吩噻嗪类药物分析
吩噻嗪类药物的分析精品PPT课件
盐酸 (1→20) 乙醇
10 256
8
264与315
A 0.46
-
- 0.65
- - - -
915
-
883~93 7 - -
553~59 3
630 -
第三节有关物质的检查
❖ 合成工艺
COOH NH2 重氮化 NaNO2,HCl
COOH
N2Cl Cu2Cl2
COOH H2N
Cl
Cl
Cu,缩合
COOH NH
H3C N CH3 CH2
N
Cl
S O 3-(2-氯-10H-吩噻嗪-10-基)-N,N-二甲基-1-丙胺S-氧化物(Ⅴ)
O H3C N CH3
CH2
N
Cl
S
3-(2-氯-10H-吩噻嗪-10-基)-N,N-二甲基-1-丙胺N-氧化物(Ⅵ)
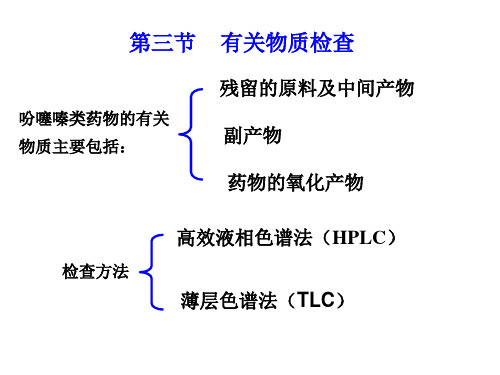
❖ 有关物质的检查方法: (ChP2010)
HPLC法 供试品:0.4 mg/ml
S
S
PdCl2
Pd2+ 2Cl-
pH2
N
R'
S
R
该反应被ChP(2010)用于吩噻嗪类药物及其制剂 的鉴别。不受氧化产物亚砜和砜的干扰
(四)含卤素取代基的反应 1.焰色反应 2.显色反应 吩噻嗪类药物2位的含氟取代基,可经 有机破坏后,分解后的氟化物,在酸性条件下,与 茜草锆试液反应呈色。
(五)氯化物的鉴别反应 1.硝酸银的沉淀反应 2.氧化还原反应
4.紫外和红外吸收:含S、N的三环共轭的大体系。S、 N与苯环形成p-共轭。紫外吸收主要由母核三环的π 系统所产生,一般具三个峰值。204~209nm(205nm附 近)、250~265nm(254nm附近)和300~325nm (300nm附近)。最强峰多在 250~265nm。
吩噻嗪类药物分析总论

《药物分析》指导教师:姜艳丽所属院系:理学院年级:09级班级:化学一班姓名:刘鑫泽学号:090521112012.04.24吩噻嗪类药物一、基本结构与化学性质(一)结构特点与典型药物吩噻嗪类药物分子结构中具有共同的硫氮杂蒽母核,结构差异:母核2位上的R‘取代基,通常为-H、-Cl、-CF3、-COCH3、-SCH2CH3等;10位上的R取代基,则为具有2-3个碳链的二甲或二乙胺基,或为含氮杂环如哌嗪和哌啶的衍生物等。
临床上使用的本类药物多为其盐酸盐。
(二)主要化学性质1.具有紫外和红外吸收光谱特征本类药物的紫外特征吸收,主要由母核三环的π系统所产生。
一般具有三个峰值,即在204nm~209nm(205nm附近)、250nm~265nm(254nm附近)、和300nm~325nm(300nm附近)。
最强峰多在250nm~265nm (ε为2.5×104~3×104);两个最小吸收峰则在220nm及280nm附近。
2位上的取代基(R‘)不同,会引起吸收峰发生位移。
例如2位上卤素的取代(-Cl及-CF3)可使吸收峰向红移2nm~4nm,同时会使250nm~265nm区段的峰强度增大。
R’引起吸收峰位移,可能是通过对位效应影响三环π系统的S,而间位效应又影响三环π系统的N所发生的。
因此,利用其紫外特征吸收可进行本类药物的鉴别。
本类药物母核的硫为二价,易氧化,其氧化产物为亚砜及砜,与未取代的吩噻嗪母核的吸收光谱有明显不同,它们具有四个峰值。
因此,可以利用紫外吸收光谱的这些特征测定药物中杂质氧化物存在的量;同时也可在药物含量测定时对氧化产物的干扰进行校正。
吩噻嗪类药物取代基R和R‘的不同,产生不同的红外吸收光谱,国内外药典已用于本类药物较多品种的鉴别。
2.易氧化呈色吩噻嗪类药物遇不同氧化剂例如硫酸、硝酸、三氯化铁试液及过氧化氢等,其母核易被氧化成自由基型产物和非离子型产物(砜、亚砜、3-羟基吩噻嗪)等不同产物,随着取代基的不同,而呈不同的颜色。
吩噻嗪类

( E1% 为910);注射液为了消除抗氧剂
1cm
维生素C(max 243nm)对测定的干扰,
测定波长改为299nm( E1% 为108)。 1cm
同理,盐酸氯丙嗪片剂测定波长
为254nm( E
1% 1cm为915);注射液测定
波长改为306nm( E1% 为 115 ) 。 1cm
6 7 8 9
S
5
4 3
N R
10
2 1
R'
S Cl N CH2CH2CH2N(CH3)2 盐酸氯丙嗪 HCl
S N CH2CHN(CH3)2 CH3 盐酸异丙嗪 HCl
S N Cl N CH2CH2OH
CH2CH2CH2 N
奋乃静
S N CF3 N CH2CH2OH 2 HCl
CH2CH2CH2 N
醋酐
橙黄Ⅳ
冰醋酸 结晶紫
奋乃静注射液
碱化游离→氯仿提取
→挥干氯仿→高氯酸滴定
注意 盐酸盐对测定有干扰
在冰醋酸中 酸性较强
S HCl HClO 4 S ClO4 HCl
需加醋酸汞以消除HCl的干扰
2 HCl Hg (Ac) 2 HgCl 2 2 HAc
难解离
(二)紫外分色
澄清度 颜色 检查游离氯丙嗪 检查氧化产物
(二)有关物质
方法 TLC法 高低浓度对比法
S样 sol样 sol对
(10mg/ml) (0.1mg/ml)
溶解
稀释
0.1 10 限量% 100% 1% 10 10
四、含量测定
(二) 显色反应
1. 氧化剂氧化显色
钯离子比色法测定吩噻嗪类药物的原理

钯离子比色法是一种常用的分析化学方法,用于测定吩噻嗪类药物中的含量。
本文将介绍钯离子比色法测定吩噻嗪类药物原理的相关知识和实验步骤。
一、钯离子比色法原理1. 吩噻嗪类药物的特性吩噻嗪类药物是一类具有含氮杂环结构的药物,常见的有卡托普利、雷米普利等。
它们对于心血管疾病和高血压等疾病有着重要的治疗作用。
2. 钯离子的特性钯离子在化学反应中常常表现出显著的催化作用,能够与含硫杂环结构中的硫原子形成络合物,并产生特有的颜色。
3. 反应原理在钯离子比色法中,吩噻嗪类药物首先与乙二胺盐酸盐溶液发生络合反应,形成深红色络合物。
然后加入氢氧化钠溶液,在碱性条件下,络合物进一步转化成难溶的沉淀。
最后加入硝酸钯溶液,沉淀中的络合物和硝酸钯发生反应,生成可溶的紫红色络合物。
通过测定紫红色络合物的吸光度,即可计算出样品中吩噻嗪类药物的含量。
二、实验步骤1. 样品制备将待测吩噻嗪类药物样品加入适量的乙二胺盐酸盐溶液中,并摇匀溶解。
2. 沉淀制备再加入氢氧化钠溶液,使溶液呈碱性,观察是否有沉淀生成,并过滤收集沉淀。
3. 离子染色将沉淀加入硝酸钯溶液中,转移至比色皿中,用紫外-可见分光光度计测定其吸光度。
4. 含量计算根据标准曲线计算吩噻嗪类药物的含量,得出样品中吩噻嗪类药物的浓度值。
三、应用与优势1. 应用钯离子比色法在药物分析、环境监测、食品安全等领域有着广泛的应用。
特别是在药物分析中,由于其灵敏度高、结果准确可靠,被广泛应用于吩噻嗪类药物的含量测定。
2. 优势与其他方法相比,钯离子比色法具有操作简便、结果准确、灵敏度高等优势。
对于含硫杂环结构的化合物具有较好的适用性,且不受样品中其他化合物的干扰。
结语钯离子比色法作为一种经典的分析方法,在吩噻嗪类药物的测定中具有重要的应用价值。
掌握其原理和实验操作步骤,有助于从事相关领域的科研工作和实验教学。
希望本文的介绍能够帮助读者加深对钯离子比色法测定吩噻嗪类药物原理的理解,对相关领域的研究和实践有所裨益。
第11篇_吩噻嗪类药物分析

• 2.问题讨论
• 适用范围 • Kb﹤10-8的有机碱盐。 Kb为10-8~10-10 选冰醋酸作溶剂 Kb为10-10~10-12 选冰醋酸和醋酐作溶剂 Kb ﹤10-12 选醋酐作溶剂
② 有机弱碱盐的酸根影响
BH A HClO 4
BH
ClO 4
HA
*置换滴定,即用强酸(HClO4) 置换出与生物碱结合的较弱的酸(HA).
课下阅读自学
四
含量测定
• 一、非水碱量法 • 在水中碱性较弱,不能顺利地进行中和 滴定(滴定突跃不明显,难以判断滴定 终点)。 • 在酸性非水介质中(如HAc中),则能 显示出较强的碱性,滴定突跃增大,可 以顺利地进行中和滴定。
• 1.测定方法 * 供试品:以消耗滴定液8ml计算。 * 溶剂:HAc,一般用量10~30ml, *滴定液:0.1mol/L HClO4/无水HAc 溶液 * 指示剂:结晶紫等 * 做空白试验
( 3 )结构上的差异主要表现在母
核2位上 R 取代基和10位上R取代基的
不同。R 通常为—H、—Cl、—CF3、 —COCH3、—SCH3等。R基团为2-3个
,
,
碳链的二甲或二乙氨基;或含氮杂环,
如哌嗪,吡啶衍生物
S N CH2CHN(CH3)2 CH3 盐酸异丙嗪 HCl
S Cl N CH2CH2CH2N(CH3)2 盐酸氯丙嗪 HCl
(一)化学法
3.与钯离子配合呈色反应
吩噻嗪类药物分子结构中的二价硫可 与金属钯离子形成红色配合物。而其
氧化产物砜和亚砜则无此反应,专属 性强。
癸氟奋乃静
ChP
[鉴别] (2)取本品约 5mg,加甲醇 2ml 溶解后,加 0.1% 氯化钯溶液
案例——吩噻嗪类药物的含量测定方法设计

案例——吩噻嗪类药物的含量测定方法设计吩噻嗪类药物是一类抗精神病药物,常用于治疗精神分裂症等精神疾病。
为了确保吩噻嗪类药物的质量和安全性,需要开发一种有效的含量测定方法。
下面是一种可能的方法设计:1.原理及仪器要求:吩噻嗪类药物的含量测定可以通过高效液相色谱法(HPLC)进行。
该方法通过测定样品中目标化合物在HPLC柱上的保留时间和峰面积来确定药物的含量。
仪器要求包括高效液相色谱仪、固定相柱、检测器(如紫外/可见光检测器)和数据处理软件。
2.样品制备:将待测样品粉碎成细粉末,并过筛以获得均匀的颗粒大小。
取适量样品,加入适量的有机溶剂(如甲醇),进行超声波提取或搅拌提取,以将药物从样品中萃取出来。
然后,使用过滤膜将提取液过滤以去除固体颗粒。
3.标准曲线制备:准备一系列浓度不同的标准品溶液,浓度范围应覆盖待测样品中吩噻嗪类药物的期望浓度。
将标准品溶液用相同方法提取并过滤,然后使用HPLC进行分析,测定各标准品的峰面积。
4.方法优化:通过调整柱温、流速、流动相组成和检测波长等参数,优化HPLC分析方法以获得最佳结果。
确保要测定的药物在柱上有良好的分离和保留,并且峰形对称、峰面积稳定。
5.含量测定:将样品溶液注入HPLC系统,通过自动进样器进行注射。
设置适当的流速和检测波长,进行药物的分析。
根据标准曲线,计算出样品中吩噻嗪类药物的含量。
重复3次测定,计算平均值和相对标准偏差(RSD)。
6.方法验证:对所开发的含量测定方法进行验证。
验证项目包括线性范围、精密度、准确度、选择性和稳定性等。
线性范围可以通过测定标准曲线来确定。
精密度可以通过重复测定样品来评估,计算出相对标准偏差。
准确度可以通过测定加标回收率来评估。
选择性可以通过测定干扰物对药物分离的影响来评估。
稳定性可以通过测定样品在不同温度、湿度和持续时间下的含量变化来评估。
7.结果分析:对测得的样品含量数据进行统计分析,如计算出均值、标准偏差和变异系数。
药物分析--吩噻嗪类含量测定

第十页,共24页。
二、分光光度法
(一)直接分光光度法
在254nm的波长处,C17H19ClN2S.HCl的吸收系数
(
E
1 )% 为915计算。
1cm
第十一页,共24页。
(二)提取后分光光度法-排除辅料干扰
【示例11-36】盐酸氯丙嗪口服液USP32NF量 2取 7 盐酸氯丙嗪适量Vml氨 水 碱化 氯丙嗪
第十六页,共24页。
A
= E C 1 %
254
1 cm (2 5 4 )
sl
A 1 % E = C 1 c m ( 2 5 4 )
254 (s) s
A 1 % E = C 1 c m ( 2 7 7 )
2 7 7 (s) s
A
= (E
1 %
1 cm (1 )
E
) C 1 %
1 cm ( 2 )
xl
碱性溶液pKb>10,冰醋酸 中滴定突跃不明显,需加
入醋酐,使碱性增强, 突跃更明显
第四页,共24页。
2.一般方法
* 供试品:以消耗标准液体积计算 * 溶剂: HAc,一般用量10~30ml
* 滴定剂:0.1mol/L HClO4 无水HAc溶液 * 指示剂:电位法
第五页,共24页。
3. HClO4在冰醋酸溶液中,具强氧化性,可氧化吩噻 嗪类药物产生红色的氧化物,干扰指示剂终点的观 察。排除方法:
冰醋酸中加入不同量的甲酸,也能使滴定突跃显著增大。
第七页,共24页。
【示例11-31】盐酸氯丙嗪测定ChP(2010)
取本品约0.2g ,精密称定,加冰醋酸10ml与醋酐 30ml,溶解后,照电位滴定法,用高氯酸滴定液 (0.1mol/L)滴定,并将滴定的结果用空白试验校 正。每1ml高氯酸滴定液(0.1mol/L)相当于 35.53mg 的C17H19N2ClS.HCl。
药物分析-吩噻嗪类抗精神病药物有关杂质的检查
解:
标示量(%)
AX D W
E1% 1cm
100W
B
100%
0.453 100100 0.5130
5
10 100% 98.6%
915100 0.0206 0.025
三、高效液相色谱法
(一)反相高效液相色谱法
在流动相中加入含氮碱性竞争试剂(扫尾剂:醋酸铵、 三乙胺、二乙胺、乙腈等),以抑制碱性药物与未硅烷化的 硅醇基作用。
计算公式: 50CAU / AS
式中:C(mg/ml)为对照品溶液的浓度;
AU和AS分别为供试品溶液及对照品溶液的吸光度。
练习题:
• 盐酸氯丙嗪片的含量测定:取本品(标示量为 25mg/片)10片,去糖衣后精密称定,重 0.5130g,研细,称取片粉0.0206g,置100ml量 瓶中,加盐酸溶液(9→1000)70ml,振摇使盐 酸氯丙嗪溶解,用溶剂稀释至刻度,摇匀,滤过 ,精密量取续滤液5ml,置100ml量瓶中,加溶 剂稀释至刻度,摇匀,于254nm波长处测定吸光 度为0.453,按吸收系数E1%1cm=915计算每片的 含量。
长)
空白对照液:0.1mol/L盐酸溶液 计算公式: 12.5C( A254 A277 )U /V ( A254 A277 )S
式中:C(mg/ml)为对照品溶液的浓度; V(ml)为所取供试品的体积; 括号内的公式分别为供试品溶液(U)和对照品溶液(S)在下标所
示波长处的吸光度差。
(四)二阶导数分光光度法(自学) 吩噻嗪类药物制剂中其他成分对含量测定的干
第三节 有关物质检查
吩噻嗪类药物的有关 物质主要包括:
残留的原料及中间产物 副产物 药物的氧化产物
吩噻嗪类药物分析

《药物分析》指导教师:姜艳丽所属院系:理学院年级:09级班级:化学一班姓名:刘鑫泽学号:090521112012.04.24吩噻嗪类药物一、基本结构与化学性质(一)结构特点与典型药物吩噻嗪类药物分子结构中具有共同的硫氮杂蒽母核,结构差异:母核2位上的R‘取代基,通常为-H、-Cl、-CF3、-COCH3、-SCH2CH3等;10位上的R取代基,则为具有2-3个碳链的二甲或二乙胺基,或为含氮杂环如哌嗪和哌啶的衍生物等。
临床上使用的本类药物多为其盐酸盐。
(二)主要化学性质1.具有紫外和红外吸收光谱特征本类药物的紫外特征吸收,主要由母核三环的π系统所产生。
一般具有三个峰值,即在204nm~209nm(205nm附近)、250nm~265nm(254nm附近)、和300nm~325nm(300nm附近)。
最强峰多在250nm~265nm (ε为2.5×104~3×104);两个最小吸收峰则在220nm及280nm附近。
2位上的取代基(R‘)不同,会引起吸收峰发生位移。
例如2位上卤素的取代(-Cl及-CF3)可使吸收峰向红移2nm~4nm,同时会使250nm~265nm区段的峰强度增大。
R’引起吸收峰位移,可能是通过对位效应影响三环π系统的S,而间位效应又影响三环π系统的N所发生的。
因此,利用其紫外特征吸收可进行本类药物的鉴别。
本类药物母核的硫为二价,易氧化,其氧化产物为亚砜及砜,与未取代的吩噻嗪母核的吸收光谱有明显不同,它们具有四个峰值。
因此,可以利用紫外吸收光谱的这些特征测定药物中杂质氧化物存在的量;同时也可在药物含量测定时对氧化产物的干扰进行校正。
吩噻嗪类药物取代基R和R‘的不同,产生不同的红外吸收光谱,国内外药典已用于本类药物较多品种的鉴别。
2.易氧化呈色吩噻嗪类药物遇不同氧化剂例如硫酸、硝酸、三氯化铁试液及过氧化氢等,其母核易被氧化成自由基型产物和非离子型产物(砜、亚砜、3-羟基吩噻嗪)等不同产物,随着取代基的不同,而呈不同的颜色。
吩噻嗪类抗精神病药物的分析

5. 红外光吸收特性 吩噻嗪类药物红外光吸收图谱的指纹特征随
取代基不同而不同, 可用于鉴别。
峰位 (cm-1) 1600~1450 1250 1100 950~700
归属
C=C (苯环) N-C (芳氨基) N-C (脂氨基) OOPC-H (苯环)
图11-1A 盐酸氯丙嗪的红外光吸收图谱
S
N
HCl
CH2CHN(CH3)2
CH3
盐酸异丙嗪
S
N
Cl
CH2CH2CH2 N
N CH2CH2OH
奋乃静
S
N
Cl
HCl
CH2CH2CH2N(CH3)2
盐酸氯丙嗪
S
N
CF3
CH2CH2CH2 N
N (CH2)2OCO(CH2)8CH3
癸氟奋乃静
S
N
CF3
2 HCl
CH2CH2CH2 N
N CH2CH2OH
R N 2 S
R′
+ PdCl2
R
N
R′
S
Pd2
2Cl
S
N
R′
R
【示例11-7 】 癸氟奋乃静及制剂 ChP(2010)
取本品约 50mg, 加甲醇2ml 溶解后, 加 0.1% 氯 化钯溶液 3ml, 即有沉淀生成, 并显红色, 再加过量的 氯化钯溶液, 颜色变深。
(四)含卤素取代基的反应 1.焰色反应 【示例11-8 】 奋乃静 ( JP15)
3. 与金属离子配合呈色
硫氮杂蒽母核的二价硫可与钯离子配合, 生成有色化合物, 可用于鉴别和 含量测定。
氧化产物亚砜和砜无此反应。
药物分析11吩噻嗪类抗精神病药物的分析

Company Logo
第四节 含量测定
HPLC法
RP-HPLC
Ion-RP-HPLC法
分离效果不理想
药物极性较强,保留时间弱
形成离子对,保留时间大大增加
分离效果改善
Company Logo
第四节 含量测定
RP-HPLC
固定相:十八烷基硅烷键合硅胶(ODS) 流动相:水-甲醇或水-乙腈系统,极性较强 极性较强的组分在分离时先流出柱子,极性较弱的组分后流出柱子,适合于共存组分极性差异较大样品的分析。杂环类药物分析一般加入扫尾剂二乙胺或三乙胺。
纯度较高,杂质、辅料无干扰或干扰易排除
测定此类药物制剂,可应用提取排除干扰
提取不能完全排除干扰,而药物的氧化物同样能被提取,这时可用
在一定条件下可以方便地消除前两种药物中所产生的干扰
钯离子比色法
钯离子配位特性
Company Logo
二阶导数分光光度法是如何消除干扰的?
朗白比耳定律
把吸收光谱写成波长的函数,那么它的导函数图像就是导数光谱
Company Logo
主要理化性质
二、易氧化性
易被硫酸、硝酸、过氧化氢、三氯化铁氧化,可用于鉴别和含量测定,过程与产物比较复杂
Company Logo
药物
硫酸
硝酸
过氧化氢
盐酸氯丙嗪
-
显红色,渐变为淡黄色
-
盐酸异丙嗪
显樱桃红色,放置后颜色渐变深
生成红色沉淀,加热即溶解,溶液由红色转变为橙黄色
-
奋乃静
有关物质检查
盐酸氯丙嗪及其制剂的有关物质检查
Company Logo
有关物质检查
盐酸氯丙嗪及其制剂的有关物质检查
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
(2)定量依据
样品在二波长下吸收度差值(A):
A
Aab 1
Aab 2
(
Aa 1
Ab 1
)
(
Aa 2
Ab 2
)
Aa 1
Aa 2
( 1
2 )Cal
Ab 1
Ab 2
2位上取代基(R′)不同,会引起吸收峰发生位移。结 构中2价硫,易氧化,产物砜及亚砜有四个吸收峰。
5.红外吸收光谱特性:由于取代基R和R’的不同,具有 不同的红外光谱,已被药典用于不同品种鉴别。
第二节 鉴别试验
一、化学法
(一)与生物碱沉淀试剂反应 吩噻嗪类药物10位的含氮取代基有碱性,可与生物碱
【 ChP盐酸氯丙嗪含量测定】 取本品约0.2g.精
密称定.加冰醋酸10ml与醋酐30ml溶解后,照电位 滴定法,用高氯酸 (0.1mol/L)滴定液滴定,并将滴 定结果用空白试验校正。
每1ml高氯酸滴定液(0.1mol/L) 相当于35.53mg盐酸 氯丙嗪。
注意:
滴定剂的稳定性 非水溶液滴定法所用的溶剂为醋
注:避光操作;溶液临用新配。
LOGO
第四节 含量测定
酸碱滴定法 非水溶液滴定法 乙醇-水溶液中的氢氧化钠滴定法
紫外分光光度法 高效液相色谱法
一、酸碱滴定法
非水溶液滴定法
吩噻嗪类原料药物含量测定大多利用其侧链脂 肪胺碱性,国内外药典多采用非水滴定法测定本类药 物及其盐酸盐原料药的含量。
氯丙嗪的碱性较弱,醋酐解离生成 醋酐合乙酰氧离子比醋酸合质子的 酸性强,更利于增加氯丙嗪的碱性
5.钯离子比色法
原理:吩噻嗪类药物可与一些金属离子(如Pd2+)在适 当pH值的溶液中形成有色的络合物,借以进行比色测定。
S
S
PdCl2
Pd2+ 2Cl-
pH2
N
R'
S
R
吩噻嗪类药物 Pd2 pH20.1红色络合物
max ~500nm 在 max 处测定 A,对照法定量
反应在 pH2±0.1 的缓冲液中进行
沉淀试剂反应
(二)氧化显色反应:
氧化剂氧化显色:H2SO4、溴水、FeCl3、H2O2
母核 氧化剂呈色(樱红 红色)
吩噻嗪环为一良好电子给予体。相继失去电子
形成几个不同的氧化阶,而形成自由基型产物(自 由基、半醌自由基)和非离子型氧化产物(亚砜、 砜、3-羟基吩噻嗪、吩噻酮等)故可被多种氧化剂
S
S
PdCl 2 Pd2+ 2Cl-
pH2
N
R'
S
R
4.紫外和红外吸收:含S、N的三环共轭的大体系。S、 N与苯环形成p-共轭。紫外吸收主要由母核三环的π 系统所产生,一般具三个峰值。204~209nm(205nm附 近)、250~265nm(254nm附近)和300~325nm (300nm附近)。最强峰多在 250~265nm。
A Ca
即,吸收度差值(A)仅与待测组分的浓度有关, 而与干扰组分无关,干扰组分的干扰被消除。
双波长分光光度法 波长选择:测定波长测,参比波长参
⊿A样
⊿A样=A测’ - A 参’
⊿A标= A测 - A参
⊿A标
测
参
⊿A样
C样=
C标
⊿A标
(3)必要条件
① 干扰组分在两个波长处吸收度相等 ② 待测组分在两个波长处吸收度相差足够大
酸,具有挥发性,膨胀系数较大,故高氯酸滴定液 的浓度受温度的影响。若滴定样品与标定HClO4溶液 时的温度不一致,温差超过10℃时,应重新标定;温 差未超过10℃时,应将高氯酸滴定液的浓度用下列 公式加以校正:
N测=N标 / [1+0.0011(t测-t标)]
乙醇-水溶液中的氢氧化钠滴定法
吩噻嗪类药物的盐酸盐的水溶液显酸性,在乙醇
( E11c%m为115)。
1、盐酸氯丙嗪注射液的含量测定,选择在 299nm波长处测定,其原因是 (C)
A、299nm处是它的最大吸收波长 B、为了排除其氧化产物的影响 C、为了排除抗氧剂的干扰 D、在299nm处,它的吸收值最稳定 E、在299nm处测定误差最小
2. 提取后分光光度法 盐酸氯丙嗪注射液
氧化而呈色。
显色反应
药物名称 硫 酸
硝酸
过氧化氢
盐酸氯丙嗪
显红色, 渐变淡黄色
—
盐酸异丙嗪 显樱桃红色,放置 生成红色沉淀,加热即溶解,
后颜色渐变深 溶液由红色转变为橙黄色
奋乃静
—
—
显深红色;放置后
红色渐褪去
盐酸氟奋乃静 显淡红色, 温热
—
—
后变成红褐色
盐酸三氟拉嗪
—
生成微带红色的白色沉淀; 放
贮藏不当产生的氧化物
H3C N CH3 CH2
N
Cl
S O 3-(2-氯-10H-吩噻嗪-10-基)-N,N-二甲基-1-丙胺S-氧化物(Ⅴ)
O H3C N CH3
CH2
N
Cl
S
3-(2-氯-10H-吩噻嗪-10-基)-N,N-二甲基-1-丙胺N-氧化物(Ⅵ)
有关物质的检查方法: (ChP2010)
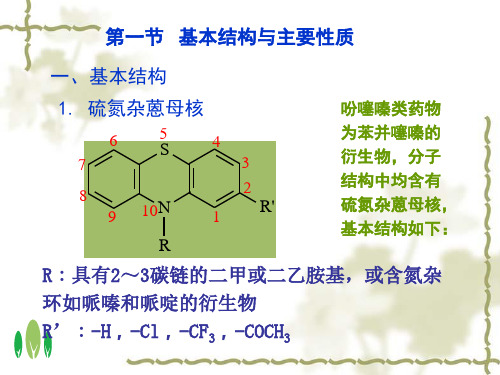
第一节 基本结构与主要性质
一、基本结构
1. 硫氮杂蒽母核
吩噻嗪类药物
6
5
S
7
8
9 10N
R
4 3
2
1 R'
为苯并噻嗪的 衍生物,分子 结构中均含有 硫氮杂蒽母核, 基本结构如下:
R:具有2~3碳链的二甲或二乙胺基,或含氮杂
环如哌嗪和哌啶的衍生物
R’:-H,-Cl,-CF3,-COCH3
S
HCl
N
Cl
盐酸氯丙嗪 氨 水碱 化 氯丙嗪 乙 醚 提 取 乙醚层 盐 酸 萃 取
盐酸氯丙嗪 max 254 1nm测定A,
以 E11c%m 为 915 计算。
3.提取后双波长分光光度法:
用于校正样品中氧化产物对测定影响 (1)原理
在待测组分(a)的最大吸收波长(测定波长, 1)处测定待测组分和干扰组分(b)吸收度的总 和;另选一适当波长(参比波长,2)测定吸收度, 并使干扰组分在测定波长和参比波长处60
10 255
盐酸 (1→20) 乙醇
10 256
8
264与315
A 0.46
-
- 0.65
- - - -
915
-
883~93 7 - -
553~59 3
630 -
第三节有关物质的检查
盐酸氯丙嗪及其制剂的有关物质检查
合成工艺
COOH NH2 重氮化 NaNO2,HCl
COOH
尾剂)如二乙胺、三乙胺等。
采用反相HPLC法测定本类药物含量时,常常会产生 拖尾现象,这是因为ODS固定相表面未被硅烷化的 硅醇基与碱性药物发生了吸附或离子交换作用。
抑制或掩蔽固定相
解决的方法有:
表面的游离硅醇基 的活性
调整流动相pH,使呈弱碱性(pH7-8)
加含氮碱性试剂(扫尾剂),最常用的扫尾剂为
优点:钯离子比色法可选择性地用于未被氧化的吩噻 嗪类药物的测定
讨论:
(1)钯离子试剂: PdCl2和十二烷基硫酸酯钯盐。 用PdCl2时,形成的有色络合物水中溶解度小,样 品量大时,产生沉淀。 十二烷基硫酸酯钯盐,其络合物溶解度大,吸收强 度增大(约2倍)。
三、高效液相色谱法: (一)反相高效液相色谱法 固定相:十八烷基键合硅胶 流动相:水-甲醇,水-乙腈中加入含氮碱性试剂(扫
HPLC法 供试品:0.4 mg/ml
主成分自身对照法
对照品:供试品稀释至2μg/ml
色谱柱:辛烷基硅烷键和硅胶
流动相:乙腈-0.5%三氟乙酸(50:50)
检测波长:254nm
离子对试剂,增加氯 丙嗪的保留
判定:供试品溶液如有杂质峰,单个杂质峰面积不得大于主峰 面积(0.5%),各杂质峰面积和不得大于对照溶液主峰面积 的2倍(1.0%)。 同时检查多个有关物质
(五)氯化物的鉴别反应 1.硝酸银的沉淀反应 2.氧化还原反应
二、光谱法 1、特征的紫外吸收:
药物 盐酸氯丙嗪
盐酸异丙嗪
奋乃静 癸氟奋乃静 盐酸氟奋乃静
盐酸三氟拉嗪 盐酸硫利达嗪
溶剂 盐酸 (9→1000)
浓度
λ max
(μg/ml) (nm)
5
254
306
盐酸 (0.1mol/L)
6
249
无水乙醇 乙醇 盐酸 (9→1000)
-水溶液中,可采用氢氧化钠滴定液测定其含量。
ChP盐酸异丙嗪含量测定采用此法
二、紫外分光光度法
1.直接分光光度法: 盐酸异丙嗪片的测定 操作:取10片,除去糖衣后,精称,研细→称取粉
末适量→加水、盐酸溶解→过滤,取滤液于249nm处
测定。 已知:盐酸异丙嗪E=910,即可计算 优点:不需标准品 缺点:仪器精密度要求高 测定中注意问题:避光、防止氧化。
N2Cl Cu2Cl2
COOH Cl
COOH H2N
Cl
Cl
Cu,缩合
COOH NH
Cl 脱羧
Fe
NH
Cl
环合
S,I2
3-氯二苯胺 (Ⅰ)
H N
S
Cl
H
N
Cl
S 2-氯-10H-吩噻嗪 (Ⅱ)
H N 分离异构体
C6H5Cl,Cl S
H3C N CH3 CH2
N
成盐 Cl HCl,C2H5OH