广东一次性使用子宫颈扩张球囊导管产品
宫颈扩张球囊的好处有什么呢

宫颈扩张球囊的好处有什么呢
我们都知道自然分娩无论是对产妇的身体恢复,还是胎儿的健康都是最好的一种生产方式,也是最符合自然规律的,但是总是会有很多女性由于体制的原因,在预产期没有任何的生产迹象,为了能够保证胎儿在妈妈肚子里面能够健康,就需要对差福进行催产,其中宫颈扩张球囊就是常用的一种催产工具,下面一起了解下宫颈扩张球囊的好处。
宫颈扩张球囊的好处
当继续怀孕对孕妇或胎儿有一定危险,如延期妊娠、过期妊娠、羊水过少、妊娠期糖尿病已超预产期等情况未能自然临产时,孕妇将面对两个选择:自然分娩或剖宫产术结束分娩。
对具备自然分娩(阴道分娩)条件的孕妇,医生会建议孕妇计划分娩,但宫颈条件不成熟又会增加阴道分娩的难度。
据阳江市妇幼保健院妇产科主治医师曾靖燕介绍,为促进自然分娩,降低孕妇剖宫产率,该院最新引进了子宫颈扩张球囊导管促宫颈成熟的技术,让计划分娩的孕妇多了一个选择及途径。
子宫颈扩张球囊为非药物助产器械,能安全、自然地、渐进式扩张宫颈,并增加引产成功率。
宫颈的成熟与扩张通过球囊在宫颈内口和外口提供温和、持久的扩张力而实现。
其优点如下:
1、在不使用药物的情况下安全地促进宫颈成熟和扩张;
2、避免了反复用药可能引起的副作用;
3、硅胶球囊能适应宫颈管的轮廓,易于扩张宫颈;
4、容易放置、取出快捷,孕妇无痛苦;
5、无菌物品,一人一管,不增加感染率;
6、使用后孕妇可以自由走动,为胎头下降及宫颈扩张创造有利条件。
上面就是对宫颈扩张球囊的好处介绍,通过了解之后我们知道它虽然是一种常见的催产方式,但是对身体没有任何的伤害,不会产生任何的副作用,所以很多选择这种催产方式的孕妇就可以不那么的顾虑,可以放心使用。
一次性使用子宫颈扩张球囊导管产品技术要求维力医疗
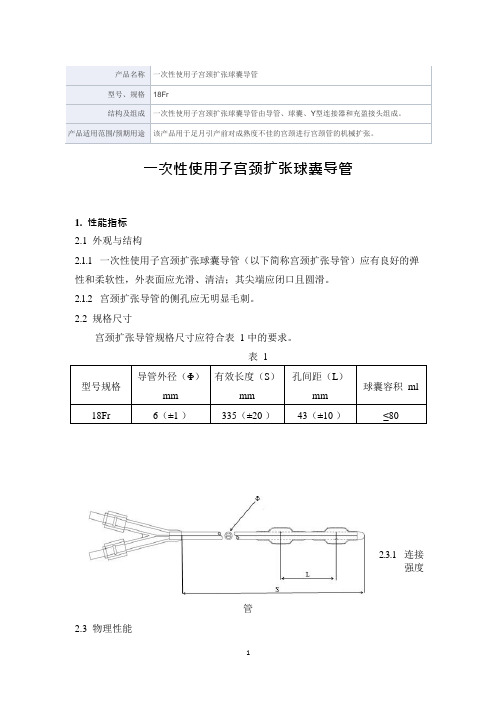
产品名称一次性使用子宫颈扩张球囊导管型号、规格18Fr结构及组成一次性使用子宫颈扩张球囊导管由导管、球囊、Y型连接器和充盈接头组成。
产品适用范围/预期用途该产品用于足月引产前对成熟度不佳的宫颈进行宫颈管的机械扩张。
一次性使用子宫颈扩张球囊导管1. 性能指标2.1外观与结构2.1.1一次性使用子宫颈扩张球囊导管(以下简称宫颈扩张导管)应有良好的弹性和柔软性,外表面应光滑、清洁;其尖端应闭口且圆滑。
2.1.2宫颈扩张导管的侧孔应无明显毛刺。
2.2规格尺寸宫颈扩张导管规格尺寸应符合表 1 中的要求。
表 1型号规格导管外径(Φ)mm有效长度(S)mm孔间距(L)mm球囊容积ml18Fr 6(±1 )335(±20 )43(±10 )≤80管2.3物理性能2.3.1连接强度1图1 子宫颈扩张球囊导当按附录A 的方法进行试验时,尖端应与管身连为一体,管身应无断裂。
2.3.2Y 型连接器分离力当按附录B 的方法进行试验时,Y 型连接器应不与充盈止逆阀分离。
22.3.3球囊可靠性2.3.3.1当按附录C 的方法进行试验时,球囊应无泄漏。
2.3.3.2球囊未充起时,其两端外形应与管身平滑地连为一体,在其周围环境温度下,球囊充入规定容积的水后,应呈现基本对称的鼓起。
2.3.4抗扭结按 3.3.4 的方法进行试验,宫颈扩张导管应没有折痕或开裂现象。
2.3.7 6%鲁尔圆锥接头的要求宫颈扩张导管的各圆锥接头应符合GB/T 1962.1-2015 的规定。
2.4化学性能2.4.1重金属按GB/T 14233.1-2008 中5.6.1 的规定进行比色试验时,试验液呈现的颜色不超过质量浓度ρ(Pb2+)=1μg/ml 的标准对照液。
2.4.2酸碱度按GB/T 14233.1-2008 中 5.4.1 方法一规定进行试验时,检验液与空白液pH 之差应不超过1.5。
2.5环氧乙烷残留量宫颈扩张导管采用环氧乙烷灭菌,出厂时环氧乙烷残留量应不大于10μg/g。
一次性子宫颈扩张球囊联合催产素对足月妊娠引产前促宫颈成熟有效

一次性子宫颈扩张球囊联合催产素对足月妊娠引产前促宫颈成熟有效性、安全性发表时间:2018-11-30T10:27:06.367Z 来源:《航空军医》2018年18期作者:王楚兰[导读] 足月妊娠引产是妇产科常见手术,是在自然临产前,通过辅助分娩方式刺激子宫收缩,使产程提前发动。
(汕尾市第二人民医院(汕尾逸挥基金医院) 516600)摘要:目的探讨一次性子宫颈扩张球囊联合催产素对足月妊娠引产前促宫颈成熟的有效性、安全性。
方法选择我院收治的足月妊娠引产孕妇62例为研究对象,均有引产指征,采用随机数字表法分为观察组和对照组各31例。
对照组静滴小剂量缩宫素引产,观察组采用一次性子宫颈扩张球囊联合催产素引产,对比两组患者促宫颈成熟率、产妇产程时间、围生儿结局、分娩结局以及不良反应情况。
结果观察组宫颈评分、促宫颈成熟率及引产成功率明显高于对照组(P<0.05);诱发临产时间、第一产程及总产程时间明显短于对照组(P<0.05);观察组新生儿窒息率、出生体质量和胎儿窘迫,产妇产后出血、宫颈裂伤、阴道血肿、盆腔感染等并发症发生率比较,差异均无显著性(P>0.05);观察组羊水污染发生率显著高于对照组,两组比较差异有统计学意义(P<0.05)。
结论一次性子宫颈扩张球囊联合催产素能有效促进宫颈成熟,缩短产程,提高产妇引产成功率,未出现严重不良结局,安全性高,值得临床推广。
关键词:引产;足月妊娠;羊水污染;宫颈扩张球囊足月妊娠引产是妇产科常见手术,是在自然临产前,通过辅助分娩方式刺激子宫收缩,使产程提前发动,早日解除胎儿不良宫内环境威胁,达到分娩目的[1]。
该手术成功与否的主要决定因素是宫颈成熟度,宫颈不成熟会延长产程,直接导致引产失败。
目前临床常用的引产药物有催产素、前列腺素制剂等,但催产素使用效果受环境的制约较大,对成熟宫颈作用不明显;前列腺素制剂易诱发子宫收缩过强等风险,因此需不断优化临床催产方案[2]。
球囊扩张导管使用说明书

球囊扩张导管使用说明书
球囊扩张导管是一种医疗设备,用于扩张或扩大人体内部管道或腔隙。
以下是球囊扩张导管的使用说明书:
1. 术前准备:
a. 确保球囊扩张导管的完整性和干净度。
b. 检查导管的尺寸和长度是否适合特定的手术需求。
c. 进行必要的消毒和清洁。
2. 植入导管:
a. 使用麻醉剂和消毒解剖技术,将球囊扩张导管插入体内。
b. 通过适当的引导工具,将导管放置在需要扩张的部位。
c. 当导管到达目标位置后,小心地将球囊扩张导管放置在目标区域内。
3. 扩张球囊:
a. 确保球囊扩张导管的球囊部分位于正确的位置。
b. 连接球囊扩张导管的球囊部分与充气设备。
c. 缓慢注入适量的液体或气体来充实球囊,在此过程中需要监测压力。
d. 根据需要逐渐增加球囊内的压力,直到达到所需的扩张程度。
4. 扩张完成后:
a. 将球囊扩张导管从体内缓慢拔出。
b. 清洁和消毒球囊扩张导管,以备下次使用。
c. 将球囊扩张导管妥善保存,避免受潮或受损。
请注意,以上仅为一般性的使用说明,具体的使用方法应根据医生的指示和操作步骤执行。
在使用球囊扩张导管时,应遵循医疗器械使用的相关法规和标准,确保患者的安全和有效治疗。
子宫颈球囊扩张导管介绍ppt课件

David MD, *Ron Auslender MD, *Haim Abramovici MD :《 Ripening and dilatation of the unfavourable cervix
for indution of labour by a double ballon device :experience with 250 cases》;妇产科系,卡梅尔医疗中心
完整版课件
10
信息现状的小结与分析
剖宫产的危害? 科学健康分娩? 解决现实的问题
----减轻疼痛、缩短产程、降低剖宫产 率、避免危害 产品的意义
完整版课件
11
子宫颈扩张球囊结构示意图
充盈止逆 阀
双腔导管
球囊
尖端
锁定环
管身有效 长
完整版课件
12
Uterus Vagina
完整版课件
13
产品材料
育和避孕方面都有严厉的 限制 ,即便3年后再次怀孕, 子宫也容易破裂,威胁产 妇生命 。
完整版课件
7
对新生儿的影响
新生命孕育在母体里时胎儿泡在 羊水内,肺泡内充满液体(医学 称肺液),而氧气由母体供应。
从胎儿方面来说,经产道自然过 程的新生儿有时间适应外界压强, 通过产道的挤压,将大部分肺液 通过口腔排出,对残留在新生儿 体内的肺液通过肺泡的自然吸收;
完整版课件
3
剖宫产只是分娩过程中碰到难产、 难处理问题时的权宜之计,不应是 分娩的正常手段。
剖宫产临床指征:孕妇有骨盆畸形、 臀位、前置胎盘、高龄初产妇、巨 大儿等。
完整版课件
4
剖宫产产妇术后出现并发症的可能 性,远远高于自然分娩。
剖宫产除了手术过程中存在麻醉风 险、脏器损伤风险以及由于手术创 伤面大,疼痛和恢复时间更长外;其 术后并发症更是大大高于自然分娩
一次性使用组织扩张球囊导管套件产品技术要求模板

7018B
12
DG-
8515B
13
DG-
X115B
14
DG-
X207B
15
DG-
X310B
16
DG-
X315B
17
DG-
X320B
30±5 mm 50±5 mm 50±5 mm 50±5 mm 50±5mm 70±5 mm 70±5 mm 70±5 mm 70±5 mm 85±5 mm 115±5 mm 120±5 mm 150±5 mm 150±5 mm 150±5 mm
20±2mm 10±2 mm 15±2mm 18±2mm 20±2mm 10±2mm 13±2mm 15±2mm 18±2mm 15±2mm 15±2 mm 7.5±2mm 10±2mm 15±2mm 20±2mm
2.2.3 球 囊 导 管 的 圆 锥 接 头 配 合 尺 寸 符 合 GB/T 1962.1-2015 或 GB/T 1962.2-2001 的规定。
21 QK-18L17XL
22 QK-22L17
23 QK-22L17L
牵开直径
12±3mm 12±3mm 12±3mm 18±3mm 18±3mm 18±3mm 22±3mm 22±3mm 22±3mm 12±3mm 12±3mm 18±3mm 18±3mm 22±3mm 22±3mm 12±3mm 12±3mm 12±3mm 18±3mm 18±3mm 18±3mm 22±3mm 22±3mm
一次性使用组织扩张球囊导管套件采用环氧乙烷灭菌,应无菌。 2.6 细菌内毒素:
一次性使用组织扩张球囊导管套件细菌内毒素应<2.15EU/件。
介入直径 总长
3.0± 0.5mm (9Fr)
2024年一次性使用子宫颈扩张球囊导管市场分析报告

2024年一次性使用子宫颈扩张球囊导管市场分析报告1. 引言本市场分析报告旨在对一次性使用子宫颈扩张球囊导管市场进行全面的分析和评估。
该报告将提供市场规模、市场竞争格局、市场发展趋势以及市场驱动因素等方面的详细信息。
通过深入研究市场数据和调查结果,我们将帮助读者了解该市场的现状和未来发展趋势。
2. 市场概览2.1 市场定义及分类一次性使用子宫颈扩张球囊导管是一种医疗器械,用于妇科手术中的子宫颈扩张程序。
它通过充气球囊来扩大子宫颈,以便进行子宫内膜刮宫、取出子宫内膜异位病灶或进行其他妇科手术。
2.2 市场历史和发展随着妇科手术的不断进步和技术的发展,一次性使用子宫颈扩张球囊导管在妇科手术过程中的应用越来越广泛。
相比传统的金属或塑料扩张器,一次性使用子宫颈扩张球囊导管更具优势,如操作简便、无需重复消毒和易于丢弃等。
2.3 市场规模根据市场调研数据显示,一次性使用子宫颈扩张球囊导管市场在过去几年中呈稳步增长的趋势。
预计在未来几年内,市场规模将进一步扩大,受益于技术的进步和医疗行业对更高质量、更方便的医疗器械的需求。
3. 市场竞争格局3.1 主要厂商和产品目前,一次性使用子宫颈扩张球囊导管市场上存在多家主要厂商。
以下是其中几家主要厂商及其产品:•公司 A: 产品 A1、产品 A2•公司 B: 产品 B1、产品 B2•公司 C: 产品 C1、产品 C23.2 市场份额分析根据市场调研数据显示,目前市场上领先的几家公司在市场上占据主导地位,其市场份额较大。
然而,由于市场竞争激烈,其他厂商也在努力提高产品质量和服务,以争夺更大的市场份额。
3.3 市场竞争策略为了保持竞争优势,各大厂商通过不断研发新产品、提高产品质量、拓展市场渠道、加强品牌推广等方式来提高市场占有率。
一些厂商还通过与医院和医疗机构的合作建立长期稳定的销售合作关系,以确保产品的稳定供应和销售。
4. 市场发展趋势4.1 技术进步驱动市场增长随着医疗技术的进步,一次性使用子宫颈扩张球囊导管的设计和性能不断提高,为医生的操作提供更多便利和精确度。
总局关于发布第二批免于进行临床试验医疗器械目录的通告(2016年第133号)-二类

止血夹
6801
由夹子、弹簧和轴等部件组成。一般采用纯钛、不锈钢材料制成。非无菌提供。用于术中临时阻断血管。
4
撑开器
6801
通常由不锈钢制成。在脊柱手术中用于撑开和扩张椎间隙。
5
外科术前备皮器
6801
一般由刀头(基座为ABS树脂、刀刃为低碳钢等材料)和手柄组成。刀头为灭菌产品,一次性使用。用于去除患者身体和头部的毛发,为需要去除毛发的医学操作做准备。
20
胸腔心血管外科用剥离器
6807
可由剥离头、杆部和柄部等部件组成。一般采用不锈钢材料制成。用于剥离软组织,或剥离静脉血管内的血栓等,使静脉畅通。
21
胸腔心血管外科用钩
6807
可由头部和柄部等部件组成。头部带弯钩。一般采用不锈钢材料制成。用于牵拉心脏组织。
22
胸腔心血管外科用夹
6807
可由夹子、弹簧和轴组成。一般采用不锈钢材料制成。用于胸腔手术中临时阻断血管或夹持血管止血。
48
脊柱手术用刮匙
6810
该类产品通常由不锈钢材料制成。用于脊柱后路手术中刮除已经捣碎的椎间盘、去除上下终板等。
49
脊柱后路手术用骨钻
6810
该类产品通常由手柄和金属部分组成。手柄通常由塑料制成,金属部分通常由不锈钢制成。用于脊柱后路手术中钻出骨道。
50
与有源器械联用钻头
6810
该类产品一般由不锈钢材料制成。骨科手术时用于钻骨。与有源器械配合使用。
60
骨取样器
6810
该类产品由套管、推杆或管芯等组成。金属部分通常采用不锈钢材料制成,手柄通常采用高分子材料制成。用于经皮椎体后凸成形术/经皮椎体成形术中骨组织取样。
61
走进子宫颈扩张球囊引产技术

走进子宫颈扩张球囊引产技术妊娠期会让女性的身体发生巨大变化,使孕妇身体各项器官的功能都会与平常状况有所不同,因此很多孕妇在妊娠期间都会出现不同的妊娠疾病,如妊娠糖尿病或者妊娠期高血压等。
对于出现特殊情况的孕妇来说,在怀孕的末期容易出现羊水过少的现象,对胎儿的身体发育以及孕妇的健康状况都有不良影响。
当孕妇出现这种状况时,需要对孕妇进行及时的引产才能保证胎儿和孕妇的生命安全。
随着医疗技术的逐渐提高,引产技术也越来越成熟,能够在对孕妇造成最小伤害的基础上成功接生婴儿。
子宫颈扩张球囊引产技术是近年来广泛应用在引产手术中的重要技术,能够大幅度的提升妊娠晚期的引产成功几率。
那么你知道什么是子宫颈扩张球囊引产技术吗?一、什么是子宫颈扩张球囊引产技术?子宫颈扩张球囊引产技术是近年来经常对孕妇使用的引产技术,并在实践的过程中取得了良好的效果,是一种实用的且值得在临床医学上推广的引产技术。
子宫颈扩张球囊引产技术与传统引产技术相比,是利用机械工具来完成引产操作的,通过在孕妇的体内放置球囊,并采取相关的压迫操作对孕妇的子宫颈进行催熟,让孕妇体内产生内源性的前列腺素,进而出现宫缩的现象,引导孕妇顺利生产。
子宫颈扩张球囊引产技术能够使孕妇和胎儿都脱离生产过程中的危险因素,避免出现新生婴儿出现窒息等不良状况,减小孕妇需要进行剖腹产的几率。
二、子宫颈扩张球囊引产技术准备工作以及操作流程子宫颈扩张球囊引产技术的有效实施需要进行科学、完整的准备工作,才能保证引产过程的顺利实施。
首先需要对孕妇的身体状况进行详细检查,观察孕妇是否出现引产指征,以宫颈Bishop的评分为依据,如果宫颈Bishop的评分小于6则代表孕妇可以接受子宫颈扩张球囊引产。
其次,要排除孕妇是否有相关的禁忌病症,如胎儿位置不正、胎盘或者血管前置、胎儿巨大、生殖道感染、妊娠高血压、妊娠糖尿病或者多胎妊娠等其他状况。
再次,对孕妇进行白带常规+BV检查,如果检查结果为阳性,则代表孕妇不适合使用子宫颈扩张球囊引产技术,需要选择其他方法进行引产。
宫腔球囊扩张导管国标

宫腔球囊扩张导管国标宫腔球囊扩张导管(简称宫腔球囊导管)是一种在妇科手术中广泛使用的器械,用于宫腔扩张、检查和操作。
国标是指该器械所需符合的国家标准。
本文将从宫腔球囊导管的定义、分类、作用、国标制定以及相关注意事项进行详细介绍,旨在加深对宫腔球囊导管的理解。
宫腔球囊导管是一种柔软的管状器械,形状类似于导尿管。
其主要由管腔和球囊两部分组成。
管腔的长度、直径和形状可以根据手术需要进行选择。
球囊位于导管末端,并可以通过充气来扩张。
宫腔球囊导管在手术中通常通过阴道插入,通过宫颈进入宫腔,然后通过充气球囊来扩张宫腔,以便进行宫腔检查、手术和操作。
根据不同的目的和要求,宫腔球囊导管可以分为多种类型。
常见的有宫腔扩张导管、宫腔镜扩张导管和宫腔插管。
其中,宫腔扩张导管主要用于宫腔扩张和手术操作;宫腔镜扩张导管则用于辅助腔镜检查和手术;宫腔插管则用于宫腔内种植和输注。
宫腔球囊导管的主要作用是扩张宫腔,使医生能够清楚地观察宫腔内的情况,并进行手术和操作。
其扩张宫腔的机制是通过充气球囊来实现的。
当球囊被充气时,会逐渐扩张,从而使导管的外径增大,宫腔的直径也相应增加。
通过宫腔扩张,医生可以更好地进行宫腔检查、子宫内膜切除、取出子宫内膜异位病变等操作。
为了确保使用安全和效果,宫腔球囊导管有一系列的国家标准制定,以规范其质量和使用。
国标要求宫腔球囊导管应符合特定的物理和化学性能要求,并且需要进行相关的技术评价和测试。
其中,物理性能要求包括尺寸、容量、压力承受能力等;化学性能要求包括材料的生物相容性和无毒性等。
通过符合国家标准,可以保证宫腔球囊导管的质量和安全性,并为临床手术提供可靠的支持。
在使用宫腔球囊导管时,还需要注意一些事项。
首先,需要选择适合手术需要的导管尺寸和类型。
其次,需要针对每位患者进行个体化的使用和操作,以确保操作的准确性和有效性。
此外,在使用过程中需要细致观察患者的生命体征,及时处理可能出现的并发症和不良反应。
中林一次性使用子宫颈扩张球囊导管

中林一次性使用子宫颈扩张球囊导管临床运用足月妊娠促宫颈成熟操作步骤:1、规范清洁、消毒,如有生殖道感染,应治疗后再放置球囊或放置前予与抗生素不预防感染;2、向两个球囊内分别注入50ml无菌水,检查球囊完整性,注射器顶住阀门注水接口,抽出球囊内液体和空气,使球囊变扁,钳夹球囊导管,防止空气进入球囊内,球囊抽扁更易放置;3、插管,避免球囊碰到阴道壁,导管突起处(10cm)与宫颈外口平行,确保球囊在宫颈内口上方膨胀;4.暴露宫颈(使用阴道窥镜),将导管球囊端插入宫颈,并保证两个球囊都通过宫颈内口。
用40ml生理盐水通过红色充盈口充盈子宫球囊(末端球囊)。
充盈后,将球囊往后拉至子宫球囊紧贴宫颈内口。
阴道球囊此时位于子宫外口,用20ml生理盐水充盈阴道球囊。
确定将球囊置于宫颈两侧后,充入生理盐水(按每次20ml的注射量,逐渐交替将各自球囊的容积增加至80ml,即红色端再注入两次,另一个端口再注入三次)。
4、如宫颈成熟度极差,插管困难,或中期引产,必要时先扩宫棒扩宫后插管;5、注水:足月妊娠促宫颈成熟80ml;6、适度牵拉导管,确保球囊固定于宫颈内口处,确保低位水囊;7、导管用胶带固定在大腿内侧;8、有条件可行B超影像学确认球囊位置;9、晚孕促宫颈成熟放置时间建议不超过12小时,特殊情况超过12小时的需抗生素预防感染。
特别注意:放置水囊引产时不可与宫缩剂同时使用!【晚孕促宫颈成熟:置管时间建议不超过12小时,具体置管时间根据宫颈评分,预判分娩时间,大致可控制在白天人手充裕时完成分娩(如:宫颈评分<4分上午9点左右置管;宫颈评分>4分下午3点左右置管),球囊如自行脱落说明宫口已经打开5-6cm!物理扩张宫口的优势是不引起强烈宫缩,非常温和,因此产妇能够充分的休息,不知不觉中宫口打开至5cm左右,迅速进入产程活跃期,后续产程进展顺利,减少产妇体力消耗,降低产后出血发生率。
因此水囊的在某种程度上可减轻疼痛,缩短宫缩痛感总产程,可替代药物或器械的镇静止痛方法,对于怕痛的产妇也是可以推荐的,不仅仅是限于有引产指征的孕妇;同时也是疤痕子宫唯一试产选择;大量临床使用经验总结还可用于计划分娩。
一次性使用输尿管球囊扩张导管套件产品技术要求深圳泰睿仕医疗科技

2.性能指标2.1外观2.1.1球囊扩张导管2.1.1.1球囊扩张导管有效长度的外表面应光滑、无杂质,不应有加工缺陷和表面缺陷。
2.1.1.2球囊扩张导管的接头应光滑、无毛刺。
2.1.1.3球囊扩张导管上应有完整、清晰的标识。
2.1.1.3.1应有在推荐压力充盈后的球囊的直径、球囊的有效长度标识,标识单位应以毫米表示。
2.1.1.3.2应有与球囊扩张导管配合使用的导丝的最大直径标识,标识单位应以in 表示。
2.1.1.3.3应有球囊扩张导管的导管有效长度标识,标识单位应以厘米表示。
2.1.2选配件导丝的附加要求如有导丝,导丝外表面应光滑,应无毛刺、机械损伤、锋棱和污斑。
2.1.3选配件球囊扩充压力泵的附加要求如有球囊扩充压力泵,产品表面光滑平整、色泽均匀、无毛刺、飞边、裂纹、伤痕等加工缺陷。
2.2尺寸产品的尺寸应符合要求,各型号规格的尺寸及允差要求见附录 A 产品配置中A.3 产品型号规格表。
2.3物理性能2.3.1球囊扩张导管2.3.1.1峰值拉力球囊扩张导管应符合 YY 0285.1-2017 中 4.6 的规定。
2.3.1.2球囊疲劳:充起时无泄漏和损坏按3.3.1.2 试验时,球囊扩张导管在额定爆破压力(RBP)下应不发生泄漏、破裂或突出变形。
2.3.1.3球囊爆破压力球囊扩张导管的爆破压力应大于其规定的的额定爆破压力(RBP),破坏模式是轴向破坏。
2.3.1.4球囊卸压时间球囊从额定爆破压力(RBP)至卸压终点所需时间应不大于 60s。
2.3.1.5球囊直径与充盈压力的关系球囊顺应性:0.3%~3.0%/atm(101.3kpa)。
2.3.1.6导丝通过性外径 0.89mm(0.035in)的导丝应能顺利通过球囊扩张导管的导丝腔,整个过程不应有堵塞现象发生。
2.3.1.7射线可探测性球囊扩张导管的球囊内两端应有标示球囊位置的标记环,标记环应能被射线探测到。
2.3.1.8接头接头能与 GB/T 1962.1-2015 规定的 6%(鲁尔)外圆锥接头配合。
低位双腔水囊联合缩宫素应用于晚期妊娠期糖尿病引产的临床效果

低位双腔水囊联合缩宫素应用于晚期妊娠期糖尿病引产的临床效果作者:晏三华万静雯熊贤海刘芳付广红高军来源:《中国当代医药》2019年第01期[摘要]目的研究分析低位双腔水囊联合缩宫素应用于晚期妊娠期糖尿病引产的临床效果。
方法选择本院2017年3月~2018年1月收治的60例晚期妊娠期糖尿病需终止妊娠的初产妇,根据自愿原则分为实验组和对照组,每组各30例。
实验组采用低位双腔水囊联合缩宫素引产,对照组采用缩宫素引产。
比较两组的引产成功率、出血量、新生儿Apgar评分及血糖控制程度。
结果实验组产妇的引产成功率明显高于对照组,差异有统计学意义(P0.05)。
结论在晚期妊娠期糖尿病中,低位双腔水囊联合缩宫素引产成功率高,是一种简单、经济、安全的有效方法,值得临床推广应用。
[关键词]低位双腔水囊;缩宫素;晚期妊娠期糖尿病;引产[中图分类号] R714.256; ; ; ; ; [文献标识码] A; ; ; ; ; [文章编号] 1674-4721(2019)1(a)-0148-03糖尿病是一种由于胰岛素分泌不足或外周组织对胰岛素不敏感引起的代谢性疾病。
妊娠期糖尿病(gestational diabetes mellitus,GDM)属于特殊类型的糖尿病,是妊娠期最常见的并发症之一。
GDM发生率世界各国报道为1%~14%,我国GDM发生率为1%~5%,近年来有明显增高趋势[1]。
选择恰当的方式终止妊娠,可避免糖尿病对母儿的危害,最大程度地减少对产妇和新生儿的影响。
目前大部分医院采用小剂量缩宫素静脉滴注引产,产程时间长,易致胎儿宫内窘迫,这提高了剖宫产率,严重时危及母婴安全[2]。
本研究选择低位双腔水囊(产品名称:益心达一次性使用子宫颈扩张球囊导管,规格型号:CVB-18F,深圳市益心达医学新技术有限公司生产)联合缩宫素引产应用于30例晚期妊娠期糖尿病,进行临床效果观察,现报道如下。
1资料与方法1.1一般资料选择2017年3月~2018年1月我院收治的60例晚期妊娠期糖尿病需终止妊娠的产妇,均为初产妇。
2024年一次性使用子宫颈扩张球囊导管市场规模分析

2024年一次性使用子宫颈扩张球囊导管市场规模分析简介随着医疗技术的不断发展和人们对个人卫生健康意识的提高,一次性使用子宫颈扩张球囊导管作为一种安全、方便、卫生的医疗器械,逐渐受到市场的青睐。
本文将对一次性使用子宫颈扩张球囊导管市场规模进行分析,并探讨其未来发展趋势。
一次性使用子宫颈扩张球囊导管市场概述一次性使用子宫颈扩张球囊导管是一种用于妇科手术中扩张子宫颈和子宫腔的器械。
相比传统的可重复使用器械,一次性使用子宫颈扩张球囊导管具有操作简单、消毒方便、减少交叉感染风险等优势。
近年来,随着人们的生活水平和卫生意识的提高,以及医疗技术的不断进步,一次性使用子宫颈扩张球囊导管市场需求逐渐增加。
2024年一次性使用子宫颈扩张球囊导管市场规模分析从全球范围来看,一次性使用子宫颈扩张球囊导管市场规模呈现稳步增长的态势。
根据市场研究数据,2019年全球一次性使用子宫颈扩张球囊导管市场规模约为X亿美元,并预计未来几年将保持每年X%的复合年增长率。
1.区域分析1.1 北美地区北美地区是一次性使用子宫颈扩张球囊导管市场的主要消费地区之一。
这主要得益于该地区的发达医疗卫生系统和高度发达的医疗技术。
根据数据统计,2019年北美地区一次性使用子宫颈扩张球囊导管市场规模约为X亿美元。
1.2 欧洲地区欧洲地区也是一次性使用子宫颈扩张球囊导管市场的重要消费地区。
该地区的医疗保健水平较高,医疗卫生设施齐全,对一次性使用子宫颈扩张球囊导管的需求较大。
根据数据统计,2019年欧洲地区一次性使用子宫颈扩张球囊导管市场规模约为X亿美元。
1.3 亚太地区亚太地区是全球一次性使用子宫颈扩张球囊导管市场增长最快的地区之一。
该地区的人口众多,经济发展迅速,医疗服务水平逐渐提高,对一次性使用子宫颈扩张球囊导管的需求呈现增长趋势。
根据数据统计,2019年亚太地区一次性使用子宫颈扩张球囊导管市场规模约为X亿美元。
2.产品类型分析一次性使用子宫颈扩张球囊导管市场根据产品类型可分为不同规格和款式。
一次性使用宫颈扩张棒产品技术要求

医疗器械产品技术要求编号:一次性使用宫颈扩张棒1.产品型号/规格及其划分说明一次性使用宫颈扩张棒的型号有A型、B型、C型、D型。
具体划分说明见下表。
型号划分说明表内径W1 内径H1 结构组成A型 4.9mm 4.5mm 由聚乙烯醇缩甲醛扩张棒、固定套、后盖和推杆组成。
固定套、后盖推杆B型 5.5mm 4.8mm材料为ABS塑料。
C型 4.9mm 4.5mm 由聚乙烯醇缩甲醛扩张棒和绳子组成D型 5.5mm 4.8mm扩张棒的原材料聚乙烯醇缩甲醛的CAS号为No.63148-64-1,绳子的材料为涤纶缝纫线。
图1 A型、B型结构示意图1-聚乙烯醇缩甲醛扩张棒2-固定套3-后盖4-推杆图2 C型、D型结构示意图1-聚乙烯醇缩甲醛扩张棒2-绳子图3 聚乙烯醇缩甲醛扩张棒图示注:H为L3中点的直径,W为L2中点的直径2.性能指标2.1 外观产品表面应无杂质、粉末或水印痕迹,扩张棒头部有自然弯曲。
A型、B型固定套、后盖和推杆表面应光洁、无毛刺;扩张棒头端应圆润、表面应光滑、无毛刺。
2.2 尺寸产品尺寸应符合表1,尺寸允差范围为±10%。
表1 产品尺寸单位:mm 规格型号W1 W2 H1 H2 L1 L2 L3A型 4.9 9.0 4.5 7.4 60.5 15.0 45.5B型 5.5 9.0 4.8 7.4 60.5 15.0 45.5C型 4.9 9.0 4.5 7.4 60.5 15.0 45.5D型 5.5 9.0 4.8 7.4 60.5 15.0 45.5 2.3 膨胀度吸水5min后,扩张棒尺寸应符合表2的要求,允差范围±10%表2 膨胀后扩张棒尺寸单位:mm规格型号W1 H2A型12.0 12.0B型14.0 14.02.4 溶解析出物2.4.1外观溶出液应无色透明、无异物。
2.4.2 pH与空白对照液对照,其pH差应小于2.0。
2.4.3可溶出的重金属总含量溶出液中可溶出的重金属总含量不超过5.0μg/ml。
2024年一次性使用子宫颈扩张球囊导管市场发展现状

2024年一次性使用子宫颈扩张球囊导管市场发展现状概述子宫颈扩张球囊导管是一种广泛应用于妇科手术中的医疗器械。
它通过充气球囊的膨胀来扩张子宫颈,便于手术器械的进入和操作。
与传统的金属扩张子宫颈器械相比,一次性使用子宫颈扩张球囊导管具有更好的安全性和易使用性,因此越来越受到医生和患者的青睐。
市场规模目前,全球一次性使用子宫颈扩张球囊导管市场呈现稳步增长的趋势。
根据市场研究机构的数据显示,2019年全球一次性使用子宫颈扩张球囊导管市场规模达到X 亿美元,并预计在2025年将增至X亿美元。
市场增长的主要推动因素包括手术医疗技术的进步、妇科手术的普及以及对安全和便利性的需求的不断增加。
市场主要参与者一次性使用子宫颈扩张球囊导管市场中的主要参与者包括国内外医疗器械制造商和供应商。
国外制造商占据了市场的主要份额,其中一些知名企业包括ABC公司、DEF公司和GHI公司。
然而,国内一次性使用子宫颈扩张球囊导管制造商也在努力提高产品质量和创新能力,逐渐在市场中占据一席之地。
市场发展趋势技术创新随着医疗技术的不断革新,一次性使用子宫颈扩张球囊导管的技术也在持续改进。
一些制造商通过引入新材料和改良设计来提高产品的性能和安全性。
例如,一些新型导管采用了独特的膨胀球囊设计,提供更好的扩张效果和更低的风险。
市场竞争加剧随着市场规模的扩大,竞争也日益激烈。
不仅来自国内外的制造商,还有其他医疗器械公司也纷纷进入这个市场。
为了在竞争中脱颖而出,制造商们努力提高产品质量、研发创新技术,并将产品价格合理化。
市场细分一次性使用子宫颈扩张球囊导管市场根据用途可以进一步细分为宫腔镜手术用导管、腹腔镜手术用导管等。
不同用途的导管在设计和性能上有所差异,以满足不同手术需求。
市场细分有助于提高产品的定位和满足不同客户群体的需求。
市场挑战一次性使用子宫颈扩张球囊导管市场面临一些挑战。
首先,市场的法规监管在不同国家和地区存在差异,对产品的准入和质量管理提出了要求。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
广东省一次性使用子宫颈扩张球囊导管产品注册技术审查指导原则(征求意见稿)本指导原则旨在帮助和指导申请人对一次性使用子宫颈扩张球囊导管产品注册申报资料进行准备,以满足技术审评的基本要求。
同时有助于审评机构对该类产品进行科学规范的审评,提高审评工作的质量和效率。
本指导原则是对一次性使用子宫颈扩张球囊导管产品注册申报资料的一般要求,申请人应依据具体产品的特性对注册申报资料的内容进行充实和细化。
申请人还应依据具体产品的特性确定其中的具体内容是否适用,若不适用,需具体阐述其理由及相应的科学依据。
本指导原则是对申请人和审查人员的指导性文件,但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其他方法,也可以采用,但是需要提供详细的研究资料和验证资料。
应在遵循相关法规的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将进行适时地调整。
一、适用范围本指导原则适用于《医疗器械分类目录》(国药监械[2002]302号) 中第二类一次性使用子宫颈扩张球囊导管产品,分类编码为:6812。
也适用于《医疗器械分类目录》(2017年第143号)中1801妇产科手术器械中一次性无菌球囊宫颈扩张器,分类编码为180105。
二、技术审查要点(一)产品名称要求产品名称应为通用名称,并符合《医疗器械通用名称命名规则》(国家食品药品监督管理总局令第19号)等相关法规、规范性文件的要求,如:一次性使用子宫颈扩张球囊导管、一次性无菌球囊宫颈扩张器。
(二)产品的结构和组成宫颈扩张球囊导管主要由导管、球囊、充盈头组成,导管、球囊、充盈头材质为高分子材料。
导管主体可设计支撑腔,配合可调式针芯(或导丝)使用。
该产品一般以无菌形式提供,一次性使用。
产品示意图见下图。
图1 宫颈扩张球囊导管(从左至右依次为双腔型和三腔型)(三)产品工作原理宫颈扩张球囊导管是一种硅胶双球囊导管,将双球囊(阴道球囊和子宫球囊)分别置于宫颈内外口,通过对宫颈内外口缓慢、持续的机械刺激作用产生渐进性扩张宫颈作用。
宫颈扩张球囊导管无药物作用,促宫颈成熟的主要原理是靠导管及宫颈口内外球囊压力,机械性刺激宫颈管,促进官颈局部内源性前列腺素合成与释放,从而促进宫颈软化成熟。
(四)注册单元划分的原则和实例按照《医疗器械注册管理办法》(国家食品药品监督管理总局令第4号)第七十四条的要求,“原则上以产品的技术原理、结构组成、性能指标和适用范围为划分依据”实施。
(五)与产品相关的标准表1 相关产品标准上述标准包括了产品技术要求中经常涉及到的标准。
企业还应根据产品的特点引用涉及到的其他行业外的标准或特殊标准。
在对产品适用及引用标准审查时,首先对引用标准的齐全性和适宜性进行审查,也就是在编写产品技术要求时是否准确参考了与产品相关的国家、行业标准。
其次,应注意标准编号、标准名称是否完整规范,年代号是否有效。
一般来说,产品技术要求应不低于相应的国家标准及行业标准。
如有新版强制性国家标准、行业标准发布实施,产品性能指标等要求应执行最新版本的国家标准、行业标准。
(六)产品的适用范围/预期用途、禁忌证用于足月引产前对成熟度不佳的宫颈进行宫颈管的机械扩张。
禁忌证:1)胎位异常、胎儿横位2)脐带脱落3)胎儿窘迫4)臀位5)正在接受或准备接受外源性前列腺素治疗的患者6)前置胎盘、血管前置或植入性胎盘7)以前有剖宫产史、疤痕子宫、子宫肌瘤切除术史或任何其他的子宫手术切开史8)宫颈结构异常9)活动性生殖道疱疹感染(阴道炎)10)浸润性宫颈癌11)孕妇有心脏病12)多胎妊娠13)羊水过多、羊水过少14)先露部位于骨盆入口之上15)孕妇妊娠高血压16)胎膜破裂17)任何引产相关禁忌证。
(七)产品的研究要求根据所申报的产品,提供适用的研究资料。
1.产品性能研究应当提供产品性能研究资料以及产品技术要求的研究和编制说明,包括功能性、安全性指标(如化学性能、物理性能、微生物性能等)以及与质量控制相关的其他指标。
2.生物相容性评价研究宫颈扩张球囊导管不应释放出任何对人体有害的物质。
正常使用条件下宫颈扩张球囊导管直接与人体接触,接触时间不超过12小时,应按GB/T 16886.1《医疗器械生物学评价第1部分:风险管理过程中的评价与试验》对宫颈扩张球囊导管进行生物学评价。
一般情况至少应评价:细胞毒性、致敏、粘膜刺激。
生物相容性评价研究资料应当包括:生物相容性评价的依据和方法;产品所用材料的描述及与人体接触的性质;实施或豁免生物学试验的理由和论证;对于现有数据或试验结果的评价。
3.灭菌工艺研究提交产品灭菌方法的选择依据及验证报告。
器械的灭菌应通过GB 18278、GB 18279或GB 18280确认并进行常规控制,无菌保证水平(SAL)应保证达到1×10-6。
灭菌过程的选择应至少考虑以下因素:产品与灭菌过程间的适应性;包装材料与灭菌过程的适应性、灭菌对产品安全有效性的影响。
如果灭菌方法是辐射,应该确定剂量。
如果用环氧乙烷灭菌,应制定环氧乙烷残留量的指标并进行检测。
4.产品包装产品包装验证可依据有关标准进行(如GB/T 19633、YY/T 0681系列标准等),提交产品的包装验证报告。
包装材料的选择应至少考虑以下因素:包装材料的物理化学性能;包装材料的毒理学特性;包装材料与产品的适应性;包装材料与成型和密封过程的适应性;包装材料与灭菌过程的适应性;包装材料所能提供的物理、化学和微生物屏障保护;包装材料与使用者使用时的要求(如无菌开启)的适应性;包装材料与标签系统的适应性;包装材料与贮存运输过程的适合性。
5.产品货架有效期产品货架有效期是指产品在一定的温度、湿度、光线等条件的影响下保持其物理、化学、生物学性质的期限。
有效期的研究应贯穿于产品研究与开发的全过程,在产品上市后还应继续进行有效期的研究。
货架有效期包括产品有效期和包装有效期。
产品有效期验证可采用加速老化或实时老化的研究,实时老化的研究是唯一能够反映产品在规定储存条件下实际稳定性要求的方法,应遵循极限试验等原则;加速老化研究试验的具体要求可参考YY/T 0681.1。
对于包装的有效期验证,建议申请人提交在选择恰当的材料和包装结构合格后的最终成品包装的初始完整性和维持完整性的检测结果。
在进行加速老化试验研究时应注意产品选择的环境条件的老化机制应与在实时正常使用环境老化条件下真实发生产品老化的机制一致。
6.其他资料证明产品安全性、有效性的其他研究资料。
(八)产品的主要风险宫颈扩张球囊导管应按照YY/T 0316-2016《医疗器械风险管理对医疗器械的应用》进行风险分析。
企业在进行风险分析时,至少应考虑表2中的主要危害,企业还应根据自身产品特点确定其他危害。
针对产品的各项风险,企业应采取应对措施,确保风险降到可接受的程度,并明确告之剩余风险。
1.与产品有关的安全性特征判定可参考YY/T 0316-2016的附录C。
2.危害、可预见的事件序列和危害处境判断可参考YY/T 0316-2016附录E、I。
3.风险控制的方案与实施、综合剩余风险的可接受性评价及生产和生产后监视相关方法可参考YY/T 0316-2016附录F、G、J。
4.风险可接收准则,降低风险的措施及采取措施后风险的可接收程度,是否有新的风险产生。
5.市场上已上市同类产品的不良事件分析总结。
审查时可参考不良事件历史记录,重点关注由于潜在设计缺陷导致的抱怨和不良事件,以及相应的风险控制措施。
表2 宫颈扩张球囊导管产品的主要危害(九)产品技术要求应包括的主要性能指标根据《医疗器械注册管理办法》(国家食品药品监督管理总局令第4号)的要求,产品技术要求应符合国家标准、行业标准和有关法律、法规的要求。
在此基础上,申请人应根据产品的特点制定保证产品安全有效、质量可控的技术要求。
产品技术要求及试验方法均应经过验证。
常见的技术指标包括以下几点(不限于此):1、外观与尺寸1.1应有良好的弹性和柔软性,外表面应光滑、无毛刺、清洁;其尖端应闭口且圆滑。
1.2 尺寸:导管、球囊、导丝尺寸。
2、物理性能2.1尖端及各个连接处连接强度2.2球囊可靠性2.3按照最大充起体积的1.5倍注入37摄氏度生理盐水,并在37摄氏度环境下下保持12小时后,球囊、充起管路及接头均无无泄漏和损坏。
2.4 球囊未充起时,其两端外形应与管身平滑地连为一体。
球囊充起时,球囊偏心度的要求:球囊最大半径应不大于最小半径的两倍。
2.5抗扭结(手持球囊导管中点分别与导管头部和顶部平行接触,持续1min后,球囊导管应没有折痕或开裂现象)2.6接头的要求2.7 设计有支撑腔配合针芯(或导丝)使用的产品,应制订相应性能指标。
三腔型所用针芯或导丝有效导丝不得超过导管长度。
2.8 若含有球囊充起组件,应制定相应的性能指标。
如:若含有球囊充起用注射器,应参考YY15810制定性能指标。
3、化学性能根据产品材料特性,由企业决定对化学性能(如重金属、酸碱度、紫外吸光度等)提出具体要求。
若用环氧乙烷灭菌的产品应规定环氧乙烷残留量的要求。
4、无菌宫颈扩张球囊导管经灭菌应使其无菌。
备注:通过广东省不良反应监测中心查询发现,此类产品有多起疑似不良事件,大部分报道为使用过程中球囊破裂,泄漏。
因此建议技术要求中着重考虑球囊可靠性、球囊爆裂体积、球囊爆破压力等性能指标。
因申报产品无相应的国标行标,建议参考YY0325、YY/T0817等相关标准进行性能指标的制定。
如有不适用的项目,请予以说明。
(十)同一注册单元内注册检验典型产品确定原则和实例同一注册单元中典型产品是指能够代表本注册单元内其他产品安全性和有效性的产品,其材料最多、或结构最复杂、或功能最齐全、或风险最高。
典型产品的确定可以通过比较同一注册单元内所有产品的技术结构、性能指标和预期用途等相应资料,说明能够代表本注册单元内其他产品的安全性和有效性。
建议登陆广东省医疗器械质量监督检验所网站下载《医疗器械产品型号覆盖差异性预评价报告(无源)》,提交型号覆盖报告。
必要时,应考虑同一注册单元中产品之间的差异性,进行相关差异性检测。
(十一)产品生产制造相关要求应当明确产品生产加工工艺,注明关键工艺和特殊过程(如挤出、粘接、球囊制备、清洗、灭菌、初包装等),并说明其过程控制点。
明确生产过程中各种加工助剂的使用情况及对杂质(如残留单体、小分子残留物、生产环境杂质颗粒等)的控制情况。
(十二)产品的临床评价细化要求根据《关于发布第二批免于进行临床试验医疗器械目录的通告》(国家食品药品监督管理总局通告2016年第133号),宫颈扩张球囊导管为第73号产品,对列入上述目录的产品,注册申请人可不必开展临床试验,在注册申请时应当提交申报产品相关信息和对比资料,资料内容应符合《医疗器械临床评价技术指导原则》(国家食品药品监督管理总局通告2015年第14号)的要求。