重症肌无力治疗指南 新
重症肌无力诊疗指南

重症肌无力诊疗指南重症肌无力(Myasthenia·Gravis,MG)是一种表现为神经肌肉接头传递障碍的获得性自身免疫性疾病,临床特征为一部分或全身横纹肌软弱和异常疲劳;通常在活动后加剧,休息后减轻。
患病率约为5/10万,女性多于男性。
各年龄组均可发病。
自青年期至40岁间发病者,以女性为多;中年以后发病者,则以男性为多,本组中胸腺瘤较多见。
【临床表现】1、眼外肌是最常受累的肌肉,可表现为眼睑下垂、斜视和复视,双侧常不对称。
其次为颅神经支配的其他肌群,颈肌,肩胛带以及髋部的屈肌。
受累肌肉呈现为易疲劳和波动性。
连续收缩后发生无力,经短期休息后好转。
早晨症状较轻而傍晚时加重。
整个病程可有波动,病程早期可有自发缓解和复发。
2、特殊类型短暂新生儿重症肌无力为女性患者所生的婴儿暂时有经胎盘输入的母体AchR抗体。
大约有10%呈现不同程度的临床症状。
患婴全身软弱,哭声轻微,吸吮无力,上脸下垂,严重者有呼吸困难,经救治后可在数日内或数周内痊愈。
3、先天性肌无力出生后或儿童期出现肌无力,持续存在眼外肌麻痹。
母亲虽无重症肌无力,但家族中或同胞兄弟姐妹有肌无力病史。
4、重症肌无力患者除上述的临床表现外,神经系统检查体征主要是疲劳试验(+)。
腱反射正常或活跃。
Osserman根据受累部位和严重程度,可分为5型。
1型单纯眼肌型,始终仅累及眼肌。
2型轻度全身肌无力型。
不伴明显延髓肌麻痹者为lla型,伴有明显延髓肌麻痹者为llb型。
3型急性进展型。
常在首次症状出现后数月之内发展至包括延髓肌、肢带肌、躯干肌和呼吸肌的严重无力。
4型为晚发型全身肌无力。
由l,lla,llb发展而来,常在首发症状出现后数年或数十年后出现全身无力。
5型肌无力伴肌萎缩者。
以上各型患者,如果急骤发生呼吸肌严重无力,不能维持换气功能时,称为危象。
可分为三种(1)肌无力危象为疾病发展所致。
多见于暴发或严重的全身型。
静脉注射腾喜龙2~10mg或肌肉注射硫酸新斯的明1.5mg可见暂时好转。
肌无力的最好治疗方法

肌无力的最好治疗方法
肌无力是一种神经肌肉疾病,其最好的治疗方法因个体差异而有所不同。
以下是一些常见的治疗方法,但请在接受治疗前先咨询医生。
1. 药物治疗:肌无力常用的药物包括乙酰胆碱酯酶抑制剂和免疫调节剂。
乙酰胆碱酯酶抑制剂可以增加乙酰胆碱在神经肌肉接头的浓度,从而改善肌肉收缩。
免疫调节剂可以调节免疫系统的异常反应,减轻免疫攻击导致的肌肉无力。
2. 物理治疗:包括康复训练、物理疗法和肌肉强化锻炼等。
这些方法可以帮助改善肌肉力量、平衡和协调能力,并提高日常生活的功能。
3. 手术治疗:对于某些严重病例,手术可能是必要的。
手术选项包括胸腺切除术和淋巴细胞清除术。
胸腺切除术可以减少免疫系统攻击神经肌肉接头的程度,而淋巴细胞清除术可以去除免疫系统中引起免疫攻击的异常细胞。
4. 辅助疗法:包括使用助行器具、呼吸器具、语音治疗等。
这些辅助疗法可以帮助患者更好地应对肌无力带来的生活影响。
除了以上治疗方法外,个体化的治疗还需要考虑患者的健康状况、病情严重程度和病因等因素。
因此,寻求专业医生的指导和个体化的治疗方案非常重要。
重症肌无力诊断和治疗指南

作为成年MG患者单一治疗药物,推荐剂量为375 mg/m2体外 表积,静脉滴注,每周1次,22 d为一疗程,共给药4次
各种治疗的起效时间
治疗方法 吡啶斯的明 血浆置换 静脉丙球
临床分类
•Osserman分型
Ⅲ 型——重度激进型,起病急、进展快,发病 数周或数月累及咽喉肌;半年内累及呼吸肌, 伴或不伴眼外肌受累,生活不能自理
Ⅳ 型——迟发重度型,隐袭起病,缓慢进展。2 年内逐渐进展,由Ⅰ型、ⅡA、ⅡB型进展而来, 累及呼吸肌
Ⅴ型——肌萎缩型,起病半年内可出现骨骼肌萎 缩、无力
预后
谢谢!
二、肌电图检查
单纤维肌电图(SFEMG) 使用特殊的单纤维 针电极通过测定“颤抖〞(Jitter)研究神经-肌肉 传递功能,“颤抖〞通常15~35 us;超过55us为“颤抖 增宽〞,一 块肌肉记录20个“颤抖〞中有2个或2个 以上大于55us那么为异常。 检测过程中出现阻滞 (block)也判定为异常。SFEMG并非常规的检测 手 段,但敏感性高。SFEMG不受胆碱酯酶抑制剂影响。 主要用于眼肌型MG或临床疑心MG但RNS〔低频重复神经电刺激〕 未 见异常的患者。
全身骨骼肌均可受累〔脑神经支配的肌肉较脊神经支配的肌 肉更容易受累〕
眼外肌受累最为多见,常以对称或非对称性上睑下垂和(或) 双眼复视为首发病症,也可出现眼球活动障碍等
咽喉肌受累出现构音障碍、吞咽困难、鼻音、饮水困难、声 音嘶哑等
面肌受累可致鼓腮漏气、眼睑闭合不全、鼻唇沟变浅、苦笑 或呈肌病面容
临床表现
咀嚼肌受累可致咀嚼困难 颈肌受累,以屈肌为著,出现头颈活动 障碍、抬头困难或不能 肢体各组肌群可累及,以近端为著 呼吸肌受累可致呼吸困难、无力,局部 患者可出现肌无力危象,需人工辅助呼 吸 特点: 表现为波动性和易疲劳性,晨轻暮重, 疲劳后加重,休息后可减轻 可从一组肌肉无力开始,在一至数年内 逐步累及其它肌群
重症肌无力诊断和治疗指南PPT课件

预防感染
保持室内空气流通,避免患者 接触感染源,预防感冒和其他 感染。
症状观察
密切观察患者的病情变化,如 出现呼吸困难、吞咽困难等症
状,应及时就医。
康复训练
物理治疗
通过电刺激、按摩等方法,促进肌肉 收缩和血液循环,改善肌肉功能。
运动疗法
在专业医师指导下,进行适当的运动 训练,如散步、游泳等,以增强肌肉 力量和耐力。
02 重症肌无力的诊断
诊断标准
临床表现
抗体检测
出现肌肉无力、疲劳、晨轻暮重等症 状,且活动后加重,休息后缓解。
检测血清中是否存在抗乙酰胆碱受体 抗体(AChR-Ab)或肌肉特异性酪氨 酸激酶抗体(MuSK-Ab)。
重复神经电刺激检查
在低频刺激时,出现肌肉动作电位幅 度递减,高频刺激时,出现肌肉动作 电位幅度递增。
最新治疗方法
免疫治疗
使用免疫抑制剂、免疫调节剂等药物,调节免疫 系统功能,控制病情发展。
胸腺切除术
对于胸腺增生或胸腺瘤的重症肌无力患者,手术 切除胸腺可缓解症状。
血浆置换疗法
通过置换血浆中的抗体,减轻症状,控制病情发 展。
未来研究方向
探索新的诊断方法
研究更快速、准确的诊断技术,提高重症肌无力的早期发现率。
作业疗法
通过日常生活活动训练,如穿衣、进 食等,提高患者的生活自理能力。
言语治疗
针对患者出现的言语障碍,进行发音、 语速等训练,以改善语言交流能力。
心理支持
心理疏导
关注患者的心理状态,给予必要 的心理疏导和支持,帮助患者树
立信心。
家属支持
鼓励家属积极参与患者的护理和 康复过程,给予患者关爱和支持。
诊断方法
01
中国重症肌无力诊断和治疗指南

中国重症肌无力诊断和治疗指南一、本文概述《中国重症肌无力诊断和治疗指南》是一份权威的医学指南,旨在为医生和医疗工作者提供关于重症肌无力(Myasthenia Gravis, MG)的标准化诊断和治疗建议。
重症肌无力是一种由自身免疫系统异常引起的神经-肌肉接头传递障碍性疾病,临床表现为部分或全身骨骼肌无力和易疲劳。
本指南的编写目的是确保患者能够得到及时、准确和个性化的医疗服务,改善其生活质量,降低疾病对社会和家庭的影响。
本指南涵盖了重症肌无力的流行病学、发病机制、临床表现、诊断方法、治疗策略以及疾病管理等方面的内容。
通过全面系统地介绍重症肌无力的最新研究进展和临床实践经验,本指南旨在为医生提供科学、规范、实用的治疗建议,以促进重症肌无力患者的康复。
本指南也为政策制定者、医学教育工作者和科研人员提供了有价值的参考信息,有助于推动我国重症肌无力诊疗水平的提高。
本指南的制定遵循了科学、规范、实用的原则,充分借鉴了国内外相关指南和研究成果,结合我国实际情况,形成了具有中国特色的重症肌无力诊断和治疗方案。
我们希望本指南能够为广大医生和医疗工作者提供有益的帮助,共同推动我国重症肌无力诊疗事业的发展。
二、重症肌无力的病因与病理生理机制重症肌无力(Myasthenia Gravis,MG)是一种慢性自身免疫性疾病,主要表现为神经-肌肉接头的传递功能障碍。
其病因复杂,涉及遗传、免疫、环境等多因素。
遗传因素在MG的发病中起着重要作用,许多研究已经证实,MG与主要组织相容性复合体(MHC)基因、乙酰胆碱受体(AChR)基因、补体基因、T细胞受体基因、免疫球蛋白基因以及细胞因子基因等相关。
在病理生理机制方面,MG主要是由于患者体内产生了针对乙酰胆碱受体(AChR)的自身抗体,导致神经-肌肉接头的突触后膜乙酰胆碱受体数量减少或功能受损,进而影响了神经冲动的传递,使骨骼肌发生易疲劳现象。
MG患者体内还可能存在针对肌肉特异性激酶(MuSK)和低密度脂蛋白受体相关蛋白4(LRP4)的自身抗体,这些抗体同样可以影响神经-肌肉接头的功能。
中国重症肌无力诊断和治疗指南(2020版)

中国重症肌无力诊断和治疗指南(2020版)中国重症肌无力诊断和治疗指南(2020版)引言:重症肌无力是一种慢性、进展性的自身免疫性疾病,患病率逐年增加。
为了提高重症肌无力的诊断和治疗水平,中国医疗专家经过多次研讨和讨论,综合国内外最新研究成果,制定了2020版的中国重症肌无力诊断和治疗指南,以指导医生进行准确的诊断和科学的治疗。
一、诊断标准:1. 病史:重症肌无力的患者常有进行性肌无力症状,疲劳易感和活动后恢复缓慢等特点。
2. 体格检查:重症肌无力患者差异较大,但大多会出现眼肌麻痹和泪液分泌减少等眼部表现,以及颈肌无力和呼吸肌无力等全身表现。
3. 神经肌肉传导检查:针对神经肌肉传导方面的检查项目,包括神经肌肉传导速度、肌肉动作电位和运动单位电活动等。
4. 免疫学检查:通过抗乙酰胆碱受体抗体、MuSK抗体和Lrp4抗体等自身抗体的检测,可以帮助确定诊断。
二、治疗原则:1. 发病初期的治疗:在确诊后的早期,应立即给予对症治疗,抗乙酰胆碱酯酶药物是最常用的治疗方法,如新斯的明和吡拉西坦等。
此外,合理的运动和心理支持对于患者恢复也十分重要。
2. 疾病稳定期的治疗:在疾病进入稳定期后,除继续维持治疗控制病情外,还需考虑减少激素使用以避免副作用。
此时,免疫抑制剂的应用可以取得较好的疗效,包括环孢素A、硫唑嘌呤和甲氨蝶呤等。
3. 疾病进展期的治疗:若患者在维持治疗后,仍然存在严重的肌肉无力和呼吸肌无力等症状,可能需要考虑开展其他治疗方法,如免疫吸附、静脉免疫球蛋白等。
4. 并发症的治疗:重症肌无力可伴随多种并发症,如肺部感染和呼吸衰竭等。
因此,及时诊断并治疗这些并发症,对提高患者生活质量至关重要。
三、其他治疗手段:1. 物理治疗:包括物理疗法、功能锻炼等,可以改善患者的肢体活动能力和肌力。
2. 急救处理:对于突然发生呼吸窘迫等急性病情的重症肌无力患者,应立即进行气管插管、机械通气和选择性免疫吸附等治疗,以保证生命安全。
眼肌重症肌无力治疗方案

一、引言重症肌无力(Myasthenia Gravis,MG)是一种自身免疫性疾病,主要特征是肌肉易疲劳和无力。
眼肌重症肌无力(Ocular Myasthenia Gravis,OMG)是MG的一种类型,主要表现为眼外肌无力。
本文将对眼肌重症肌无力的治疗方案进行详细介绍。
二、药物治疗1. 抗胆碱酯酶药物抗胆碱酯酶药物是治疗OMG的首选药物,可增加神经肌肉接头的乙酰胆碱浓度,改善肌肉力量。
常用药物有:(1)新斯的明(Neostigmine):口服、皮下注射或静脉注射均可,起始剂量为15-30mg/日,分3-4次给药。
(2)吡啶斯的明(Pyridostigmine):口服,起始剂量为60-120mg/日,分3-4次给药。
(3)安贝氯铵(Ambenonium):口服,起始剂量为5-10mg/日,分3-4次给药。
2. 免疫抑制剂免疫抑制剂用于治疗对抗胆碱酯酶药物反应不佳或需要减少药物剂量的患者。
常用药物有:(1)糖皮质激素:如泼尼松、甲泼尼龙等,起始剂量为0.5-1mg/kg/日,逐渐减量至维持剂量。
(2)环磷酰胺:口服或静脉注射,起始剂量为1-2mg/kg/日,逐渐减量至维持剂量。
(3)硫唑嘌呤:口服,起始剂量为1-2mg/kg/日,逐渐减量至维持剂量。
3. 免疫球蛋白免疫球蛋白用于治疗对药物治疗反应不佳或病情危重的患者。
常用药物有:(1)人免疫球蛋白(IVIG):静脉注射,剂量为0.4g/kg,每4周1次。
(2)人免疫球蛋白(IVIG):静脉注射,剂量为0.5g/kg,每2周1次。
三、手术治疗1. 神经肌肉接头移植术神经肌肉接头移植术是将患者的神经肌肉接头移植到其他部位,以改善肌肉力量。
适用于对药物治疗反应不佳或病情危重的患者。
2. 腺体摘除术腺体摘除术是指切除患者的胸腺,以降低自身免疫反应。
适用于伴有胸腺瘤的患者。
四、康复治疗1. 物理治疗物理治疗包括电刺激、热疗、按摩等,可改善肌肉力量和运动功能。
重症肌无力的治疗方法有哪些

重症肌无力的治疗方法有哪些重症肌无力(Myasthenia Gravis,MG)是一种慢性进展性自身免疫性疾病,主要表现为肌肉疲劳和乏力。
此病通常通过治疗可以得到控制,而治疗的目标是减轻症状、提高生活质量、减少并发症的发生。
下面是一些常见的重症肌无力治疗方法:1. 胆碱酯酶抑制剂:胆碱酯酶抑制剂是最常用的治疗重症肌无力的药物,可增加乙酰胆碱在突触间隙的浓度,从而提高神经肌肉传导。
常用的胆碱酯酶抑制剂包括乙酰胆碱酯酶抑制剂(如内斯托尔、吡拉负极),和胆碱脂酶抑制剂(如吡呋喃酮)。
这些药物通常需要定期调整剂量以达到最佳效果。
2. 免疫调节疗法:免疫调节疗法主要包括环孢素、甲强龙和类固醇等免疫抑制剂。
这些药物通过减少免疫系统的活性来抑制自身免疫反应,从而减少对神经-肌肉接头的攻击。
然而,这些药物的使用可能会导致一些副作用,如免疫抑制和感染的风险。
3. 血浆置换:血浆置换是一种治疗重症肌无力的常见方法,通过将患者的血浆置换为新鲜血浆来清除有害的自身抗体,并为患者提供正常的血浆成分。
这种治疗方法能够快速改善症状,但效果持续时间相对较短,通常需要每周进行多次置换。
4. 静脉免疫球蛋白(IVIg):IVIg是通过静脉注射提供的免疫球蛋白制剂,可中和破坏神经-肌肉接头的自身抗体。
IVIg治疗能够迅速改善重症肌无力症状,但其效果仅持续数周至数个月,因此需要定期进行治疗。
5. 手术治疗:对于某些重症肌无力患者,手术治疗可能是一种有效的选择。
例如,对于长时间使用胆碱酯酶抑制剂仍无法控制症状的患者,可考虑切除胸腺。
胸腺切除手术可以减少胸腺中产生有害自身抗体的细胞数量,从而减轻症状。
6. 康复治疗和支持性护理:康复治疗包括肌肉锻炼、推拿和物理疗法等,可帮助患者恢复肌力,改善肌肉功能。
此外,支持性护理也很重要,包括提供合适的营养、充足的休息和精神支持等。
总之,重症肌无力的治疗方法多种多样,选择合适的治疗方案应根据患者的具体情况、病情严重程度和治疗效果来决定。
重症肌无力诊疗指南

重症肌无力诊疗指南
重症肌无力主要是一种自身的免疫疾病,主要就是说自身产生了针对自己本身的乙酰胆碱受体的抗体,导致了神经递质间的传递障碍,患者因为这种发病的病型不同,可以临床表现是不一样的。
它可以有这种单纯的眼肌型患者,主要表现为双侧眼睑上抬费力,眼球的运动障碍,会出现复视的症状。
也有些患者是轻度的全身型,就是周身的乏力不适,拿东西拿不起,走路费力这些情况。
也有些患者会比较重,出现重症的全身型,患者可以出现行走障碍,然后会出现吞咽困难,饮水呛咳,构音障碍情况。
我们在治疗方面,就需要针对的治疗,可以用激素,用溴吡斯的明,以及一些免疫性的药物治疗,在重症肌无力的指南上面,就是有一些急性期的患者,还是推荐给他用人免疫丙种球蛋白的治疗,假如以上这些方法治疗效果都不好,患者还合并有胸腺瘤的话,我们指南上面推荐进行胸腺瘤的切除,还可以进行血液置换,血浆置换这些治疗,所以说,我们对于重症肌无力的治疗,还是要依据指南规范来进行诊治。
解放军第309医院重症肌无力治疗指南(

重症肌无力治疗总原则提高神经肌肉接头处传导的安全系数:主要是胆碱酯酶抑制剂;2. 免疫治疗:肾上腺糖皮质激素、免疫球蛋白、血浆交换、免疫抑制剂、胸腺摘除术、胸腺放射治疗,及中药治疗等;3. 避免用乙酰胆碱产生和/或释放的抑制剂。
一、胆碱酯酶抑制剂1.适应证适用于除胆碱能危象以外的所有重症肌无力病人,尤其适用肌无力症状较轻的患者。
2.作用机制该类药能抑制胆碱酯酶对乙酰胆碱的降解,使神经肌肉突触间隙乙酰胆碱量增多,增强与乙酰胆碱受体抗体竞争乙酰胆碱受体的能力,有利于神经-肌肉接头处的传导,而使肌力有所恢复。
3.常用药物新斯的明 (Prostigmine) 包括注射甲基硫酸新斯的明用0.5mg,口服溴化新斯的明15-30mg,3-4次/日。
溴吡啶斯的明 (Pyridostigmine) 60-120 mg;3-4次/日4.副作用主要为胃肠道不良反应,一般用硫酸阿托品、654-2以对抗。
心率过慢,心律不齐,机械性肠梗阻以及哮喘患者均忌用或慎用。
二、肾上腺皮质激素治疗1. 适应症:1、单纯眼型重症肌无力患者。
2、病情恶化又不适于或拒绝做胸腺摘除的重症肌无力患者。
3、胸腺摘除术后的重症肌无力患者。
2. 作用机制(1)免疫抑制抑制乙酰胆碱受体抗体合成,使神经-肌肉接头处突触后膜上的乙酰胆碱受体免受或少受自身免疫攻击所造成的破坏。
也抑制针对骨骼肌其它成分的抗体。
(2)容易化作用使突触前膜易释放乙酰胆碱,使兴奋易于传递。
(3)再生使终板再生,使突触后膜乙酰胆碱受体数目成倍增加。
3. 治疗方法:一般主张自每日大量(强的松60-80,甚至100mg/d)开始,当出现连续好转后逐渐减量,于病情好转后,尽快减量乃至完全停用胆碱酯酶抑制剂。
优点:短期内达满意疗效,减用乃至停用胆碱酯酶抑制剂;明显好转2个月后行胸腺摘除术可减少并发症。
冲击疗法用于危重病例,已经用气管插管及人工呼吸器者。
甲基强的松龙1000mg/d,静脉滴入,连续3天,第4天500mg,连续3天,以后每3天减半量,直到停药。
重症肌无力严重症状的治疗有哪些

重症肌无力严重症状的治疗有哪些引言重症肌无力(Myasthenia Gravis,MG)是一种极具特征性的自身免疫性疾病,其主要特征是肌肉无力和疲劳,特别影响眼部、咀嚼和吞咽肌肉。
本文将详细介绍重症肌无力严重症状的治疗方案。
药物治疗1.抗胆碱酯酶药物(抗AChE药物): 乙酰胆碱酯酶(AChE)是降解乙酰胆碱的酶,而乙酰胆碱在神经突触处起到传递神经冲动的作用。
抗AChE药物通过抑制乙酰胆碱的降解,增加神经传递量,从而改善肌肉无力。
常用的抗AChE药物包括新斯的明(Pyridosquidin),雷米封(Pyridostigmine)等。
2.免疫抑制药物: 免疫抑制药物用于抑制免疫系统的异常活动,减少自身免疫反应,从而延缓病情进展和减轻严重症状。
常用的免疫抑制药物包括环孢素A(Cyclosporine A),甲氨蝶呤(Methotrexate)等。
这些药物需要在医生的监测下使用,因为它们可能带来严重的副作用。
3.全身性糖皮质激素: 糖皮质激素可以通过抑制免疫细胞的活性,减少自身免疫反应。
然而,副作用较多,如易感染、血糖升高、激素依赖等。
4.免疫球蛋白: 免疫球蛋白(IVIg)是从血浆中提取的免疫球蛋白,可以通过减少抗体产生、清除异常抗体以及改善神经传导功能来改善重症肌无力的症状。
IVIg的治疗效果较好,但是需要大量的血浆,费用较高。
5.补体抑制剂: 补体是免疫系统中的一组蛋白质,参与炎症反应和免疫反应。
补体抑制剂主要通过抑制补体的激活和清除异常的补体来改善症状。
手术治疗1.胸腺切除术: 胸腺切除术是治疗重症肌无力的有效方法之一。
该手术可在切除胸腺后减少自身抗体的产生和释放,从而改善症状。
此外,胸腺切除术也可用于中度至重度的病例,特别是起病较早、体重较轻的患者。
2.眼部手术: 对于MG患者中严重眼睑下垂或眼球运动障碍的情况,可以考虑进行眼部手术,如提眉手术、眼睑重建手术等,以改善视力和外观。
支持性治疗1.重症肌无力危象的救治: 重症肌无力危象是指因呼吸肌麻痹引起的突发性、严重的呼吸困难,是一种危及生命的急性并发症。
中国重症肌无力诊断和治疗指南2020(全文版)
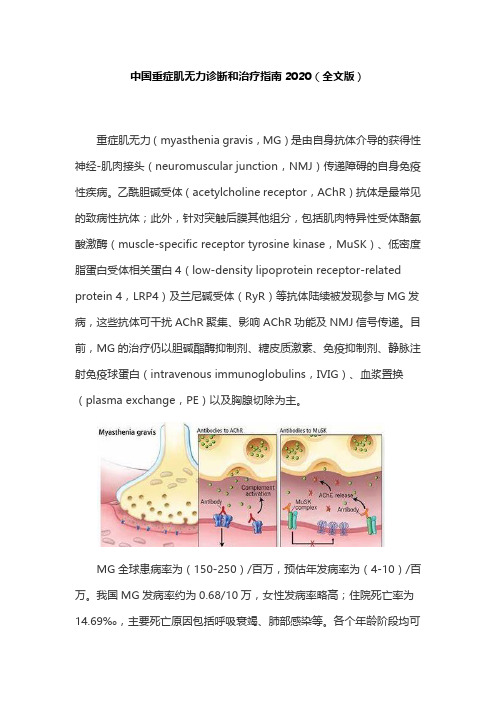
中国重症肌无力诊断和治疗指南2020(全文版)重症肌无力(myasthenia gravis,MG)是由自身抗体介导的获得性神经-肌肉接头(neuromuscular junction,NMJ)传递障碍的自身免疫性疾病。
乙酰胆碱受体(acetylcholine receptor,AChR)抗体是最常见的致病性抗体;此外,针对突触后膜其他组分,包括肌肉特异性受体酪氨酸激酶(muscle-specific receptor tyrosine kinase,MuSK)、低密度脂蛋白受体相关蛋白4(low-density lipoprotein receptor-related protein 4,LRP4)及兰尼碱受体(RyR)等抗体陆续被发现参与MG发病,这些抗体可干扰AChR聚集、影响AChR功能及NMJ信号传递。
目前,MG的治疗仍以胆碱酯酶抑制剂、糖皮质激素、免疫抑制剂、静脉注射免疫球蛋白(intravenous immunoglobulins,IVIG)、血浆置换(plasma exchange,PE)以及胸腺切除为主。
MG全球患病率为(150-250)/百万,预估年发病率为(4-10)/百万。
我国MG发病率约为0.68/10万,女性发病率略高;住院死亡率为14.69‰,主要死亡原因包括呼吸衰竭、肺部感染等。
各个年龄阶段均可发病,30岁和50岁左右呈现发病双峰,中国儿童及青少年MG(juvenile myasthenia gravis,JMG)患病高达50%,构成第3个发病高峰;JMG 以眼肌型为主,很少向全身型转化。
最新流行病学调查显示,我国70-74岁年龄组为高发人群。
近年来,在MG诊疗方面取得了众多进展,积累了更多循证医学证据。
为此,中国免疫学会神经免疫分会基于近5年国内外文献中的最新证据,参考相关国际指南,反复讨论,在对中国MG诊治指南(2015)更新修订的基础上编写了本指南。
新指南采用MGFA临床分型替代Osserman 分型,旨在对疾病严重程度进行量化评估;提出MG亚组分类,指导精准化治疗;对治疗目标进行了定义;针对胸腺切除,利妥昔单抗、依库珠单抗等生物制剂的应用,眼肌型MG(ocular MG,OMG)早期免疫抑制治疗以及免疫检查点抑制剂(immune check point inhibitors,ICIs)治疗相关MG等方面提出了新的建议。
老年人重症肌无力怎么治疗

老年人重症肌无力怎么治疗背景介绍老年人重症肌无力(Myasthenia Gravis, MG)是一种以肢体无力、易疲劳、眼肌麻痹等为主要表现的自身免疫性疾病。
该病主要是由于自身免疫系统出现问题,导致免疫细胞攻击和破坏神经肌肉连接处,进而导致神经肌肉传导功能失常。
老年人重症肌无力的发病机制尚不完全清楚,但与遗传、环境、免疫调节失衡等因素有关。
老年人重症肌无力的治疗一般包括药物治疗、手术治疗和改善生活习惯等综合措施。
治疗方法1. 药物治疗① 抗胆碱酯酶药物抗胆碱酯酶药物是老年人重症肌无力治疗的首选药物。
它们通过抑制胆碱酯酶的活性,增加乙酰胆碱在神经肌肉接头的浓度,从而改善神经肌肉传导。
常用的抗胆碱酯酶药物包括:新斯的明(Pyridostigmine)、苯扎贝特(Neostigmine)等。
患者需要根据医生的指导进行逐渐增加剂量的治疗。
② 免疫抑制剂免疫抑制剂主要用于抑制免疫系统对自身神经肌肉连接处的攻击,以改善病情。
常用的免疫抑制剂包括:糖皮质激素(如泼尼松)和免疫抑制剂(如环孢素A)。
用药时需注意副作用,如感染、肝脏损害、肾脏损害等,并定期进行监测。
2. 手术治疗在一些病情较重或对药物治疗无效的老年人重症肌无力患者,手术治疗可能是一种有效的治疗方式。
手术治疗主要包括胸腺切除术和胸腺旁淋巴结切除术。
胸腺切除术通过手术切除胸腺来减少免疫系统攻击自身神经肌肉连接处的机会;而胸腺旁淋巴结切除术则可进一步减少免疫系统攻击的机会。
手术治疗需根据患者的具体情况和医生的建议进行决定。
3. 改善生活习惯老年人重症肌无力患者在治疗过程中,也需要注意改善生活习惯,以减轻病情和提高生活质量。
① 避免过度疲劳过度疲劳可能加重肌无力症状,患者需要合理安排休息时间,避免过度劳累。
② 调整饮食合理的饮食可以提供足够的能量和营养,提高免疫力。
建议患者多摄入富含维生素、矿物质和蛋白质的食物,如蔬菜、水果、鱼类、瘦肉等。
③ 定期运动和康复治疗适当的运动可以预防肌无力的进一步恶化,并有助于康复。
最新重症肌无力诊断和治疗

2024重症肌无力诊断和治疗要点(全文)摘要重症肌无力是一种自身免疫性疾病,其特征是在神经肌肉接头的突触后膜中存在针对乙酰胆碱受体的自身抗体,从而损害肌肉收缩,并导致患有该疾病的患者出现各种肌肉缺陷,包括眼睑下垂、视力模糊或复视、呼吸急促、吞咽困难、四肢无力。
重症肌无力被称为老年男性和年轻女性的疾病,但与全球情况相反,在印度,重症肌无力在男性中占主导地位,比例为2.70:1。
虽然这种疾病已经研究了几个世纪,但我们仍然无法找到疾病的真正原因及其病理生理学。
但是,分子生物学和诊断工具的最新进展使我们能够确定许多药物治疗和早期诊断的靶点。
因此,改善了患者的发病率和生活质量。
在本文中,我们将讨论该疾病诊断和治疗方面的最新进展。
关键词:重症肌无力,自身免疫,肌肉障碍,免疫障碍,治疗引言免疫力是人体抵抗生物或非生物病原体的攻击及其有害影响表达的能力。
因此,免疫通过对抗、中和和清除这些病原体,来维持人体的完整性和功能。
一个由屏障、器官、细胞成分和分子组成的复杂网络共同构成了一个人的免疫系统。
抗体是由病原体侵入人体时特殊细胞产生的。
但在自身免疫性疾病中,这些抗体开始攻击自身的细胞,导致各种免疫介导的疾病。
这种自身免疫性疾病的一个例子是重症肌无力(MG)。
重症肌无力(MG)是一种免疫介导的疾病,由攻击骨骼肌神经肌肉接头(NMJ)中的烟碱(胆碱能)受体或在神经肌肉接头信号传导中起作用的其他蛋白质(突触后膜中的功能相关分子)的自身抗体引起。
在美国,重症肌无力(MG)的患病率每100,000 人中增加了约20 人。
患病率和发病率都随着年龄的增长而增加。
MG导致无痛性肌无力和自发性肌肉疲劳,这是唯一的疾病表现。
1672年,Thomas Willis首次在医学上记录了重症肌无力,当时他指出患者患有一种罕见的疾病,其特征是肌肉疲劳,尤其是四肢肌肉疲劳,且随着时间的推移,这种疲劳会缓慢增加。
Dale、Otto Loewi和Feldberg 对乙酰胆碱在神经肌肉接头的运动终板中的作用进行了研究,这在重症肌无力的病理生理学、诊断和管理方面取得了重大进展。
目前国内外如何治疗重症肌 无力?

目前国内外如何治疗重症肌无力?重症肌无力(Myasthenia Gravis,简称MG)是一种以肌肉无力为主要表现的自身免疫性疾病,常见于青壮年人群。
该病主要由于人体免疫系统攻击和损害神经肌肉接头处的信号传导,导致患者肌肉无力、易疲劳等症状。
目前,国内外在重症肌无力治疗方面取得了一定的进展,主要包括药物治疗、手术治疗以及康复治疗等多种治疗手段。
药物治疗是重症肌无力的首选治疗方法。
目前常用的药物包括抗胆碱酯酶药物和免疫调节剂两类。
抗胆碱酯酶药物如吡唑嗪、新斯的明等可以增加乙酰胆碱在神经肌肉接头处的浓度,从而增强肌肉的收缩力,减轻肌肉无力症状。
免疫调节剂如泼尼松和环孢素A等可以抑制免疫系统的异常反应,减少对神经肌肉接头的攻击,缓解病情发展。
此外,还有其他辅助药物如抗炎药、免疫球蛋白等,可以提供对症治疗和缓解病情。
手术治疗主要适用于一些药物治疗效果不佳、病情进展较快或合并其他疾病的患者。
目前常用的手术方法有胸腺切除术和胸腔镜辅助下的胸腺切除术。
胸腺切除术是通过切除胸腺来减少抗体的产生,从而改善病情。
胸腔镜辅助下的胸腺切除术相较于传统的胸腺切除术更加微创,患者术后恢复快,创伤小。
手术治疗虽然对重症肌无力有一定疗效,但也存在一定的风险,因此必须根据患者的具体情况和医生的建议来决定是否需要手术治疗。
康复治疗在重症肌无力患者中起到了重要的辅助作用。
康复治疗主要包括物理治疗和功能锻炼两个方面。
物理治疗包括电疗、热疗、按摩等手段,可以促进肌肉的血液循环,缓解疼痛和肌肉紧张等不适症状。
功能锻炼主要通过锻炼肌肉力量和改善肌肉协调性来提高患者的日常生活能力和运动能力。
康复治疗需要与药物治疗和手术治疗相结合,共同达到减轻肌肉无力症状、提高生活质量的效果。
除了上述常规治疗方法之外,目前还有一些新的治疗手段在研究和探索中。
例如,近年来免疫检查点抑制剂类药物在免疫治疗中的应用取得了一些进展。
免疫检查点抑制剂如尼伦帕切、帕卢珠单抗等可以调节免疫系统的平衡,增强免疫应答,从而提高患者的治疗效果。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
人工辅助呼吸的MG患者需加强护理,定时雾化、拍背、吸痰,防 止肺部感染,通过辅助呼吸模式的逐步调整等尽早脱离呼吸机。
治疗MG过程中需注意的事项
MG患者
:部分激素类药物,部分抗感染
药物(如氨基糖苷类抗生素、喹诺酮类等以及二性霉素等
Ⅲ型:(重型)全身混合性多肌群受累,但无呼 吸
肌受累, 药物治疗效果不佳,生活不能自理。
Ⅲa:发病时间长,病情进展慢; Ⅲb:发病时间短,病情进展快; Ⅳ型:(潜在危象型)曾有过胸闷或呼吸困难症 状,但无发生肌无力危象病史;
Ⅴ型:(危象型)正处于或有急性呼吸困难肌无 力危象病史。
分期
发作期/进展期:临床病 情在短期内迅速加重 或在 稳定一段时间后又加重。
在具有MG典型临床特征的基础上,具备药理学特征和( 或)神经电生理学特征,临床上则可诊断为MG。
有条件的单位可检测患者血清AChR抗体等,有助于进 一步明确诊断。
。
鉴别诊断
①Miller–Fisher综合征; ②慢性进行性眼外肌麻痹; ③眼咽型肌营养不良; ④眶内占位病变; ⑤Graves眼病; ⑥Meige综合征、吉兰–巴雷综合征、慢性炎性脱髓鞘
二、免疫抑制药物治疗
1.
:
是治疗MG的一线药物,可使70%~80%的MG患者症状
得到显著改善。糖皮质激素由于其强大的抗炎及免疫抑
制作用,被广泛应用于MG的治疗[9]。目前常用于治疗重
症肌无力的糖皮质激素包括:醋酸泼尼松、甲泼尼龙、
地塞米松。
使用方法:醋酸泼尼松0.5~1.0 mg/kg,每日晨顿服; 或20 mg/d晨顿服(糖皮质激素剂量换算关系为:5.0 mg 醋酸泼尼松=4 mg甲泼尼龙=0.75 mg地塞米松),每3 天增加醋酸泼尼松5.0 mg直至足量(60~80 mg)。通常2 周内起效,6~8周效果最为显著。
著。呼吸肌无力可致
,部分患者可出现
,需行人工辅助呼吸[3,4,5]。
改良Osserman 分型
Ⅰ型:眼肌型,病变仅局限于眼外肌,两年之内其他 肌群不受累。
Ⅱ型:全身型,有一组以上肌群受累。 ⅡA 型:轻度全身型,四肢肌群轻度受累,伴或不伴眼
外肌受累,通常无咀嚼、吞咽和构音障碍,生活能自理。 :中度全身型,四肢肌群中度受累,伴或不伴眼
如
,在经良好医患 并做好充分
下,可用糖
皮质激素冲击治疗,其使用方法为:甲泼尼龙1 000 mg/d,连续静
脉滴注3 d,然后改为500 mg/d,静脉滴注2 d;或者地塞米松10~
20 mg/d,静脉滴注1周;冲击治疗后改为醋酸泼尼松或者甲泼尼龙
,晨顿服。
视病情变化调整药物剂量,醋酸泼尼松或甲泼尼龙减量需要根据患者 病情改善情况个体化,如病情稳定并趋好转,可维持4~16周后逐渐 减量;一般情况下逐渐减少醋酸泼尼松用量,每2~4周减5~10 mg ,至20 mg左右后每4~8周减5 mg,酌情隔日服用最低有效剂量。 过快减量可致病情反复、加剧。
的患者,为防止患者手术后出现肌无力危象,术前可
予
等药物。
三、MG危象[14,15]
呼吸肌功能受累导致严重呼吸困难,危及生命者,应积 极行人工辅助呼吸,包括正压呼吸、气管插管或气管切 开,监测动脉血气分析中血氧饱和度和二氧化碳分压, 并进一步判断MG危象的类型(表1)。
如为肌无力危象,应酌情
,直
手术时机的选择:在稳定期/缓解期,肌 无力症状最轻,尤其是呼吸功能改善, 用药量最小时手术。对于女性病人,应 选择在月经周期初、中期手术。
外科MG临床分型和分期
分型
Ⅰ型:(轻型)眼肌型,仅仅眼部肌群受累; Ⅱ型:(中型)全身混合性多肌群无力,但无呼
吸肌受累, 药物治疗后症状可部分缓解,影 响劳动;
抗真菌药物),部分心血管药物(如利多卡因、奎尼丁、β–
受体阻滞剂、异搏定等),部分抗癫痫药物(如苯妥英钠、
乙琥胺等),部分抗精神病药物(如氯丙嗪、碳酸锂、地西
泮、氯硝西泮等),部分麻醉药物(如吗啡、度冷丁等),
部分抗风湿药物(如青霉胺、氯喹等)。
其他注意事项包括:禁用肥皂水灌肠;注意休息、保暖 ;避免劳累、受凉、感冒、情绪波动等。
。但在发病 可单独出现眼
外肌、咽喉肌或肢体肌肉无力;脑神经支配的肌肉较脊
神经支配的肌肉更易受累。
经常从一组肌群无力开始,逐渐累及其他肌群,直到全 身肌无力。
部分患者短期内出现全身肌肉收缩无力,甚至发生
骨骼肌无力表现为
和
,
,活动
后加重、休息后可减轻。
眼外肌无力所致对称或非对称性 是MG最常见的首发症状,见于80%以上的患者[2];
广泛使用免疫抑制药物治疗之前,MG的病死率高达30%,而随着 机械通气、重症监护技术以及免疫抑制剂广泛应用于MG的治疗, 目前病死率(直接死于MG及其并发症的比例)已降至5%以下。
一般术前护理主要包括重视心理、生理 因素对MG的影响,减少MG危象发生的 诱因,教会患者咳嗽、排痰方法。术后 因吞咽肌无力、吞咽困难、气管插管刺 激及抗胆碱酯酶药的应用,均可使喉部 分泌物增多,所以要彻底清除呼吸道分 泌物,保持气道通畅,不易清除者应采 用雾化吸入。
使用糖皮质激素期间须严密观察病情变化,40%~50%的
MG患者肌无力症状会在4~10 d内一过性加重并有可能
促发肌无力危象,因此,对病情危重、有可能发生肌无
力危象的MG患者,应
糖皮质激素;同时应注意
类固醇肌病,补充钙剂和双磷酸盐类药物
,使用抗酸类药物
。
长期服用糖皮质激素可引起食量增加、体重增加、向心 性肥胖、血压升高、血糖升高、白内障、青光眼、内分 泌功能紊乱、精神障碍、骨质疏松、股骨头坏死、消化 道症状等,应引起高度重视。
到安全剂量范围内肌无力症状改善满意为止;如有比较
严重的
,应酌情使用
;如不
能获得满意疗效时考虑用
;部分患者还可
考虑同时应用,应尽快减少或者停用胆碱酯酶抑制剂,一般5
~7 d后再次使用,从小剂量开始逐渐加量,并可酌情使用阿托
品;同时给予甲泼尼龙冲击、血浆交换或静脉注射免疫球蛋白
。
随着医学科学技术的发展,目前胆碱酯酶抑制剂的使用剂量有限( ),胆碱能危象已极为少见。若血气分析
性多发性神经病等
治疗
1.一般治疗 治疗
此类药物是治疗所有类型MG的一线药物,用于改善临床 症状,特别是新近诊断患者的初始治疗,并可作为单药 长期治疗轻型MG患者[8]。不宜单独长期使用胆碱酯酶抑 制剂,其剂量应个体化,一般应配合其他免疫抑制药物 联合治疗。胆碱酯酶抑制剂中溴化吡啶斯的明是最常用 的胆碱酯酶抑制剂。不良反应包括:恶心、腹泻、胃肠 痉挛、心动过缓和口腔及呼吸道分泌物增多等。国内一 般最大剂量为480 mg/d,分3~4次口服。
胸腺摘除手术后通常在2~24个月病情逐渐好转、稳定,用药剂量亦 减少。 MG患者经胸腺摘除手术治疗后可完全 ;也有 MG患者胸腺摘除术后几年甚至数年后MG症状 ,但总体来说
一般选择手术的年龄为
。MG
的患
者,除非怀疑高度恶性胸腺瘤,可以
,如丙
种球蛋白冲击等,待病情改善、稳定后再行手术治疗,
有助于减少、防止手术后发生肌无力危象。需要
诊断
一、诊断依据
1.临床表现:某些特定的横纹肌群肌无力呈斑片状分
布,表现出波动性和易疲劳性;
晨轻暮重,
持续活动后加重,休息后缓解、好转。通常以眼外肌受
累最常见。
2.药理学表现:
。
3.
低频刺激波幅递减10%以上;SFEMG测定
的"颤抖"增宽、伴或不伴有阻滞。
4.抗体:多数全身型MG患者血中可检测到 ,或在极少部分MG患者中可检测到抗MuSK抗体、抗 LRP 4抗体。
中毒量较接近,用药过程中应密切观察药物疗效
,及时发现药量不足和药物过量,随时调整药物
剂量及服药时间。
术后继续服用强的松,2 月后逐步减量 ,直到 4-5 个月逐步停药,吡啶斯的明则根据症状服用更长 时间。
所有临床病人
用量因人而异;术前用量以能控制病
人的症状、维持良好肌力的最小剂量为
宜。
是,先从小剂量开
其主要临床表现为骨骼肌无力、易疲劳,活动后加重, 休息和应用胆碱酯酶抑制剂后症状明显缓解、减轻。
年平均发病率为(8.0~20.0)/10万人[1]。MG在各个年龄 阶段均可发病。
在40岁之前,女性发病率高于男性;40~50岁男女发病 率相当;50岁之后,男性发病率略高于女性。
临床特点
患者
外肌受累,通常有咀嚼、吞咽和构音障碍,生活自理困难 。 Ⅲ型:重度激进型,起病急、进展快,发病数周或数月内 累及咽喉肌;半年内累及呼吸肌,伴或不伴眼外肌受累, 生活不能自理。 Ⅳ型:迟发重度型,隐袭起病,缓慢进展。两年内逐渐
进展,由Ⅰ、ⅡA、ⅡB 型进展,累及呼吸肌。 Ⅴ型:肌萎缩型,起病半年内可出现骨骼肌萎缩、无力 。
始逐渐加大到 治疗量,使肌力明显
增强、症状明显改善或消失,然后再将
剂量 1/5~1/6,且达到
激素类药物
强的松,1次晨服1-2㎎/㎏,两种疗法:①直接治疗量 ;②半量疗法+钙片+氯化钾(对于比较重的患者),疗 程为3月;
其他激素类药物和环孢素、环磷酰胺、硫唑嘌呤、 他克莫司等免疫抑制剂治疗方法,在此不再赘述。
其他治疗
硫唑嘌呤 环孢菌素A 他克莫司 环磷酰胺 吗替麦考酚酯(MMF) 抗人CD20单克隆抗体(利妥昔单抗,rituximab) 静脉注射用丙种球蛋白 血浆置换 胸腺放射治疗
成年全身型MG和部分眼肌型MG患者,为尽快减少糖皮质激素的用 量或停止使用、获得稳定而满意的疗效、减少激素不良反应,应早期 联合使用免疫抑制剂,如硫唑嘌呤、环孢素A或他克莫司等。