噬血细胞综合症
噬血细胞综合征治疗进展

噬血细胞综合征治疗进展噬血细胞综合征,也被称为噬血细胞淋巴组织细胞增多症(HLH),是一种罕见而严重的疾病。
这种疾病是由于机体的免疫系统异常激活引起的、导致炎症反应过程持续并且无法控制的一种疾病。
噬血细胞综合征可以发生于各个年龄阶段,包括新生儿和成人。
如果不及时治疗,患者的预后往往十分严峻。
噬血细胞综合征的病因目前尚不完全清楚,一部分患者是由于遗传突变导致的,而其他患者则是由于其他原因引起的,如感染、免疫缺陷、先天代谢异常等。
无论是遗传性还是获得性的噬血细胞综合征,都表现出类似的免疫系统高度激活所致的临床表现。
目前,治疗噬血细胞综合征的标准方法是免疫抑制治疗。
这种治疗方法的目的是抑制免疫系统的过度激活,并与治疗原发病因相结合。
常见的免疫抑制药物包括糖皮质激素、环孢素A、环磷酰胺等。
糖皮质激素被认为是治疗噬血细胞综合征的首选药物,通过抑制炎症反应和免疫系统的异常激活来达到控制疾病的目的。
对于某些患者,单独使用糖皮质激素可能效果有限,这时可以考虑联合使用其他免疫抑制药物进行治疗。
近年来,随着对噬血细胞综合征认识的深入和治疗方法的不断改进,治疗疗效有了显著的提高。
目前,早期诊断和早期治疗是提高患者生存率的关键。
早期诊断依赖于对患者全面的体格检查和病史采集,以及相关实验室检查的结果。
疾病严重程度的评估对于选择恰当的治疗方案也非常重要。
除了免疫抑制治疗,造血干细胞移植也被认为是治疗噬血细胞综合征的有效方法之一。
这种治疗方法通常适用于药物治疗无效或疾病复发的患者。
造血干细胞移植通过重新建立免疫系统的功能来控制疾病,并提供长期的治愈效果。
除了传统的治疗方法,目前还有一些新的治疗方法正在研究中。
通过抑制噬血细胞综合征发生和发展的关键信号通路,如干扰素γ信号通路、补体信号通路等。
这些新的治疗方法有望为噬血细胞综合征的患者提供更有效和更安全的治疗选择。
噬血细胞综合征是一种罕见而严重的疾病,治疗方法主要是免疫抑制治疗和造血干细胞移植。
噬血细胞综合征治疗进展

噬血细胞综合征治疗进展噬血细胞综合征(Hemophagocytic lymphohistiocytosis,HLH)是一种罕见但严重的疾病,其特征是免疫系统异常激活和炎症反应过度。
HLH可以是原发性的,也可以是继发于其他疾病的。
无论是原发性还是继发性HLH,都需要及时治疗以避免严重的并发症和死亡。
近年来,随着对HLH病理生理机制的深入了解和治疗技术的不断进步,HLH的治疗也取得了一些进展。
本文将对HLH的治疗进展进行概述,以便提高对HLH治疗的认识和理解。
一、免疫抑制剂治疗:免疫抑制剂是治疗HLH的主要手段之一。
常用的免疫抑制剂包括环孢霉素、依托泊苷、他克莫司和米胪骚。
这些药物可以抑制T细胞和自然杀伤细胞的活性,从而减少炎症反应和细胞毒作用。
在多中心临床研究中发现,免疫抑制剂能有效控制HLH的症状,并且可以改善患者的预后。
二、干细胞移植治疗:对于原发性HLH患者,干细胞移植是一种有效的治疗手段。
通过干细胞移植,可以替换异常的造血系统,重建患者的免疫系统。
近年来,随着干细胞移植技术的不断进步,干细胞移植已经成为原发性HLH患者长期生存的关键因素。
三、靶向治疗:近年来,针对HLH发病机制的深入研究,使得一些靶向治疗药物成为治疗HLH的新选择。
IL-6抑制剂托珠单抗和雷珠单抗可以抑制炎症反应,减少组织损伤,改善患者的预后。
抗CD52单克隆抗体阿雷替伏也显示出了一定的疗效。
四、抗病毒治疗:HLH的发作与病毒感染有一定的关联,因此及时清除病毒感染对于HLH的治疗非常重要。
目前,常用的抗病毒药物包括阿昔洛韦、利巴韦林和奈替尼,这些药物可以有效抑制病毒复制,减轻病毒感染对机体免疫系统的刺激。
五、支持性治疗:除了针对HLH本身的治疗外,对于HLH患者来说,良好的支持性治疗也非常重要。
包括营养支持、液体管理、疼痛控制、并发症预防等方面的治疗都非常关键。
六、个体化治疗:近年来,随着个体化医疗的发展,越来越多的研究表明,个体化治疗对于HLH的治疗非常重要。
嗜血细胞综合征

再次发病
患者在完全缓解期间再次出现新的疾病活动, 可能与原发疾病不同。
长期随访
对完全缓解的患者进行长期随访,监测疾病 是否复发或出现其他并发症。
感谢您的观看
THANKS
病因
不同的病因对预后也有影响。 继发性嗜血细胞综合征的预后 通常优于原发性嗜血细胞综合 征。
治疗反应
对治疗反应良好的患者通常预 后较好。
并发症
严重的并发症,如感染、器官 功能衰竭等,可能影响预后。
转归与结局
01
02
03
04
完全缓解
在接受治疗后,患者的症状和 实验室检查恢复正常,无疾病
迹象。
部分缓解
其他血液系统疾病
如骨髓增生异常综合征、再生障碍性 贫血等,可根据临床表现和实验室检 查进行鉴别。
如淋巴瘤、白血病等,可通过组织病 理学检查进行鉴别。
实验室检查
血常规检查
全血细胞减少,尤其是 血小板和白细胞减少明
显。
血清铁蛋白检测
血清铁蛋白水平升高, 可作为诊断的重要参考
指标。
凝血功能检查
组织病理学检查
纤维蛋白原等凝血因子 水平升高,凝血功能异
常。
骨髓、淋巴结等组织病 理学检查发现噬血现象,
是确诊的重要依据。
03 嗜血细胞综合征的治疗
药物治疗
药物治疗是嗜血细胞综合征的常见治 疗方法,主要用于控制症状和缓解病 情。
药物治疗过程中需密切监测患者的病 情变化和药物副作用,及时调整治疗 方案。
常用的药物包括免疫抑制剂、糖皮质 激素、细胞毒药物等,具体药物选择 需根据患者的病情和医生的建议。
避免接触有害物质
避免接触化学物质、放射线等有害 物质,减少对身体的损伤。
噬血细胞综合征的治疗及护理

03
肝脾肿大:肝 脏、脾脏肿大, 压迫周围器官
04
贫血:面色苍 白,头晕乏力,
心悸气短
05
出血:皮肤黏 膜出血,鼻出 血,牙龈出血
等
06
神经系统症状: 头痛、头晕、
意识障碍等
07
呼吸系统症状: 咳嗽、胸闷、
呼吸困难等
08
消化系统症状: 食欲不振、恶 心呕吐、腹泻
等
09
心血管系统症 状:心悸、心 律失常、心力
生活护理
01
饮食护理: 注意营养均 衡,避免刺 激性食物
02
皮肤护理: 保持皮肤清 洁,避免感 染
03
口腔护理: 保持口腔清 洁,预防口 腔感染
04
心理护理: 关注患者心 理状态,给 予心理支持
预防感染
保持个人卫生, 勤洗手,避免
接触感染源
定期进行环境 消毒,保持室
内空气流通
避免去人群密 集的地方,减
康复指导
01
保持良好的生活习惯,如规律 的作息、合理的饮食等
02
定期进行身体检查,及时发 现并处理病情变化
03
保持良好的心理状态,避免焦 虑、抑郁等负面情绪
04
加强锻炼,提高身体素质, 增强免疫力
定期复查
01
定期复查的重要性:及时发现病
情变化,调整治疗方案
02
复查频率:根据病情严重程度和
治疗效果,制定合适的复查频率
演讲人
01
02
03
04
1
疾病定义
噬血细胞综合征是 一种罕见的血液系
1 统疾病,主要表现 为发热、肝脾肿大、 淋巴结肿大等。
病因:主要是由于 免疫系统异常,导
噬血细胞综合征0630PPT课件

流行病学
★ 以儿童多见,男性多于女性。 ★ 儿童原发性HLH(FHL)的年发病率约为0.12/1O万。
在日本和亚洲国家发病率较高。本病来势凶险, 东方患者的死亡率约为45%。
病因和发病机制
※病因: ★ HPS可以看作细胞因子病或巨噬细胞激活 综合征。
1、反响性T细胞〔Th1和Tc〕和单核吞噬细胞 过度分泌淋巴、单核因子〔巨噬细胞增生的 诱导因子〔PIF〕〕激活巨噬细胞。 2、恶性细胞亦可直接刺激组织细胞,或由 肿瘤细胞产生释放细胞因子〔如γ-干扰素〕, 诱发临床综合征,称之为副新生物综合征。
HPS 的 发 病 机 制
① 存在免疫调节障碍或免疫失衡;
② 淋巴和单核因子持续产生,作为免疫应答的反响性T 细胞分泌淋巴因子可活化巨噬细胞, 尤其如γ-干扰素 不仅能抑制造血,而且亦能活化巨噬细胞,淋巴因子 GM-CSF亦激活巨噬细胞;
③ 遗传因素影响机体对感染的反响方式,如家族性噬红 细胞性淋巴组织细胞增生症和X联淋巴增殖综合征的 儿童可发生类似的血液学异常;
④ 存在单克隆性T细胞增殖,在EB病毒相关噬血细 胞综合征(EBV-AHS)的患者采用PCR法检测10/11 例呈TCR γ链重排,亦有报道TCR β基因的单克隆 性重排,显示EB病毒感染T 细胞引起单克隆增殖 的可能,或许是末梢T细胞“肿瘤〞的一种特殊 类型。EBV-AHS患者EBV整合入宿主T细胞染色体 基因组造成单克隆T 细胞增生〔从良性到新生物 前期或明显的恶性增殖〕伴异常的T细胞。为何 异常的T细胞反响导致组织巨噬细胞的吞噬行为 改变,可能由T细胞过度分泌的淋巴因子所介导 隆性T细胞增殖,在EB病毒相关噬血细胞综合征 (EBV-AH。
噬血细胞综合征按其病因,除儿童期发病的
家族性HPS〔FHL〕之外,可分为原发性〔原因 不明〕或继发性,继发性HPS常见病因为感染、
噬血细胞综合征

XX
THANKS
感谢观看
REPORTING
未来研究方向探讨
深入研究发病机制
01
进一步深入研究噬血细胞综合征的发病机制,探索新的治疗靶
点和治疗策略。
开发新的诊断方法
02
继续开发新的诊断方法,提高诊断的准确性和效率,为患者提
供更好的诊疗服务。
优化治疗手段
03
进一步优化现有的治疗手段,提高治疗效果和患者生活质量,
同时探索新的治疗方法,为患者提供更多的治疗选择。
提高诊疗水平和患者生活质量
加强医生培训
加强对医生的培训和教育,提高医生对噬血细胞综合征的认知和诊 疗水平。
完善患者教育体系
建立完善的患者教育体系,帮助患者和家属更好地了解疾病和治疗 方案,提高患者的自我管理能力和生活质量。
加强科研与临床合作
加强科研与临床的合作,推动科研成果转化为临床应用,为患者提供 更好的诊疗服务。
年龄分布
HPS可发生于任何年龄 ,但以儿童期和青少年
期多见。
性别分布
男女均可发病,无明显 性别差异。
地域分布
HPS在全球范围内均有 报道,无明显的地域差
异。
临床表现及分型
临床表现
HPS的临床表现多样,主要包括发热、肝脾肿大、全血细胞 减少、高甘油三酯血症和/或低纤维蛋白原血症等。此外,患 者还可出现淋巴结肿大、皮疹、中枢神经系统症状等。
XX
噬血细胞综合征
汇报人:XX
REPORTING
• 疾病概述 • 诊断与鉴别诊断 • 治疗与预后 • 并发症与风险管理 • 患者教育与心理支持 • 研究与展望
目录
XX
PART 01
疾病概述
REPORTING
58_噬血细胞综合征

•XIQI
•30
¡ 5.2000年WHO鉴于真正的“恶性组织细胞病” 极少见,分类中为避免混淆,未再提出
真正的恶组,而将组织细胞肉瘤(histiocytic
Sarcoma)作为单独一类。已放弃“恶组”作为 一种疾病名称。但是,有些学者提出,为便于 临床处理,在无充分把握时,可暂冠以“意义 未明的组织细胞增生综合征”
•XIQI
•26
¡ 3.尤其是患者出现进行性器官肿大,血细胞及生化指 标改变时,就提醒医生这是机体对病原体不同寻常的 反应,想到HLH的可能。如果不治疗,机体出现不恰当 的炎症反应,最终导致中性粒细胞减少,患者可能死 于细菌或霉菌感染,或者脑功能紊乱。
•XIQI
•27
六、鉴别诊断及相关问题
•XIQI
¡ 髓造血干细胞移植。一般于治疗开始后8周进行 疗效评估。
•XIQI
•42
¡ 中医药治疗
¡ 高热,躁动不安,神昏谵语,衄血者,清开 灵30-60ml 静滴。气营两燔,热盛动血型以清瘟败毒饮加减: 生石 膏,生地,知母,玄参,白花蛇舌草,紫草,丹皮,赤 芍,连翘,黄 芩,黄连,竹叶,甘草等水煎服。
¡
¡ 丙球蛋白 0.4g/kg·d,连用5天。
•XIQI
•40
维持治疗:
¡ VP16 150mg/m2, 静注, 每2周1次。 ¡ Dex 10mg/m2, 静注, 连续3天, 每2周重复1 次。 ¡ CSA 6mg/kg, 每日口服6~12月。
•XIQI
•41
强化治疗:
¡ 对于耐药/复发病例, 给予联合化疗(如 CHOP、 CHOPE方案等)/或骨
噬血细胞综合征
噬血细胞综合征噬血细胞综合征(hemophagocytic syndrome,HPS),是一组骨髓、脾脏、淋巴结等造血组织中良性、反应性增生的组织细胞吞噬自身血细胞而引发的一系列临床病症。
临床表现为持续高热,肝、脾、淋巴结大,外周血细胞减少,肝功能异常,凝血功能障碍等。
1979年首先由Risdull等报告,多发于儿童。
可由感染病毒,或是因为用药不当,以及体内有肿瘤(对于小孩来说肿瘤几率低)等诱发,由于婴幼儿自身抵抗能力弱,调节不当,致使单核巨噬细胞增生而且对自身血细胞发生攻击并消除,使得婴儿贫血导致死亡。
嗜血细胞综合征 - 分类原发性HPS,或称家族性HPS,为常染色体隐性遗传病,其发病和病情加剧常与感染有关.继发性HPS分为感染相关性HPS(infection—associatedhemophagocytic syndrome,IAHS),此型多与病毒感染有关,由病毒引起者称病毒相关性HPS (virus-associated hemophagocyticsyndrome,VAHS);由肿瘤引起者称肿瘤相关性HPS(malignancy-associated hemophagnocytic syndrome,MAHS)。
嗜血细胞综合征 - 流行病学本病以儿童多见,男性多于女性。
儿童原发性HLH(FHL)的年发病率约为0.12/1O 万,80%的患者在2岁以前发病.继发性HPS在任何年龄均可发病.所以,一般认为2岁前发病者提示的原发性可能性大,8岁后发病者则提示继发性的可能性大,2-8岁发病者则根据临床表现进行判断.在日本和亚洲国家发病率较高。
本病来势凶险,东方患者的死亡率约为45%。
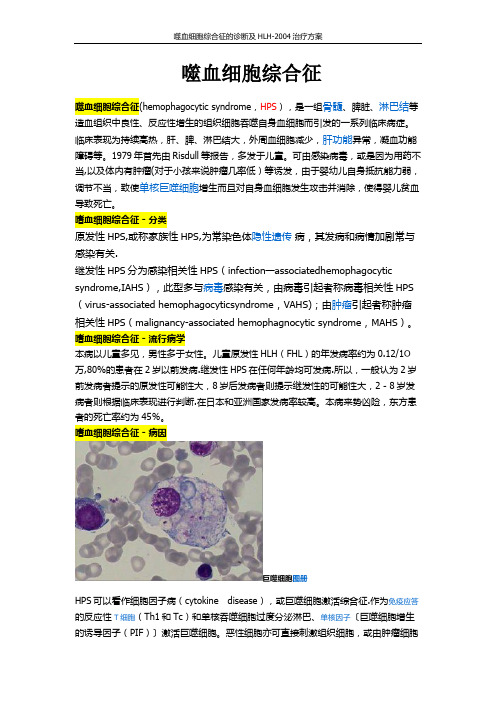
嗜血细胞综合征 - 病因巨噬细胞图册HPS可以看作细胞因子病(cytokine disease),或巨噬细胞激活综合征.作为免疫应答的反应性T细胞(Th1和Tc)和单核吞噬细胞过度分泌淋巴、单核因子〔巨噬细胞增生的诱导因子(PIF)〕激活巨噬细胞。
嗜血细胞综合征资料

并发症
►出血、感染、多脏器功能衰竭和DIC。
治疗
► 原发性HPS或病因不明未检出明显潜在疾患者除加强支持治疗和并发症的治疗外, 目前尚无特效治疗,根本性治疗是同种异体造血干细胞移植。继发性HPS应作病 因探索,治疗应以基础病与HPS并重。
►
家族性噬血细胞综合征
►
a.化学疗法:常用的化疗药物有细胞毒性药物,如长青花碱或长春新碱与肾
或抗胸腺细胞球蛋白;
►
④直接拮抗细胞因子的抗TNF抗体和IL-1受体拮抗剂;
►
⑤为抑制或减少淋巴因子的供应源可采用化疗。包括CHOP、CHOPE
方案或缓慢静滴长春新碱。屡已报道应用依托泊甙(VP16)治疗原因不明
的重症HPS、EBV-AHS或LAHS奏效。预后分析表明,对于不易与MH鉴别
的HPS患者启用化疗是必需的;
► 流行病学以儿童多见,男性多于女性。儿童原发性 HLH(FHL)的年发病率约为0.12/1O万。在日本和亚 洲国家发病率较高。本病来势凶险,东方患者的死亡 率约为45%。
实验室和其他检查
► 血象:多为全血细胞减少,以血小板减少为明显,白细胞减 少的程度较轻;观察血小板的变化,可作为本病活动性的一 个指征。病情缓解时,首先可见到血小板上升;而在病情恶 化时,亦首先见到血小板下降。
► 免疫学检查:家族性HPS常有自然杀伤细胞及T细胞活性降低。 ► 影像检查:部分病人胸片可见间质性肺浸润,晚期病人头颅CT或MRI
检查可发现异常,其改变为陈旧性或活动性感染,脱髓鞘,出血,萎 缩或(及)水肿。有时亦可通过CT检查发现脑部钙化。B超提示肝脾 肿大。
诊断
► 国内很多医院诊断HPS 系采用日本Histiocyte SocietyFHL 研究组制定 的诊断标准。
噬血细胞综合征治疗进展

噬血细胞综合征治疗进展噬血细胞综合征(Hemophagocytic lymphohistiocytosis, HLH)是一种罕见而严重的疾病,其特点是免疫系统异常激活导致噬血细胞(histiocyte)过度增殖和异常激活,进而对正常组织器官造成损害。
HLH患者常常表现为高热、肝脾肿大、淋巴结肿大、低血小板和白细胞减少等症状,并可伴有中枢神经系统损害、出血和多器官功能障碍等严重并发症。
目前,HLH的治疗主要包括免疫抑制剂的使用和造血干细胞移植。
针对免疫抑制剂的治疗,糖皮质激素是首选药物,而机制不明确的HLH患者常常需要接受更强效的免疫抑制剂,例如环孢素A、羟基脲、甲氨蝶呤等。
化疗药物如顺铂和环磷酰胺也被用于HLH的治疗。
近年来,针对HLH的治疗方案逐渐丰富和完善。
在早期干预方面,研究者提出了以血清铁蛋白水平作为指标进行快速诊断和治疗的策略。
该策略能够在早期筛查出HLH患者,并通过及时使用免疫抑制剂来控制疾病的进展。
近年来关于HLH的分子基础研究也取得了一定的进展。
目前已发现与HLH发生相关的多个基因突变,并通过对这些基因突变进行分析,研究者们对HLH的发病机制有了更深入的了解。
这些研究成果为HLH的治疗提供了新的思路,例如通过针对特定突变的基因治疗。
通过干细胞移植治疗已成为HLH的标准疗法。
随着技术的发展,HLH的干细胞移植治疗效果逐渐提高。
研究者们利用HLH患者的同胞造血干细胞进行移植,以重建其免疫系统,取得了较好的疗效。
基因治疗和干细胞基因编辑技术的发展也为HLH患者提供了新的治疗选择。
随着对HLH病理生理机制的深入研究和治疗技术的发展,HLH的治疗进展日益显著。
通过早期诊断和干预,选择合适的免疫抑制剂和干细胞移植方案,以及探索基因治疗等新的治疗策略,有望提高HLH患者的生存率和治愈率,改善其生活质量。
噬血细胞综合征医学课件

鉴别诊断
1 2
淋巴瘤
淋巴结肿大更为显著,无发热和脾大等症状。
系统性红斑狼疮
有皮疹、肾脏损伤、血液系统损伤等临床表现 。
3
病毒感染
如EB病毒感染、巨细胞病毒感染等,可伴有发 热、淋巴结肿大等症状,但无脾大和血液系统 损伤等表现。
03
治疗方案与用药
治疗方案
诱导治疗
通过使用依托泊苷、地塞米松等药物诱导缓解, 迅速控制病情。
输血支持
当患者出现贫血、出血等情况 时,可适当输血纠正症状。
抗感染治疗
对于存在感染的患者,应及时 使用抗生素进行治疗。
营养支持
患者食欲不振、消化不良时, 应给予营养支持,如静脉营养
、肠内营养等。
04
预后与并发症
预后情况
良性过程
01
某些类型的噬血细胞综合征,如家族性噬血细胞综合征,病情
较轻且预后较好,经过治疗可完全康复。
再发风险
部分患者治愈后可能再次发生噬血细胞综合征或其他类型的 血液系统疾病,应长期关注并采取预防措施。
05
预防措施与生活建议
预防措施
疫苗接种
鼓励人们接种各种疫苗,包括流感疫苗、肺炎球菌疫苗等,以增 强免疫力,预防感染。
健康生活方式
保持健康的饮食、适当的运动、充足的休息和睡眠,以增强身体 免疫力。
发病率与死亡率
发病率
噬血细胞综合征在人群中的发病率约为0.005‰~0.04‰,可发生于任何年 龄阶段,以青少年和成年人为主。
死亡率
病情危重,如不及时治疗,死亡率极高,五年生存率不到10%。
病因与病理机制
病因
噬血细胞综合征的病因多样,包括感染、免疫异常、药物反应、肿瘤等多种因素 。其中,感染是最常见的病因之一。
噬血细胞综合征医学课件

05
噬血细胞综合征的预防与控制
宣传教育与筛查策略
宣传教育
开展针对高危人群的宣传教育,提高对噬 血细胞综合征的认知和预防意识。
筛查策略
针对高危人群制定筛查策略,包括定期检 查、症状监测和影像学检查等,以及时发 现并确诊病例。
疫情监测与控制措施
疫情监测
建立健全疫情监测和报告体系,及时掌握噬血细胞综合征的发病和流行情况 。
输血、补充血小板和抗 感染等治疗措施,以维 持患者的生命。
使用免疫抑制剂如依托 泊苷、环孢素A等,抑制 过度免疫反应,缓解病 情。
采用针对血液肿瘤的联 合化疗方案,如依托泊 苷+长春新碱+环磷酰胺 等,以达到长期生存的 目的。
根据患者病情和治疗反 应,定期进行实验室和 影像学检查,评估疗效 和调整治疗方案。同时 关注患者的生活质量和 心理健康状况,给予必 要的支持和关怀。
个性化治疗方案
通过对患者基因、病情的全面分析,制定出更加个性化的治疗方案,提高治疗效果并降低 不良反应。
药物研发与临床试验
通过大量实验和临床验证,将有更多新型药物和治疗手段应用于噬血细胞综合征的治疗中 ,为患者带来更多福音。
THANKS
谢谢您的观看
控制措施
制定并实施针对性的控制措施,如隔离治疗、密切接触者追踪、疫苗研发和 应用等,以遏制疫情的传播和扩散。
公共卫生政策与干预手段
公共卫生政策
制定有利于预防和控制噬血细胞综合征的公共卫生政策,如健康教育和健康促进 、高危人群管理、医疗保障等。
干预手段
采取多种干预手段,如社会宣传、行为干预、环境改善等,提高公众的预防意识 和健康水平,从而有效控制噬血细胞综合征的传播。
02
噬血细胞综合征的病因与发病机制
噬血细胞综合征医学

xx年xx月xx日
噬血细胞综合征医学
CATALOGUE
目录
噬血细胞综合征概述噬血细胞综合征的诊断噬血细胞综合征的治疗噬血细胞综合征的预后和预防噬血细胞综合征的最新研究进展
噬血细胞综合征概述
01
噬血细胞综合征是一种由淋巴组织异常增生引起的疾病,其特征为大量异常淋巴细胞在体内积聚并破坏正常组织。
定义
感染诱发
临床表现
患者常出现持续性发热,且难以用常规方法治疗。
发热
肝脾肿大
淋巴结肿大
神经系统症状
部分患者可能出现肝脾肿大,伴有肝功能异常和黄疸。
多数患者有淋巴结肿大,部分患者可能伴有疼痛。
部分患者可能出现神经系统症状,如意识障碍、抽搐等。
噬血细胞综合征的诊断
02
符合HLH-2004诊断标准,包括以下8项指标发热;脾大;全血细胞减少;骨髓增生减低或增生异常;肝功能异常;凝血功能障碍;铁代谢异常;噬血细胞在骨髓或外周血中比例明显增高。
与自身免疫性疾病相关,患者可出现全血细胞减少、骨髓增生减低或增生异常等症状,但无发热、脾大等表现。
1
诊断流程
2
3
1. 进行血常规、骨髓检查、淋巴结活检等相关检查,初步判断是否符合噬血细胞综合征的诊断标准。
2. 进行感染、肿瘤、自身免疫性疾病等相关疾病的鉴别诊断。
3. 根据患者的临床表现、实验室检查等结果,制定治疗方案。
定期监测和治疗建议
噬血细胞综合征的最新研究进展
05
研究针对特定基因突变或蛋白质的药物,以更精确地治疗噬血细胞综合征。
靶向治疗
新药研发
开发能够调节免疫系统功能的新型药物,以减轻噬血细胞综合征的症状和体征。
免疫调节药物
《噬血细胞综合症》课件

免疫治疗
免疫抑制剂
如糖皮质激素、环孢素等,用于 调节免疫系统,减轻异常免疫反 应。
细胞因子治疗
如干扰素、白介素等,通过调节 细胞因子网络,控制炎症反应。
造血干细胞移植
01 供体选择
选择HLA配型相合的供体,确保移植成功并降低 排斥反应的风险。
05
展望与未来研究方向
新药研发与临床试验Fra bibliotek针对噬血细胞综合症的特异性药物
开发针对该疾病的特异性药物,以提高治疗效果和减少副作用。
临床试验的标准化和规范化
建立统一的临床试验标准和方法,以确保试验结果的可靠性和可重复性。
基因治疗与精准医疗
基因突变与疾病关系的研究
深入探究基因突变与噬血细胞综合症发病机制的关系,为精 准医疗提供依据。
病因与发病机制
病因
噬血细胞综合症的病因多样,包括感染、自身免 疫性疾病、肿瘤等。
发病机制
该病的发病机制涉及到免疫系统的异常激活,导 致大量炎症细胞和噬血细胞增殖,对正常组织造 成损害。
临床表现与诊断标准
临床表现
噬血细胞综合症的临床表现多样,常见的症状包括高热、肝脾肿大、全血细胞减少、肝功能异 常等。
诊断标准
根据临床表现和实验室检查,如血常规、肝功能、骨髓检查等,可以确诊噬血细胞综合症。同 时,还需要排除其他类似的疾病,如淋巴瘤、骨髓增生异常综合征等。
02
噬血细胞综合症的治疗
药物治疗
01
化疗药物
常用的化疗药物包括依托泊苷、地塞米松、环磷 酰胺等,用于抑制异常免疫反应和杀灭异常细胞
。 02
生物制剂
02 定期体检
《噬血细胞综合征》课件

注意个人卫生,避免接触感染源,减少不必要的创伤。
对于已经存在的慢性疾病,如糖尿病、高血压等,应积极治疗并控制病情。
向患者及家属介绍噬血细胞综合征的病因、症状、治疗和预防等方面的知识。
疾病知识教育
指导患者正确使用药物,包括药物的种类、剂量、使用方法和注意事项等。
详细描述
总结词
常见、症状多样、诊断困难
详细描述
成人噬血细胞综合征是一种较为常见的疾病,症状多样,包括发热、淋巴结肿大、肝脾肿大等。由于该病表现多样,因此诊断较为困难。该病例中,患者经过多次检查和排除其他疾病后,最终确诊为成人噬血细胞综合征。经过积极治疗,病情得到缓解,但容易复发。
05
CHAPTER
特点
噬血细胞综合征的病因较为复杂,可能与遗传、免疫缺陷、感染等多种因素有关。
该病的发病机制主要涉及免疫系统的异常激活,导致大量炎症因子和细胞因子的释放,引发全身性的炎症反应和组织损伤。
发病机制
病因
临床表现
噬血细胞综合征的临床表现多样,常见的症状包括持续高热、肝脾肿大、全血细胞减少、肝功能异常等。病情严重时可能出现多器官功能衰竭。
《噬血细胞综合征》PPT课件
目录
噬血细胞综合征概述噬血细胞综合征的治疗噬血细胞综合征的预防与护理噬血细胞综合征的案例分析噬血细胞综合征的未来ห้องสมุดไป่ตู้究方向
01
CHAPTER
噬血细胞综合征概述
噬血细胞综合征是一种严重的疾病,由于免疫系统异常激活导致大量炎症和组织损伤。
定义
该病通常表现为持续高热、肝脾肿大、全血细胞减少等症状,病情进展迅速,死亡率较高。
噬血细胞综合征的未来研究方向
噬血细胞综合征发病机制诊疗和治疗

病因与发病机制
Infection-associated HLH
Develops as the result of infection Viral (most common), bacterial, fungal, parasites Often in immunocompromised hosts (HIV, oncologic, Crohn’s disease)
Syntaxin- -20% 3 11
Monobranolytic protein expressed in the cytoplasmic granules of cytotoxic T cells and NK cells.
Responsible for the translocation of granzyme B from cytotoxic cells into target cells; granzyme B then migrates to target cell nucleus to participate in triggering apoptosis.
related donors. The majority were unrelated.
小朋友HLH疗效比较
不治疗组(n=121;1983年):1年OS 5% HLH-94治疗组(n=113;2023年):5年OS 55% 其他治疗组(n=122;1996年): 5年OS 22%
HLH-94治疗EBV-HLH
白原血症(≤1.5g/L) E.脾、肝、淋巴结、骨髓或CNS中噬血细胞增多,无肿瘤细胞 F.NK细胞(CD56+/16+)活性减弱或缺乏(在SHLH缓解时可恢复
正常),数量一般正常 G.高铁蛋白血症(≥500μg/L) H.血浆sIL-2R(CD25)≥2400U/mL
噬血细胞综合征

噬血细胞综合征的社会 影响
对患者和家庭的影响
心理压力:噬血 细胞综合征对患 者和家庭造成巨 大的心理压力, 影响生活质量。
经济负担:治疗 噬血细胞综合征 需要高额的医疗 费用,给家庭带 来沉重的经济负 担。
社交障碍:疾病 可能导致患者和 家庭成员出现社 交障碍,影响人 际关系。
家庭关系:疾病 对家庭关系产生 影响,可能导致 家庭成员之间的 矛盾和冲突。
挑战和机遇
单击此处添加标题
挑战:噬血细胞综合征的病因和发病机制仍不完全清楚,诊断和治疗难度较大。
单击此处添加标题
机遇:随着科研的深入,越来越多的研究成果为噬血细胞综合征的诊断和治疗提供 了新的思路和方法,为患者带来更好的治疗和预后。
单击此处添加标题
科研进展:新型药物和治疗手段的研究不断取得突破,为临床治疗提供了更多选择。
单击此处添加标题
未来展望:随着科研的进一步发展,相信噬血细胞综合征的治疗将会取得更大的突 破和进步。
噬血细胞综合征国际研究联盟的成 立
国际合作与交流
国际学术会议和研讨会是交流最新 研究成果的重要平台
添加标题
添加标题
添加标题
添加标题
跨国合作开展临床试验和药物研发
跨国合作研究项目,共同攻克噬血 细胞综合征难题
发者
优点:治愈率 高,可有效延
长生存期
注意事项:需 寻找匹配的供 体,严格筛选 供体和预处理
方案
其他治疗方法
化疗:使用化学 药物杀死或抑制 肿瘤细胞生长
放疗:利用放射线 治疗肿瘤,破坏肿 瘤细胞生长和繁殖
免疫治疗:通过调 节免疫系统来增强 机体对肿瘤细胞的 抵抗力
靶向治疗:针对肿 瘤细胞表面的特定 蛋白或基因进行治 疗
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
传染性单核细胞增多症
恶性组织细胞病(MH)
本病无血脂改变,外周血或骨髓中可发现异常组织细胞: 外周血、骨髓、淋巴结、肝、脾等组织中可找到恶性组织细
胞,支持MH; 恶性组织细胞对乙酸萘酚脂酶、酸性磷酸酶染色阳性,免
疫组化是细胞内链及链均阳性; HPS的中性粒细胞碱性磷酸酶活性可增高; MH血清铁蛋白和血管紧张素转换酶增高,组化染色见巨噬
脑脊液:压力升高,细胞数增多,5-20×106/L,但以淋巴细 胞为主,可有单核样细胞,蛋白升高,但也有脑炎症状明显 而脑脊液正常者。
免疫学检查:家族性HPS常有自然杀伤细胞及T细胞活性降低。 ANA、Coomb’s test可阳性
实验室检查
骨髓检查:骨髓多数增生活跃,在骨髓涂片的尾部可发现 单独存在或成团分布的噬血细胞性组织细胞增生,阳性率 为75%。但有少数病例早期仅表现为增生活跃,并无嗜 血细胞,病程晚期出现增生低下。
与其他疾病鉴别—
与全血细胞减少相关疾病鉴别: 恶性组织细胞病:骨髓见异常组织细胞,多无血脂改变。 再生障碍性贫血:肝脾不大,骨髓无噬血细胞。 白血病:骨髓及外周血可见原始细胞 其他疾病: 传染性单核细胞增多症 败血症 类风湿关节炎 系统性红斑狼疮等。 以上疾病均可合并继发HLH疾病,需鉴别。
4. 高 甘 油 三 脂 血 症 和 ( 或 ) 低 纤 维 蛋 白 原 血 症 , 甘 油 三 脂 ≥3.0mmol/L或纤维蛋白原<1.5g/L; 5.骨髓、肝、脾、淋巴结组织细胞非恶性增生伴嗜血细胞现象; 6.NK细胞活性降低或完全缺少; 7.血清铁蛋白≥500mg/L; 8.可溶性CD25 (IL-2受体)≥2400u/ml;
细胞中大量抗核蛋白酶; 部分MH含有特异性染色体组织细胞增生症
LCH发病是以婴儿多见,2岁以上少见。 表现为发热、皮疹、肝脾淋巴结肿大、肺部浸润、N-S受
累,但LCH的皮疹为特异性皮疹,多分布于躯干、胸腹部、 发际、耳后及颈部,可与HPS的一过性皮疹鉴别, LCH 常 出 现 骨 骼 破 坏 , 活 检 组 织 在 电 镜 下 见 到 含 有 Birbeck颗粒的郎格汉斯细胞是诊断LCH的主要依据。
维持治疗:
足叶已甙(VP16):150mg/m2,每2周1次,共一年。
地塞米松(Dex): 每日10mg/m2 ,每2周用3天,共一年。
作用机制
足叶已甙较好的细胞凋亡始动因子 环孢菌素A:降低T细胞活性 地塞米松:抗炎、促凋亡及很强的脑膜穿透作用
HLH-1994方案
HLH-1994方案
肺部症状:与肺部淋巴细胞和巨噬细胞浸润有关; 其它:可有乏力、厌食、体重增加、关节痛、胃肠道症状。
实验室检查
全血细胞减少
转氨酶 胆红素 铁蛋白 甘油三酯 纤维蛋白原
巨噬细胞吞噬现象
(吞噬RBC、WBC、PLT)
注:输血、外科术后亦可见吞噬RBC现象
吞噬现象
实验室检查
凝血检查:纤维蛋白原降低、部分凝血活酶时间(APTT)延长, 肝功能损害者,凝血酶原时间(PT)延长。
诊断
诊断指南(HLH-2004)
符合下列2项中1项者即可诊断: 1. 分子生物学诊断符合HPS 2. 符合下列8项中5项者以上者
1.发热; 2.脾肿大; 3.全血细胞减少(外周血2或3系细胞减少)
Hb<90g/L,新生儿<100g/L; 血小板<100×109/L; 中性粒细胞<1.0×109/L;
有条件尽早行HSCT(造血干细胞移植)方能 根治
继发性噬血细胞综合征
病情稳定、症状轻,先选择糖皮质激素,如不能控制再 加用CSA及VP-16.
EBV相关者早期使用VP-16效果较好 持续病毒感染者每4周输注丙球0.5g/(kg.次) 重症病例行血浆置换 巨噬细胞活化综合征相关者建议强免疫抑制治疗(大剂量
后很差。7天—5年,平均存活时间8周。
嗜血细胞综合征
分类
根据触发噬血细胞综合征的病因不同分为两类型 原发性噬血细胞综合征,或称家族性噬血性
淋巴组织细胞增生症(FHL) 继发性噬血细胞综合征
1.感染相关性噬血细胞综合征 2.肿瘤相关性噬血细胞综合征 3.药物相关性噬血细胞综合征 4.风湿免疫相关性噬血细胞综合征
淋巴细胞介导溶细胞作用示意图
靶细胞
杀伤性 T细胞
穿孔素 “打孔”
介导性颗粒 (穿孔素、 颗粒酶)
细胞膜损伤
调亡 或坏死
细胞凋亡或坏死
穿孔素 (Perforin) 的作用机制
噬血细胞综合征时溶解细胞效应缺失示意图
遗传缺陷 T细胞、NK细胞功能缺陷
病原体来源的抗原持续刺激淋巴细胞
感染因素
活化的T细胞、NK细胞持续增多
药物
苯妥英钠
发病机制- 先天性或获得性免疫缺陷
PRF1基因缺陷与穿孔素功能异常相关。 MUNC13-4、STX11、STXBP2、RAB27A、LYST、 AP3B1基因缺陷可导致NK细胞和CTL脱颗粒异常,细胞表 面CD107a表达水平降低。 NK细胞和CTL细胞数量与功能受到损害,导致溶细胞颗粒 (穿孔素和颗粒酶)释放障碍,不能“杀死”受感染的靶细 胞,使感染持续存在并不断加重以及多脏器损伤 (uncontrolledinflammatory process); 由于巨噬细胞的过度活化、增殖,致巨噬细胞侵润和吞噬作 用“泛化”,导致持续性的全血细胞减少、感染、出血和多 脏器功能障碍。
注意:在2-8岁之间发病者,则要根据临床表现来判断,如 果还难肯定,则应按家族性HPS处理。其次要与恶性组织细 胞病(恶组)相鉴别,二者在骨髓片上很难鉴别,但HPS要 比恶组常见得多。但如临床上呈暴发经过、严重肝功能损害、 骨髓中组织细胞恶性程度高,特别是肝、脾或其他器官发现 异常组织细胞浸润,则先考虑为恶组为宜;否则应诊断为 HPS。
CSA(环孢菌素A):口服,5mg/(kg・d),分2次,每12小时1次,自化 疗第15天起。血药浓度(谷浓度)不超过200ug/L
儿童噬血细胞综合征
噬血细胞综合征
亦称噬血细胞性淋巴组织细胞增生症 (hemophagocytic lymphohistiocytosis,
HLH) 一种以淋巴细胞、巨噬细胞非恶性增生伴噬
血细胞增多。 多器官、多系统受累,呈进行性加重。 临床表现为发热、肝脾肿大、全血细胞减少
为特征的综合征。 病情常发展迅速,若不及时诊断及治疗则预
原发性噬血细胞综合征
原发性噬血细胞综合征,或称家族性噬血细胞综合征。为常染色 体或X连锁隐性遗传,伴有相关基因异常。多(70-80%)于1岁 内发病,高峰为1-6个月。
已知基因缺陷: 穿孔素( perfrin )基因 、 Munc 13-4 基因、 syntaxin 11 基因 免疫缺陷: Chediak-Higashi syndrome
(1)诱导治疗(8周)
甲泼尼龙(MP):静脉滴注,10mg/(kg・d)x3d,5mg/(kg・d)×3d, 2mg/(kg・d)×8d,1mg/(kg・d)×2周,0.5mg/(kg・d)×2周, 0.25mg/(kg・d)×1周,继于1周内减停,疗程共8周
VP-16(足叶已甙):静脉滴注,100mg/(m2:次),2次/周x1周,1次/周 ×7周。
这些细胞因子源自过度活化的T细胞和巨噬细胞,又进一步 促进巨噬细胞的持续性激活、增殖、侵潤、吞噬现象,同时 进一步造成杀伤 T 细胞和 NK 细胞功能缺陷;受感染的细 胞(靶细胞)不能溶解和清除而持续存在,导致多脏器功能 损害,产生一系列临床症状。
临床表现
发热:早期多见发热,为高热-稽留热、驰张热或不规则热。 体温峰值>38.5℃,持续7天以上,可自行下降。
Th1细胞活化 胞
分泌细胞因子 (IFN-γ、GM-CSF)
肿瘤细
活化巨噬细胞
活化的T细胞、巨噬细胞浸润组织同时分泌 细胞因子(TNF-α、IL-1、IL-6等)
组织损伤
“ 细胞因子风暴导致所有临床表现” 干扰素-γ(IFN- γ) 白介素-6,-10,-12,-16,-18 sIL-2R(sCD25) 肿瘤坏死因子-α ( TFN-α)
并发症
出血 感染 多脏器功能衰竭 弥散性血管内凝血
治疗
治疗原则
原发性或病因不明、未检出明显潜在疾病者 1.支持治疗 2.并发症治疗 3.同种异体造血干细胞移植
继发性噬血细胞综合征 1.查明病因 2.治疗基础病与噬血细胞综合征并重
原发性噬血细胞综合征
一旦确诊,尽早HLH-2004(1994)方案 治疗。
(白细胞颗粒异常综合征) Griscelli syndrome
(部分白化伴有多种免疫异常) X-linked prliferative Syndrme , XLP
(X-连锁淋巴增殖综合征)
继发性噬血细胞综合征
外源性因素(病原微生物、毒素) (感染相关性噬血细胞综合症,IAHS) (病毒相关性噬血细胞综合症,VAHS)
鉴别诊断
原发与继发鉴别
原发:家族遗传性,(近亲结婚)基因缺陷,2岁前发病多 见,病情重,易于反复,NK细胞活性进行性减低,HSCT为 目前唯一根治性治疗手段。在2岁前发病者多提示为家族性 HPS,而8岁后发病者,则多考虑为继发性HPS。
继发:无家族史,无基因缺陷,多有明确诱因或基础疾病, 8岁后发病者多,病情轻,NK细胞活性阶段性减低一般不需 要HSCT治疗。
影像检查:部分病人胸片可见间质性肺浸润,晚期病人头 颅CT或MRI检查可发现异常,其改变为陈旧性或活动性 感染,脱髓鞘,出血,萎缩或(及)水肿。有时亦可通过 CT检查发现脑部钙化。B超提示肝脾肿大。
实验室检查
病理学检查:肝、脾、淋巴结、骨髓、N-S、甲状腺、 肺、心、肠、肾和胰腺均可受累。在单核-巨噬细胞系 统良性的淋巴组织细胞浸润、组织细胞有吞噬现象, 以吞噬红细胞为主。
肝、脾肿大:明显肿大,且进行性加重;可出现黄疸、腹 水。