次方
(这从四个指标的双电子积分不难看到)成正比
分子轨道函数是描述电子在分子中的运动状态的,它属于整个分子,并 由分子的结构所决定
由于一个分子的结构与组成这个分子的那些原子的结构密切相关,所以 把分子中各个原子所属的某些原子轨道函数适当地线性组合起来,就有 可能得到反映电子在整个分子中运动的轨道函数
LCAO方法的成功也反过来说明了上述想法的合理性。
在里德堡单位中的哈密顿算符为:
N
Hˆ
i 1
A
2
1
u i P1 P
N
2
A 2ZP N
1
A
ZPZq
r P i1 P1 iP
r r i j1 ij Pq1 Pq
(1.2)
1.2、波恩—奥本海默(Born-Oppenheimer)近似
式(1.1)的哈密顿算符所决定的分子波函数Ψ,同时反映各个不同的原 子核和电子的运动状态。
(1-4)
1.4 LCAO自洽场方法和罗汤(Roothaan)方程
H-F方程的严格求解仍是很困难的,即使采取迭代自洽的办法进行求 解,也是相当繁复的
人们设计了若干近似方法来求解,其中一种就是分子轨道用原子轨道 的线性组合来逼近,即LCAO(Linear Combination of Atomic Orbitals) 方法。
描述这类体系的最常用的方法是假设自旋向上的电子( 自旋)和自旋向下 的电子(β自旋)所处的分子轨道不同,即不限制自旋相反的同一对电子 填入相同的分子轨道。
这样得到的HFR方程称为非限制性的HFR方程,简称UHF方程。
(3)从头计算法:原则上讲,有了HFR方程(不论是RHF方程或是UHF方 程),就可以计算任何多原子体系的电子结构和性质 真正严格的计算称 之为从头计算法。
量子化学
分子动力学
计算机的发展
计算化学
计算化学能解决的问题
预测分子结构
↓
预测分子的化学性质
给定一个化 学式,通过 理论计算定 出它的最优 结构
单分子性质 集团性质(容量性质) 不可测量性质
分 振红 电
子 动外 多
的 频和 极
能 量 和
率
拉 曼
矩
结光
构谱
电热 NMR 成
子化 屏 键
亲学 蔽 和
和性 和 化
4. 从头算法
从头算,在解薛定鄂方程的过程中,只采用了几个物理 常数,包括光速,电子和核的质量,普朗克常数,在求解薛 定鄂方程的过程中采用一系列的数学近似,不同的近似也就 导致了不同的方法。
最经典的是 Hartree-Fock方法,缩写为 HF。 HartreeFock方法对于稳定的分子和一些过渡金属的处理能够得到很 好的结果, 是一个很好的基本的理论方法。
a11 a22 ann
φ1,φ2,···,φn表示原子轨道,它们是已知的
分子轨道和原子轨道一样,它也是相应的单电子哈密顿算符 Ĥ的本征函数:
Hˆ E
但在HMO法中,并不去考虑如何求解哈密顿算符的本征方程, 而是以原子轨道的线性组合作为尝试波函数,用线性变分法 来求得近似的分子轨道波函数。
(2) 这些积分一般都是较难处理的多中心积分,多中心积分必须经过变换 之后方可进行积分,这种变换和随后的积分都是很繁杂的
所以,对于罗汤方程的求解,往往也采取各种近似的方法进行处理
后文将介绍的各种半经验方法都是以不同的近似程度和方法来求解罗 汤方程而建立的。
2、组态相互作用(Configuration Interaction,CI)
(2)UHF 方程:开壳层体系,是指体系中有未成对的电子(即有的
壳层未充满)
体系的特点:描述开壳层体系的波函数一般应取斯莱特行列式的线性组 合,这样,计算方案就将很复杂。
然而对于开壳层体系的对应极大多重度(所谓多重度,指一个分子因总 自旋角动量的不同而具有几个能量相重的状态)的状态(即自旋角动量最 大的状态)来说,可以保持波函数的单斯莱特行列式形式(近似方法)。
组态相互作用方法在计算时考虑了电子相关能。
Φk N! 1/2 ψk1 x1 ψk2 x2 ψkN xN
任何多电子波函数都可以用它来展开 一般{Ψk(x)}称为轨道空间, {Φk}称为组态空间。
3、各种半经验量子化学近似方法
3.1 休克尔(Hückel)分子轨道法
分子轨道理论中最简单的一种处理, 最初由Hückel提出,因此称Hückel分子轨道法, 缩写为HMO法。
➢用以解释一些具体的化学问题。
经典力学
1687 Newton
量子力学
1926
Heisenberg SchrÖ dinger Born
统计力学
Before 1900 Boltzmann
1946 Westheimer Mayer
分子力学
分子和物质结构问题
1927
Heitler London
1957 Alder Wainwright
计算简单,并可预测一些简单的分子,有时也可作为其它 近似方法进行迭代的基础
因此,HMO法仍为一种重要的分子轨道方法。
后来,Hoffmann把HMO法推广到包括σ电子的体系中去, 称为推广的休克尔分子轨道法(简称EHMO法)
虽然EHMO法的某些计算结果说明它是有前途的,但由于 它忽略了电子之间的排斥作用而受到激烈批评
子的薛定谔方程的问题。
利用变分原理,单粒子薛定谔方程中的单电子哈密顿算符可以被明确地 给出,这样得到的单电子薛定谔方程就称为哈特利—福克方程,或简称 为H-F方程,它是在总能量准确到一级近似的情况下得到的,具有如下形 式:
Hˆ (r1 ) K (q1 ) K K (q1 )
其中,Ĥ(r1)可称为H-F哈密顿算符。
运动的问题
而电子又都是电荷、质量、自旋等特征完全相同的粒子,因此,分子结 构问题的研究就转化为N个全同粒子体系的问题的研究
1.3 哈特利—福克(Hartree-Fock)方程(H-F方程)
引入单粒子模型近似(又称轨道近似) 该近似模型假定:每个电子都是在一个平均场中独立运动,平均场是稳
定不变的,每个电子在平均场中的势能只是单电子坐标的函数。 这样,求解N个粒子体系的薛定谔方程的问题,就可归结为求解一个单粒
狄拉克的宣言: “大部分物理和全部化学问题的基本定律已经找到,唯一的困难
是这些定律导出的数学方程过于复杂而无法求解。” The underlying physical laws necessary for the mathematical theory
of a large part of physics and the whole of chemistry are thus completely known, and the difficulty is only that the exact application of these laws leads to equations much too complicated to be soluble.
Paul. A. M. Dirac, Proc. Roy. Soc. London Ser. A 123, 714 (1929).
一、计算化学基本原理 二、Gaussian使用方法及实例
第一部分:基本原理
量子化学:应用量子力学的基本原理和方法研究化学问题的化学分支 学科。
理论基础:量子力学 计算工具:计算机 研究内容:物质的微观结构与宏观性能的关系 研究目的:解释物质和化学反应具有的特性的内在本质及其规律性 研究范围:稳定和不稳定分子的结构、性能及其结构与性能之间的关
(3) 假定分子中任何两个原子轨道之间都不发生重叠,即若 m≠n,则Smn=0。
事实上,相邻成键碳原子的2pz轨道之间的Smn≈0.25,然而,考虑了这种 非正交性就加大了计算工作量,而并不能使计算结果的准确性有明显的 提高。
HMO法有时仍与实验结果相差很大,归根结底是由于它忽 略了电子之间的排斥能------致命的弱点。
RHF方程的极限能量与非相对论薛定谔方程的严格解之差称为相关能。 对于某些目的,还需要考虑体系的相关能。
UHF方程考虑了相关能的一小部分,更精密的作法则须取多斯莱特行列 式的线性组合形式的波函数,由变分法求得这些斯莱特行列式的组合系 数。
这些由一个斯莱特行列式或数个斯莱特行列式按某种方式组合所描述的 分子的电子结构称为组态,所以这种取多斯莱特行列式波函数的方法称 为组态相互作用法(简称CI)。
但是,以上方程一般是很难求解的。
为使方程易于求解,经常采取的一个近似就是波恩—奥本海默近似。这 个近似的主要点,就是假定原子核不动。
代入式(1.1),则:
(Hˆ 'I ) E
即:
Hˆ ' E'
(1-3)
其中,
E' E I
可见: 在波恩—奥本海默近似下,方程(1.1)的求解问题就归结为求解式(1.3) 而式(1.3)的哈密顿Ĥ΄以及波函数Ψ都仅仅是电子坐标ri的函数 这样,研究一个分子内部运动的问题,就变为讨论N个电子在固定核场中