序列组装的过程
RNA-seq(转录组学)的分析流程和原理

RNA-seq(转录组学)的分析流程和原理在开始详细讲解RNA测序之前,我们先来了解一下它的基本步骤:1.建库:提取RNA,富集mRNA或消除rRNA,合成cDNA和构建测序文库。
2.测序:然后在高通量平台(通常是Illumina)上进行测序(每个样本测序reads在DNA测序中,读数是对应于单个DNA片段的全部或部分的碱基对(或碱基对概率)的推断序列。
深度为10-30 Million reads。
)3.分析:先比对/拼装测序片段到转录本,通过计数、定量,样本间过滤和标准化,以进行样本组间基因/转录本统计差异分析。
大致了解这个过程之后,我们就先从建库开始了解建库的难点在于提纯出mRNA, 一般在我们抽离出的RNA中rRNA占比很大,其他还会有tRNA、microRNA等。
我们需要从抽离出的RNA中提取出mRNA,并建立cDNA文库。
这里以应用最广泛的Illumina公司的Truseq RNA的建库方法为例来进行介绍。
首先,利用高等生物的mRNA通常有poly(A)尾的(使mRNA更稳定,翻译不容易出错)特点,用带有poly(T)探针的磁珠与总RNA进行杂交,这样磁珠就和带poly(A)尾巴的mRNA结合在一起了。
接下来,就回收磁珠,把这些带poly(A)的mRNA从磁珠上洗脱下来。
再用镁离子溶液(或者超声波)进行处理,把mRNA打成小段。
然后,利用这些被打断的mRNA片段,以随机引物进行逆转录,得到第一链cDNA。
再根据第一链cDNA合成出ds-cDNA。
对cDNA在平末端进行3’端加A碱基(腺苷酸)(adapter接头上带了T碱基头,为了和adapter配对)在双链cDNA的两端加分别上Y型接头再经PCR扩增经筛选的目的基因,就得到可以上机的测序文库了。
这个建库方法对RNA的完整度有较高的要求。
也就是说,只有在mRNA大部分是完整的状态下,才能得到比较好的效果。
因为带Poly(T)的磁珠,它所吸附的是带有Poly(A)的那些序列。
现代分子生物学第3版【第二章】课后习题答案

第二章染色体与DNA一、染色体具备哪些作为遗传物质的特征?1、分子结构相对稳定;2、能够自我复制,使亲子代之间保持连续性;3、能够指导蛋白质的合成,从而控制整个生命过程;4、能够产生可遗传的变异。
二、什么是核小体?简述其形成过程。
核小体是染色质(体)的基本结构单位,由DNA和组蛋白组成。
形成过程:四种组蛋白H2A、H2B、H3、H4各两个分子生成八聚体,约200bp的DNA 盘绕在八聚体外面,形成一个核小体,而H1则在核小体的外面,每个核小体只有一个H1,该过程使分子收缩之原尺寸的1/7。
三、简述真核生物染色体的组成及组装过程。
组成:蛋白质+DNA组装过程:①核小体形成:DNA盘绕组蛋白形成的八聚体外,该阶段分子尺寸压缩7倍。
②染色质细丝及螺线管:前者由核小体串联形成,后者为细丝盘绕而成,该阶段分子尺寸压缩6倍。
③螺线管压缩为超螺旋:该阶段分子尺寸压缩40倍。
④超螺旋形成染色单体:该阶段分子尺寸压缩5倍。
四、简述DNA的一、二、三级结构特征。
1、一级结构:脱氧核苷酸排列顺序,相邻核苷酸通过磷酸二酯键相连。
2、二级结构:双螺旋结构。
a)两条平行的脱氧核苷酸链螺旋盘绕而成。
b)外侧骨架为脱氧核苷和磷酸,内侧为碱基序列。
c)两条链通过碱基之间形成氢键而结合,碱基结合遵循互补配对原则,A-T,G-C。
3、三级结构:空间结构,即超螺旋,包括正超螺旋和负超螺旋,二者可以在拓扑异构酶的作用下相互转变。
四、原核生物DNA具有哪些不同于真核生物DNA的特征?整体特点概括为以下三点:1、一般只有一条染色体,且大多为单拷贝基因。
2、整个染色体DNA几乎全部由功能基因加调控序列所组成。
3、几乎每个基因序列都与编码的蛋白质序列呈线性对应关系。
从基因组的组织结构来看,原核细胞DNA有如下特点:1、结构简练:绝大部分都用来编码蛋白质,与真核DNA的冗余现象不同。
2、存在转录单元:功能相关的RNA和蛋白质基因往往丛集在基因组的特定部位,形成功能单位或转录单元,可以被一起转录为含多个mRNA的分子,称为多顺反子mRNA。
蜜蜂序列组装分析及SNP位点检测

蜜蜂序列组装分析及SNP位点检测蜜蜂是我们非常熟悉的昆虫之一,也是非常重要的生态系统组成部分。
在蜜蜂的研究中,基因组学技术也越来越受到关注和应用。
本文将介绍蜜蜂基因组组装和SNP位点分析的相关内容。
一、蜜蜂基因组组装基因组组装是将测序数据转换为完整的基因组序列的过程。
蜜蜂基因组组装的过程和其他生物物种的基因组组装类似,但由于其基因组大小较小,组装过程相对较容易。
蜜蜂基因组组装的第一步是建立一个高质量的基因组序列库。
这包括用不同的方法制备高质量的DNA样品、建立测序文库并进行高通量测序等。
蜜蜂的基因组测序是高度复杂的过程,需要通过多个测序平台(如Illumina HiSeq、PacBio等)进行组合。
在获得测序数据后,需要对数据进行预处理,如去除低质量序列、去除冗余序列、纠正测序错误等。
然后,将这些清洗后的序列通过不同的软件进行组装,并利用其他评估工具对组装质量进行评估。
最终的基因组序列可以通过验证和加工来达到最终的精度。
二、SNP位点检测SNP(single nucleotide polymorphism)是指基因组中的单个碱基差异。
SNP是生物基因组中最常见的组成成分之一,也是进化研究和基因组组装等生物信息学研究中广泛应用的工具之一。
在蜜蜂研究中,SNP位点分析可以帮助我们了解种群群体、家系和探测基因功能等。
SNP位点检测的步骤包括:(1)基因组序列和基因序列的比对;(2)确立SNP位点;(3)SNP位点筛选和统计;(4)SNP位点功能分析。
首先,需要将测序数据比对到参考基因组序列上,然后使用SNP检测软件如SAMtools、GATK等,通过生物统计学方法筛选SNP位点。
接下来,使用过滤器将SNP位点进行分组和筛选,去除无效SNP位点,比如低质量位点。
最终,SNP位点的功能分析可以通过注释工具进行。
这包括检测SNP位点是否对蛋白质编码区域有影响、是否为突变位点等。
三、应用和展望蜜蜂基因组组装和SNP位点检测技术对于我们了解蜜蜂适应性进化、抗逆性、基因结构和基因功能都有着重要的意义。
TALEN技术原理及应用

Nuclease domain: DNA剪切域
▲FokI,发现于海床黄杆菌 Flavobacterium okeanokoites
▲Type IIs 限制性内切酶
▲采取其非特异性DNA结合域
▲形成二聚体时发生剪切
(三)TALEN基本原理
TALE靶点识别模块构建;
至雄原核中,另一半注射至受精卵胞质中
Chimera小鼠
Nat Biotechnol 2013 Jan; 31(1):23-4
比对(氨基酸blast)
获得最终测序正确的TALEN质粒
左臂序列比对(L3)
右臂序列比对(R3)
五、TALEN质粒活性检测
活性检测确定TALEN打靶的效率
细胞培养、铺板准备(MEFs、3T3-L1)
Fugene HD共转染eGFP-Puro质粒(24H—48H?)
嘌呤霉素筛选(kill curve)
DNA质粒抽提
酶切鉴定
质粒测序
测序正
确质粒
Step 7
Step 8
Step 9
Step 10
受精卵注
射mRNA
Chimer
a小鼠
F1杂合
子小鼠
F2代纯合
子小鼠
三、TALEN打靶载体设计策略
确定打靶的基因以及靶位点
① 输入基因名点击
search
②搜索结果中选择物种,如Mus musculus
③点击打开该基因序列 选择
电泳检测mRNA是否有降解(防止Rnase酶污染)(3300-3400 nt)
mRNA可以长保存在Nuclease-free Water (-20°C)1~2个月
无参转录组序列组装及实际操作
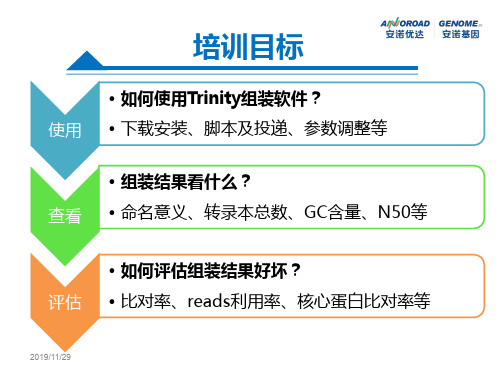
2019/11/29
Trinity使用—输入及输出
输入文件: fa或者fq文件
创建一个文件存放输出结果的目录:
mkdir assemble
框移错误导 致的缺口以 及过早终止
的比例。
2019/11/29
组装评估
判断标准: ① 无外源物种污染。
② 比对率大于80%。
组装评估
物种近缘性 良好 CDS序列相 对完整 60%以上
注释比率 核心蛋白 比对率
80%以上
准确性
2019/11/29
Stop Codon比率20%以下
Trinity参数调整
2019/11/29
实际操作
本地运行:sh triniy.sh
任务运行
本地挂起运行:nohup sh triniy.sh &
投递运行:qsub –cwd –l vf=10G –l p=5 triniy.sh
任务查看:qstat/qstat –j job_number/jobs
2019/11/29
full_cleanup
只保留组装结果文件,并以Trinity.fasta命 名。
group_pairs_distance 双端reads比对的最大长度(超过该长度认为 没有比对上)
min_kmer_cov
最小k-mer覆盖值。
2019/11/29
Trinity使用—任务及运行
生成组装任务脚本:vi trinity.sh
转录本数目过多,但是N50低,怎么办? 数据量太大,如何提高组装速度? 物种类型是真菌,参数需要注意什么?
组装基本流程
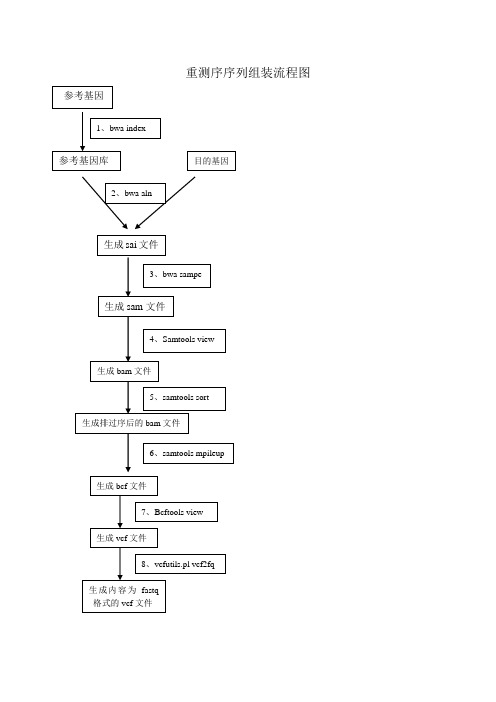
重测序序列组装流程图用户名:student 密码:1234561、bwa index就是给参考序列建索引,参数用的是默认参数。
bwa index ref.fa2、bwa aln 是把目标序列和参考基因序列进行比对bwa aln –t 6 ref.fa reads.fq > reads.sai3、bwa sampe 比对pair-end reads,输出SAM格式文件。
因为我们的基本上是pair-end reads,所以用sampe。
如果是比对single-end reads,就要用bwa samse了。
用的参数用的是默认参数。
bwa sampe ref.fa reads_1.sai reads_2.sai reads_1.fq reads_2.fq > reads.sam4、samtools view把sam文件转换成bam文件。
samtools view -bS reads.sam -o reads.bam5、samtools sort对bam文件进行排序,默认按染色体名称和位置大小顺序。
samtools sort reads.bam reads.sort6、samtools mpileup生成位点信息文件,可以通过不同的参数来的到不同的结果,mapping用的参数是-uS,之后找过snp,用的参数是-DSugf。
把bam文件转换成bcf格式文件。
samtools mpileup –us reads.sort.bam > reads.bcf7、bcftools view是把bcf文件输出为vcf格式。
bcftools view -cg reads.bcf >reads.vcf8、vcfutils.pl vcf2fq把结果的一致性序列整理成fastq文件。
vcfutils.pl vcf2fq reads.vcf >result.fq。
RNA序列拼接与组装分析的步骤与技巧

RNA序列拼接与组装分析的步骤与技巧随着高通量测序技术的发展,RNA序列在生物学研究中扮演着重要的角色。
在进行RNA测序后,我们需要将这些短片段的序列拼接起来,以便进行进一步的分析。
RNA序列的拼接与组装分析对于研究基因表达调控、发掘新的转录本、研究剪接变异等具有重要意义。
本文将介绍RNA序列拼接与组装分析的步骤与技巧。
1. 数据预处理在进行RNA序列拼接与组装分析前,需要对测序数据进行预处理。
常见的预处理步骤包括去除测序引物序列、去除低质量序列、去除接头序列等。
这些步骤可以使用专业的测序数据分析软件进行,如Trimmomatic、Fastp等。
预处理后的数据更适合进行后续的分析。
2. 数据质量评估在进行RNA序列拼接与组装分析之前,需要对数据质量进行评估。
这可以通过软件工具进行,如FASTQC、Nanoplot等。
数据质量评估有助于判断测序数据的可靠性,并进一步了解实验中的潜在问题,如测序深度是否足够、样品间的比较是否可靠等。
评估结果有助于优化后续的分析步骤。
3. 序列拼接序列拼接是将测序得到的短片段序列拼接成完整的转录本序列。
对于无刺激(unstimulated)的RNA测序数据,可以使用常规的拼接方法,如TGICL、CAP3等。
对于有刺激(stimulated)的RNA测序数据,由于存在剪接变异等复杂情况,通常需要使用更高级的拼接方法,如Trinity、StringTie等。
这些软件提供了多种算法和参数选项,可以根据实际情况选择适合的方法。
4. 异常削减与错误修复在进行序列拼接后,可能会存在部分异常序列或序列错误的情况,如插入缺失、碱基替换等。
为了消除这些异常序列的影响,可以使用异常削减(abundance filtering)和错误修复(error correction)的方法。
异常削减通过比对已知转录本或参考基因组来过滤掉异常或假阳性的序列。
错误修复可以根据测序深度和碱基质量分数来对序列进行修正,常见的错误修复工具有Rcorrector、BFC等。
一讲基因组测序与序列组装

感谢您的观看
THANKS
03
基因组序列组装
序列组装的基本流程
序列读取
通过测序技术获取基 因组序列的原始数据。
序列质量评估
对原始数据进行质量 评估,去除低质量序 列和错误序列。
序列比对
将高质量序列比对到 参考基因组或组装到 独立的基因组上。
序列拼接
将比对或独立基因组 上的序列片段拼接成 完整的基因组。
组装后验证
对组装得到的基因组 进行验证,确保其完 整性、准确性和一致 性。
下一代测序技术
总结词
更高通量、更低成本、更短周期的测序技术。
详细描述
下一代测序技术是一种尚未完全成熟的测序技术,目 前正处于研究和发展阶段。相比于前几代测序技术, 下一代测序技术将具有更高的通量、更低的成本和更 短的周期等特点。它可能采用更加先进的纳米技术、 光学技术和生物信息学技术等手段,以提高测序的准 确性和速度。下一代测序技术的出现将为基因组学和 生物医学领域的研究提供更加高效装得到的基因组的完整性,包括染色 体水平的完整性和基因水平的完整性。
准确性评估
评估组装得到的基因组的准确性,包括单核苷酸水 平上的准确性和结构变异上的准确性。
一致性评估
评估组装得到的基因组的一致性,包括不同 组装方法或不同数据集之间的一致性和内部 的一致性。
04
基因组测序与序列组装的挑 战与前景
例如,通过研究水稻基因组,科学家们发现了与抗旱、耐盐等抗逆性状相关的基因,为培育抗逆性更强的水稻品种提供了重 要的理论依据。
病原微生物基因组研究
病原微生物基因组研究是利用基因组测序和序列组装技术来了解病原微生物的基因组结构和功能,旨 在发现新的药物靶点、疫苗候选基因和诊断标记物等。
基因组组装的几个阶段

基因组组装的几个阶段1.引言1.1 概述基因组组装是一项重要的生物信息学任务,旨在将原始的DNA片段重新组合成完整的基因组序列。
在这个过程中,需要经历几个关键阶段。
本文将详细介绍基因组组装的几个阶段及其重要性。
基因组组装的第一阶段是数据质量控制和预处理阶段。
由于测序技术等因素的限制,原始DNA序列可能包含错误或低质量的片段。
因此,在组装之前,需要对原始数据进行质量控制和预处理,以去除噪声和提高数据的准确性和可靠性。
这一步骤包括去除低质量的碱基,修剪适配器序列,过滤重复的片段等等。
通过数据质量控制和预处理,我们可以获得高质量的数据,为下一阶段的组装提供可靠的基础。
基因组组装的第二阶段是序列拼接阶段,也被称为contig拼接。
在这个阶段,通过将大量的短序列片段(reads)按照其重叠关系进行拼接,得到长度更长的连续序列(contig)。
这个过程依赖于计算机算法和数学模型,例如格拉布斯算法和De Bruijn图。
通过序列拼接,我们可以在一定程度上重建原始DNA序列,但仍然存在一些空缺和不确定性。
基因组组装的第三阶段是contig的连接和填充,也被称为scaffolding。
在这个阶段,利用额外的信息,如配对的reads间的距离和方向关系,对contig进行进一步的排序和连接,填补contig之间的空缺。
这些额外的信息可以来自于配对的短序列片段(paired-end reads)或长读长度的第三代测序技术。
scaffolding可以提高基因组组装的连续性和准确性,从而得到更接近真实基因组序列的结果。
综上所述,基因组组装可以分为数据质量控制和预处理、序列拼接以及contig的连接和填充三个阶段。
每个阶段都具有其独特的重要性和挑战,但它们共同协作以实现高质量的基因组组装。
随着测序技术的不断发展和算法的改进,基因组组装的效果和精确度也将不断提高,为生物学研究和应用提供更精准和全面的基因组信息。
1.2 文章结构文章结构部分的内容如下:文章结构本文主要讨论基因组组装的几个关键阶段。
基因组序列组装的理论与方法简介

CAP3(1999)
• 特点:
– 删去read两端低质量部分; – 利用质量数据,识别重叠序列;进行多序列比
对,得到一致序列; – 利用正反向数据纠正组装错误,构建scaffold。
错误组装的Contig: 测序数据组装中出现的错误。由定义, 它涉及的片段一般大于500-bp。包括与参考序列相比,插入、 删除,以及在方向和次序上不同的片段。
错误组装的Scaffold:把非重叠contig连接在一起时出现的错误。 包括嵌套,错误的方向和顺序等。
Shotgun Sequencing Assembler Concepts
one Euler Path solution
RePS: 全基因组鸟枪法 测序数据组装软件包
特点:通过屏蔽在鸟枪法测序数据中 发现的重复序列来完成组装。
RePS的 流程图
RePS2的新流程图
scaf f ol d const r uct
super - scaf f ol d const r uct
Maynard V. Olson , The maps: Clone by clone by clone , Nature 409, 816 818 (2001)
Shotgun法序列拼接
Low Base Quality
Single Stranded Region
Sequence Gap
Consensus
exact 20-mer repeats fraction masked, by size fully-masked reads
sequence assembly total contig size [Mb] N50 contig size [Kb] total scaffold size [Mb] N50 scaffold size [Kb]
SeqMan进行序列拼

宏基因组序列拼接
总结词
将多个微生物的测序数据拼接成更完整的基因组,用于 研究微生物群落结构和功能。
详细描述
在宏基因组研究中,由于测序数据来自多个微生物,需 要将这些数据拼接成更完整的基因组,以便更好地了解 微生物群落的结构和功能。这个过程需要解决不同微生 物基因组的拼接问题,以及可能的基因重排和倒位等结 构变异。宏基因组序列拼接有助于深入了解微生物群落 的生态学和进化,为环境科学、农业和医学等领域提供 有价值的信息。
保存的拼接结果可以用于后续的分析和实验验证。
04
序列拼接的质量控制
拼接准确率的评估
准确率
评估拼接序列与原始序列的一致性,计算拼接序列中正确碱基的比例。
错误率
计算拼接序列中错误碱基的比例,反映拼接过程中的误差水平。
拼接效率的评估
拼接时间
评估拼接过程所需的时间,分析拼接效率。
内存使用
评估拼接过程所需的时间,分析拼接效率。
质量控制的方法和标准
质量控制标准
设定拼接准确率、错误率和拼接效率等 质量控制标准,确保拼接结果的质量。
VS
质量控制方法
采用多种质量控制方法,如统计检验、可 视化分析和重复实验等,对拼接结果进行 全面评估和验证。
05
序列拼接的应用实例
基因组序列拼接
要点一
总结词
将测序得到的短读段(reads)拼接成长度更长的序列,用于 基因组组装。
序列编辑
SeqMan软件提供了丰富的编辑 功能,如删除、替换、添加等, 方便用户对序列进行修改和调整。
序列比对
SeqMan软件支持多种序列比对 算法,能够快速比对新旧序列或 不同来源的序列数据。
软件应用领域
基因组学
序列组装的原理

序列组装的原理序列组装(Sequence Assembly)是将DNA或RNA测序结果中的短序列片段(short reads)通过一系列的算法与技术手段,拼接成完整的长序列。
这个过程在基因组学和转录组学研究中具有重要的意义,可以帮助人们理解生物体的基因组结构、功能和进化。
序列组装的原理有以下几个关键步骤:1. 数据预处理:对原始测序数据进行预处理是序列组装的第一步。
这包括去除低质量的测序数据(如含有过多的测序误差或未知碱基)、去除接头序列、剪除冗余序列和序列去重等。
这样可以提高拼接的准确性和效率。
2. 序列拼接:序列拼接是序列组装的核心步骤,目的是将短序列片段按照它们在原始DNA或RNA序列中的相对位置正确拼接在一起。
最简单的方法是比对(align)序列片段,然后根据它们的局部重叠关系来进行拼接。
常用的比对算法包括最长公共子序列(Longest Common Subsequence, LCS)、最长公共前缀(Longest Common Prefix, LCP)等。
通过比对,我们可以找到片段之间的相似性和重叠区域,并判断它们能否被正确拼接。
3. 误差修正:测序数据中存在着不可避免的测序误差,这些误差可能来自于实验本身的误差(如测序仪器的噪音)或者样本本身的特性(如DNA或RNA的修饰)。
为了减少这些误差对序列组装结果的影响,通常需要进行误差修正。
根据片段之间的重叠关系,可以使用图模型(如De Bruijn图)或者统计学方法来对测序误差进行修正。
4. 重复序列解决:基因组中存在着很多重复序列,这给序列组装带来了很大的挑战。
由于重复序列在测序数据中往往会有多个匹配位置,这会导致拼接时的不确定性。
为了解决这个问题,可以通过构建一些特殊的数据结构(如重叠图、De Bruijn图或DBG、字符串图等)来对重复序列进行建模。
通过对这些图进行分析和遍历,可以尽可能地确定长序列的连接方式,提高拼接结果的准确性。
基因重叠群测序与序列组装原理

基因重叠群测序与序列组装原理
基因重叠群测序(overlap-layout-consensus sequencing,OLC)是一种基于序列片段的重叠关系来组装基因组的方法。
其原理主要包括以下几个步骤:
1. 序列建图:将所有输入的序列片段构建成一个序列图,并找出序列片段之间的重叠关系。
重叠关系可以通过比对序列片段之间的相似性来确定,例如使用Smith-Waterman算法。
2. 构建重叠图:根据序列片段之间的重叠关系,构建一个图结构,其中每个节点代表一个序列片段,边表示两个序列片段的重叠关系。
一般使用无向图表示。
3. 寻找最长路径:在重叠图中寻找一条最长路径,这条路径上的节点代表组装出的序列的片段,路径中的重叠部分可以进行序列的拼接,形成更长的序列。
常用的算法是根据图的拓扑排序和动态规划算法。
4. 生成序列:将最长路径上的序列片段进行拼接,生成组装出的序列。
序列组装是一项复杂的任务,涉及到大量的计算和优化算法。
常用的序列组装方法除了基因重叠群测序外,还包括序列重叠图(overlap graph)、de Bruijn图等方法,不同的方法适用于不同的数据类型和实验设计。
在实际应用中,需要根据具体情况选择合适的组装方法。
序列组装的过程

序列组装的过程序列组装是指将测序得到的短序列片段重新组装成完整的序列的过程。
在基因组学研究中,序列组装是一项重要的任务,它可以帮助我们理解基因组的结构和功能。
本文将介绍序列组装的过程,包括数据预处理、序列比对、重叠图构建和序列拼接等步骤。
序列组装的第一步是数据预处理。
测序技术通常会产生大量的短序列片段,这些片段可能包含噪音和错误。
为了减少噪音的影响,我们需要对序列数据进行质量控制。
常见的质量控制方法包括去除低质量的碱基、过滤掉包含接头序列的片段等。
接下来,我们需要将预处理后的序列片段与参考序列进行比对。
序列比对是将短序列片段与已知序列进行匹配的过程。
通过比对,我们可以确定每个片段在参考序列中的位置,进而确定它们之间的相对顺序。
在得到序列片段的比对结果后,我们可以利用重叠图构建算法将它们组装成一个长序列。
重叠图是一种用于描述序列片段之间重叠关系的图形模型。
在重叠图中,每个节点表示一个序列片段,边表示两个序列片段之间的重叠关系。
通过分析重叠图,我们可以找到最长的路径,从而确定序列片段的顺序。
根据重叠图的结果,我们可以进行序列拼接。
序列拼接是将序列片段按照重叠关系连接起来的过程。
在拼接过程中,我们需要解决序列片段之间的重叠区域,通常使用最优拼接算法来寻找最佳的拼接方案。
最终,我们可以得到一个完整的序列,它代表了原始基因组的信息。
总结起来,序列组装是将测序得到的短序列片段重新组装成完整序列的过程。
它包括数据预处理、序列比对、重叠图构建和序列拼接等步骤。
通过序列组装,我们可以获得基因组的结构和功能信息,为基因组学研究提供重要的工具和方法。
序列组装的过程

序列组装的过程
序列组装是将从高通量测序仪中得到的短序列片段(reads)通过计算方法拼接成原始DNA或RNA序列的过程。
以下是序列组装的一般过程:
1. 数据预处理:对从测序仪获得的短序列片段进行质量控制和去除低质量的reads,同时还需要去除适配体序列、重复序列和污染序列等。
2. 序列比对:将清洗后的reads与参考基因组或已知参考序列进行比对。
这可以通过多种算法和工具实现,如Burrows-Wheeler Transform (BWT) 算法、BLAST、Bowtie等。
比对的目的是找到reads在参考序列上的位置,从而为后续的组装提供依据。
3. 碎片组装:根据比对结果,将相互之间有重叠区域的reads拼接在一起形成碎片(contig)。
这个过程就是使用图论算法和启发式策略来将reads进行拼接,生成可能的序列碎片。
4. 空隙填补:在组装过程中,有些区域可能由于读长不够而无法拼接,或者有未知序列导致无法组装。
通过采用测序技术或者利用长读长的第三代测序技术进行填补,获得更完整的序列。
5. 错误校正:根据reads的拼接位置和质量信息来修复一些可能存在的错误。
这可以通过多种方法实现,如使用参考序列进行校正、利用更长的reads校正等。
6. 组装验证和评估:对组装结果进行验证和评估,检查组装序列的准确性和完整性。
通常会与参考基因组或已知序列进行比较,使用统计学方法评估组装质量。
以上是序列组装的一般过程,需要注意的是,在不同的组装策略和算法中,可能会有一些细微的差异和额外的步骤。
同时,对于大规模基因组的组装,可能需要结合其他分析手段和高级算法来提高组装质量和效率。
基因序列表的制作过程

基因序列表的制作过程
基因序列表是描述特定基因或基因组的核苷酸序列的详细记录,是现代分子生物学的基石。
其制作过程涉及以下几个关键步骤:
1. 样品制备
从目标生物体收集基因组 DNA 或 RNA 样品。
使用特定酶(如限制性内切酶)将大 DNA 分子切成较小的片段。
2. 文库构建
将 DNA 片段插入到克隆载体(如质粒或噬菌体)中。
载体随 DNA 片段一起转化到宿主细胞,如大肠杆菌。
3. 测序
使用 DNA 测序技术,如桑格测序或二代测序(NGS),逐个确
定 DNA 片段的核苷酸序列。
每个片段的序列数据通过计算机组装,形成基因组序列的粗略图。
4. 组装
将重叠的序列片段对齐,使用算法拼接到一起,形成连续的基
因组序列。
该过程利用计算方法和人工验证相结合,以确保序列的准确性。
5. 注释
在组装好的序列上标识基因、调控区域和其他功能元件。
注释涉及使用数据库、比较基因组学和功能预测工具。
6. 质量控制
检查基因序列表的准确性,寻找错误、缺失和重复。
使用统计方法和计算工具评估序列的质量。
7. 发布
将最终的基因序列表提交到公共数据库,如 GenBank 或 EMBL。
公布的数据可供研究人员和公众使用,促进生物学研究和医学
发展。
基因序列表的制作是一项复杂而漫长的过程,涉及多种技术和
分析方法。
然而,它对我们的生物学理解和医学应用至关重要,使
我们能够深入了解基因、疾病和进化。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
序列组装的过程
序列组装是一种重要的生物信息学技术,它能够将测序得到的DNA 片段按照其在基因组中的顺序进行拼接,从而获得完整的基因组序列。
下面将从样本准备、测序、质控、序列拼接和结果分析等几个方面介绍序列组装的过程。
一、样本准备
在进行序列组装之前,首先需要从生物样本中提取DNA,并进行适当的处理。
常见的样本包括细菌、真菌、病毒、植物和动物组织等。
提取DNA的方法有多种,常见的方法包括CTAB法、酚-氯仿法和商用基因提取试剂盒等。
提取的DNA需要经过质量检测,确保其完整性和纯度。
二、测序
测序是序列组装的基础,通过测序可以得到DNA序列的碱基信息。
目前常用的测序技术包括Sanger测序、454测序、Illumina测序和Ion Torrent测序等。
这些技术在原理和操作上有所不同,但都能够高效地获取DNA序列信息。
在测序过程中,需要将DNA样本进行文库构建、PCR扩增和测序仪器读取等步骤。
三、质控
测序得到的数据可能存在测序错误、低质量碱基和接头序列等问题,因此需要进行质控处理。
常见的质控方法包括去除低质量碱基、去
除接头序列、去除重复序列和去除人类污染等。
质控处理能够提高数据的质量,减少后续序列组装的误差。
四、序列拼接
序列拼接是序列组装的核心步骤,通过将测序得到的短序列片段按照其在基因组中的顺序进行拼接,从而获得完整的基因组序列。
序列拼接可以采用多种算法,常见的方法包括重叠法、de Bruijn图法和重复序列图法等。
这些算法能够根据短序列片段之间的重叠关系,将其拼接成长序列。
五、结果分析
拼接得到的序列需要进行进一步的结果分析。
分析的内容包括序列的长度、GC含量、SNP(单核苷酸多态性)和Indel(插入缺失)等变异信息。
此外,还可以对序列进行基因注释,获得基因的功能和结构等信息。
结果分析能够帮助研究人员深入了解基因组的特征和变异情况。
序列组装是一项复杂而关键的生物信息学技术,涉及到样本准备、测序、质控、序列拼接和结果分析等多个步骤。
只有在每个步骤都严谨地进行操作,才能获得高质量的基因组序列。
序列组装的结果对于基因功能研究、进化分析和疾病诊断等领域具有重要意义,为生命科学研究提供了强大的工具和平台。