生物信息学作业-序列查找与分析
生物信息学-课堂练习作业生物信息学蛋白质序列分析-课堂练习
生物信息学蛋白质序列分析-课堂练习ZNF395, 全称为Zinc Finger Protein395, 又被称为PBF ,PRF1,DBP2,PRF-1,Si-1-8-14或DKFZp434K1210。
其氨基酸序列为(一)分析蛋白质的一级结构ZNF395蛋白的理论等电点为7.17,分子式C 2417H 3775N 679O 741S 23,原子总数为7635,总平均亲水性(GRA VY )为-0.451,脂肪指数64.54,不稳定指数69.57,序列N 末端是M (Met ),估计半衰期是:30小时(哺乳动物网状细胞,离体);>20小时(酵母,体内);>10小时(大肠杆菌,体内)。
在编码的513个氨基酸中,包括48个带负电的氨基酸(天冬氨酸+谷氨酸),33个带正电荷的氨基酸(精氨酸+赖氨酸)。
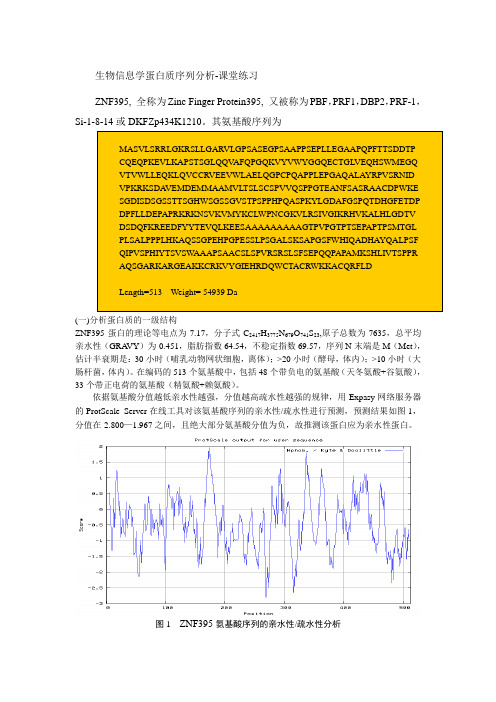
依据氨基酸分值越低亲水性越强,分值越高疏水性越强的规律,用Expasy 网络服务器的ProtScale Server 在线工具对该氨基酸序列的亲水性/疏水性进行预测,预测结果如图1,分值在-2.800—1.967之间,且绝大部分氨基酸分值为负,故推测该蛋白应为亲水性蛋白。
图1 ZNF395氨基酸序列的亲水性/疏水性分析(二)分析蛋白质的二级结构利用SOPMA在线工具对二级结构进行预测,如图2,α螺旋99个占19.30%,延伸链66个占12.87%,β-转角18个占3.51%,无规卷曲330个占64.33%,其二级结构主要由无规卷曲组成。
图2 ZNF395蛋白二级结构预测注:蓝色表示α螺旋;红色表示延伸链;紫色表示无规则卷曲(三)分析膜蛋白质利用在线分析工具TMHMM Server 2.0,对ZNF395氨基酸跨膜结构域进行在线预测和分析,结果表明,该序列编码的蛋白非跨膜蛋白(见图3)。
利用Signal P 3.0 Server在线预测工具对ZNF395蛋白质进行信号肽预测,无信号肽存在(图4)。
生物信息学实验报告3(三)蛋白质序列分析

⽣物信息学实验报告3(三)蛋⽩质序列分析(三)蛋⽩质序列分析实验⽬的:掌握蛋⽩质序列检索的操作⽅法,熟悉蛋⽩质基本性质分析,了解蛋⽩质结构分析和预测。
实验内容:1、检索SOX-21蛋⽩质序列,利⽤ProParam⼯具进⾏蛋⽩质的氨基酸组成、分⼦质量、等电点、氨基酸组成、原⼦总数及疏⽔性(ProtScale⼯具)等理化性质的分析。
2、利⽤PredictProtein、PROF、HNN等软件预测分析蛋⽩质的⼆级结构;利⽤Scan Prosite软件对蛋⽩质进⾏结构域分析。
3、利⽤TMHMM、TMPRED、SOSUI等⼯具对蛋⽩质进⾏跨膜分析;采⽤PredictNLS进⾏核定位信号分析;利⽤PSORT进⾏蛋⽩质的亚细胞定位预测;利⽤CBS(http://www.cbs.dtu.dk/services/ProtFun/)⽹站⼯具预测蛋⽩的功能,将序列⽤Blocks、SMART、InterProScan、PFSCAN等搜索其保守序列的特征,进⾏motif 的结构分析。
4、利⽤Swiss-Model数据库软件预测该蛋⽩的三级结构,结果⽤蛋⽩质三维图象软件Jmol查看。
CPHmodels 也是利⽤神经⽹络进⾏同源模建预测蛋⽩质结构的⽅法和⽹络服务器I-TASSER预测所选蛋⽩质的空间结构。
5、分析蛋⽩质的翻译后修饰:分析信号肽及其剪切位点: SignalIP http://www.cbs.dtu.dk/services/SignalP/;分析糖链连接点:分析O-连接糖蛋⽩,NetOGlyc,http://www.cbs.dtu.dk/services/NetOGlyc/;分析N-连接糖蛋⽩,NetNGlyc,http://www.cbs.dtu.dk/services/NetNGlyc/。
6、利⽤检索的序列,进⾏同源⽐对,获得并分析⽐对结果。
实验步骤(⼀)1、在NCBI 蛋⽩质数据库中查找SOX-21蛋⽩质序列分别选择⽖蟾(Xenopus laevis)、⼩家⿏[Mus musculus]、猕猴[Macaca mulatt a]的SOX-21蛋⽩质序列,并保存其FASTA格式。
生物信息学中的基因序列分析方法与技巧

生物信息学中的基因序列分析方法与技巧生物信息学是研究生物学数据的存储、检索、分析和解释的学科领域,其中基因序列分析是生物信息学的重要组成部分。
基因序列分析帮助科学家理解基因的组成和功能,并揭示生物体内的生物学过程。
在本文中,我们将介绍生物信息学中常用的基因序列分析方法和技巧。
1. 基因序列获取和处理在进行基因序列分析之前,我们首先需要获取正确的基因序列。
这可以通过多种方式来实现,例如从数据库中下载已知的基因序列,使用测序技术获得新的基因序列,或者通过在线工具从物种基因组中提取基因序列。
获取基因序列后,我们需要对其进行处理。
最常见的处理方式是去除序列中的空白字符和特殊字符,并将所有字母转换为大写或小写,以确保一致性和准确性。
此外,还可以利用生物信息学软件和工具进行序列长度修剪、质量评估和碱基配对修正等操作。
2. 序列比对和比对工具基因序列比对是将一个或多个基因序列与参考序列进行比较的过程,以便确定它们的相似性和差异性。
这对于研究基因组结构和功能非常重要。
目前,有许多比对工具可供选择,包括BLAST(Basic Local Alignment Search Tool)、Clustal Omega、Bowtie和BWA(Burrows-Wheeler Aligner)等。
BLAST 是最常用的工具之一,它可以在数据库中快速搜索相似的序列并进行比对。
Clustal Omega可以用于多序列比对,它可以同时比对多个序列并生成序列间的进化树。
Bowtie和BWA则主要用于高通量测序数据的比对。
3. 寻找开放阅读框(ORFs)开放阅读框是基因序列中的编码区域,通常由起始密码子(通常是ATG)和终止密码子(TAA,TAG或TGA)组成。
通过寻找ORFs,科学家可以确定基因的位置和可能的编码蛋白质序列。
在寻找ORFs时,可以使用生物信息学工具,如ORFfinder或EMBOSS中的getorf函数。
这些工具可以自动确定基因序列中的ORFs,并提供基因的位置、长度和推测的蛋白质序列。
生物信息学 序列分析

AGCACACA ACACACTA (B)
对于(A), score=7-2=5 对于(B), score=5-2=3 序列比对的目的是寻找一个得分最大(或代价 最小)的比对。
所以必须对序列的相似性做定量分析,然后将序列进
行排比,在排比中要用到 gaps,insertions,substitutions。
打(记)分矩阵(Scoring Matrices)
对gaps打分可用较简单的扣分方案,而 substitutions的打分则比较复杂,必须先构建出 一个计算机的算法矩阵(Matrix),再根据此方案 对序列中核苷酸/氨基酸残基之间的差异或相似 进行打分。
打分矩阵(Scoring Matrices)
Bioinformatics
1
REVIEWS
Sequence Similarity Searching
Basic Local Alignment Search Tool
REVIEWS
BLAST:
Finds regions of local similarity between two sequences: Only aligned regions are shown, such as BLAST
序列比较的基本操作是比对(Alignment)
两个序列的比对是指这两个序列中各个字符的一种 一一对应关系,或字符的对比排列 。
设有两个序列: GACGGATTAG,GATCGGAATAG
Alignment1:
GACGGATTAG GATCGGAATAG
Alignment2:
GA CGGATTAG GATCGGAATAG
(a)
(b)
(a)对人类(Homo sapiens)与黑猩猩(Pongo pygmaeus)的β球蛋白基因 序列进行比较的完整点阵图。(b)利用滑动窗口对以上的两种球蛋白基因序列进 行比较的点阵图,其中窗口大小为10个核苷酸,相似度阈值为8。
生物信息学原理与DNA序列分析方法

生物信息学原理与DNA序列分析方法生物信息学是生物学、计算机科学和数学交叉的学科,是研究生物大数据的收集、存储、管理、分析和应用的一门科学。
生物信息学在遗传学、基因组学、蛋白质组学、生物多样性等领域有广泛应用,尤其在DNA序列分析领域中得到了广泛的应用。
本文将介绍生物信息学原理与DNA序列分析方法。
一、生物信息学的原理和意义生物信息学的原理是基于生物序列和结构数据的计算机分析和处理。
生物序列包括DNA、RNA和蛋白质序列,而生物结构包括蛋白质二级结构、三级结构和配体结合结构等。
利用计算机技术对生物序列和结构进行分析,可以比较、匹配、搜索、预测和模拟,从而解决生物学研究中的许多问题。
生物信息学的应用有很多,它可以帮助人们理解生物体内的基因调控、蛋白质结构和功能、病毒和微生物的进化、生态系统的演化等生物学问题。
此外,生物信息学还可以应用于药物设计、基因工程和生物能源等领域,大大促进了生物学和其他相关学科的发展。
二、DNA序列分析方法DNA是生物体内的遗传信息载体,它在生物进化和遗传遗传传递中起到重要作用。
DNA序列分析是生物信息学中的重要领域,其研究内容包括DNA序列比对、基因预测、SNP分析等。
1. DNA序列比对DNA序列比对是将两个或多个DNA序列进行比较,找出相似性和差异性的过程。
DNA序列比对可以用于基因组的比较、基因家族的分析、SNP位点的检测、进化关系的研究等方面。
DNA序列比对方法包括Smith-Waterman、Needleman–Wunsch等局部比对算法和BLAST、FASTA等全局比对算法。
2. 基因预测基因预测是将DNA序列中的基因区域和非基因区域进行预测的过程。
基因预测的主要目的是识别DNA序列中的编码区域,推断出蛋白质的氨基酸序列。
基因预测的方法有基于机器学习的方法、基于序列比对的方法、基于统计模型的方法等。
3. SNP分析SNP是单核苷酸多态性,是DNA序列中的最小变异形式,经常用于分子生物学研究和医学诊断中。
生物信息学中的序列分析方法

生物信息学中的序列分析方法生物信息学是研究生物体在遗传、基因表达、蛋白质结构和功能等方面的信息学科学。
其中,序列分析作为生物信息学研究的核心内容之一,包括DNA序列、RNA序列和蛋白质序列等方面的分析。
DNA序列分析方法DNA序列分析通常包括基因识别、同源性搜索、基因组组装等几个方面。
其中,基因识别是指在一个DNA序列中自动鉴别出基因区域。
这个问题由于基因和非编码区域序列的相似性往往很小,因此解决比较困难。
系统采取两种方法:直接方法和间接方法。
直接方法主要基于序列特征和基因序列内的一些功能序列来预测基因组定位,如加州大学圣迭戈分校所开发的GeneFinder,可以识别核酸“ATG”起始密码子、终止密码子及剪切参考信号。
间接方法则是通过其他外部数据来做基因匹配的预测,在人类基因组的缺陷被观察到后,一些新的科技被引入,如EST (表达顺定标签)。
EST提供第一手的基因表达证据,当EST的配对普遍存在于一则DNA序列中时,也就证明此处有一个基因区域所在。
然后根据序列特性,结合同源性比对和其他信息以预测序列功能。
基因组组装问题是指如何利用突变的测序、比对和同源性簇的组装方法来解决不同物种的序列数据组装问题。
但是,细菌的基因序列比较短,而其组装是相对简单的。
在比较大的基因组问题中,通常采用高通量DNA测序和高级组装软件来进行组装。
同源性搜索方法同源性比对是基因组学和生物信息学中的一个重要问题,即根据已知的基因家族或同源蛋白的序列特征来发现既有群体的新成员,从而更好地理解它们的结构和功能,进而研究生物进化的过程。
基础的同源性比对算法包括序列比对、基因族/蛋白族建立等等。
序列的比对可以采用Smith-Waterman算法、Needleman-Wunsch算法和FASTA算法等系列算法。
大规模的序列比对则采取最简单和快速的方法(如BLAST)来应对。
而基因族和蛋白族搜索的方式主要是形成一个统计学模型,模型中有一些参数可以从大量的疑似序列中优化得到。
生物信息学中的序列分析方法

生物信息学中的序列分析方法生物信息学是一门综合性的学科,它将计算机科学和生物学相结合,用计算机技术和统计学方法来研究生物学问题。
在生物信息学中,序列分析是一种重要的方法,它可以帮助我们理解生物分子的结构和功能。
序列分析是指对生物分子的序列进行分析和解读的过程。
生物分子的序列可以是DNA、RNA或蛋白质的序列。
通过对这些序列进行分析,我们可以揭示生物分子的结构、功能和进化关系。
在序列分析中,最基本的任务是序列比对。
序列比对是将两个或多个序列进行对比,找出它们之间的相似性和差异性。
比对的结果可以帮助我们识别共同的序列特征,如保守区域和突变位点。
常用的序列比对方法有全局比对、局部比对和多序列比对。
全局比对适用于相似性较高的序列,局部比对适用于相似性较低的序列,而多序列比对可以同时比对多个序列,用于研究序列之间的共同演化关系。
除了序列比对,序列分析还包括序列搜索和序列分类等任务。
序列搜索是指通过已知的序列信息来寻找和该序列相关的其他序列。
常用的序列搜索方法有基于序列相似性的搜索和基于序列模式的搜索。
序列分类是指将一组序列分成若干个互相关联的类别。
序列分类可以帮助我们理解序列之间的功能和结构差异,以及它们的进化关系。
常用的序列分类方法有聚类分析和机器学习方法。
在序列分析中,我们还经常使用一些特定的工具和数据库。
例如,BLAST (Basic Local Alignment Search Tool)是一种常用的序列比对工具,它可以帮助我们快速地找到相似的序列。
NCBI(National Center for Biotechnology Information)是一个重要的生物信息学数据库,它收集和提供了大量的生物分子序列和相关信息。
随着生物学研究的深入和高通量测序技术的发展,生物信息学在序列分析方面的应用也越来越广泛。
例如,基因组学研究中的基因预测、蛋白质组学研究中的蛋白质结构预测,都离不开序列分析的方法。
生物信息学中的序列数据分析与挖掘研究

生物信息学中的序列数据分析与挖掘研究随着生物学研究的不断深入,生物信息学逐渐成为热门研究方向。
其中,序列数据分析与挖掘是生物信息学研究的重要领域之一。
本文将介绍生物信息学中序列数据分析与挖掘的相关知识,包括序列数据的预处理、特征提取、分类识别和分子演化等方面。
第一部分:序列数据的预处理序列数据是指DNA、RNA或蛋白质序列。
在进行序列数据分析和挖掘之前,需要对原始数据进行预处理。
其目的是去除杂质数据和错误序列,以减少对后续研究的影响。
常见的序列数据预处理方法包括序列对齐、质量控制、去除低质量序列等。
其中,序列对齐是指将不同样本的序列进行比对,以获得共同特征和差异。
质量控制是指剔除与高质量要求不符的序列。
而去除低质量序列则是在质量控制的基础上,将质量较差的序列直接去除。
第二部分:序列数据的特征提取序列数据的特征提取是生物信息学中的核心问题之一。
它可以将复杂的序列数据转化为易于理解和处理的特征向量,以便进行后续的分类和预测。
常见的特征提取方法包括基于频率的方法、基于结构的方法和基于序列的方法。
其中,基于频率的方法是指通过计算碱基或氨基酸出现的频率,得到一个数量化的指标。
基于结构的方法是通过预测序列的复杂维度结构或二级结构,得到一个特征向量。
而基于序列的方法是通过分析序列的特定模式或规律,得到一个更加明确的指标。
第三部分:序列数据的分类识别序列数据的分类识别是指将序列数据进行分类,并对其进行识别和预测。
它是生物信息学研究的重要领域,也是数据挖掘的重要应用之一。
在序列数据分类识别中,机器学习和深度学习是最常用的方法之一。
通过构建一个基于训练集建立的分类模型,对待分类数据进行分类。
其中,重点需要考虑的问题是高准确率与高鲁棒性之间的平衡。
第四部分:序列数据的分子演化序列数据的分子演化是指通过比较多个序列的共同点和差异性,推断它们的进化历史和演化模式。
在生物学研究中,序列数据的分子演化极为重要,可以揭示生物物种之间的关系和进化历史。
生物信息学中的基因序列分析与挖掘研究

生物信息学中的基因序列分析与挖掘研究生物信息学是一门集合了计算机科学、统计学、生物学和数学等学科的交叉学科,它利用计算机和统计学的方法来存储、管理和分析生物学相关的数据,其中基因序列分析与挖掘是生物信息学中的一个重要研究领域。
基因序列是生物体内负责遗传信息传递和蛋白质合成的重要分子,它们以DNA或RNA的形式存在于细胞中。
通过对基因序列的分析与挖掘,我们可以揭示基因的功能、结构和演化,为生物学研究和生物技术应用提供重要的信息。
首先,基因序列分析与挖掘可以帮助我们理解基因的结构与功能。
基因在DNA序列中以一定的顺序编码着蛋白质的氨基酸序列,通过分析基因序列中的密码子,我们可以推断出编码的蛋白质的氨基酸序列,进而预测蛋白质的结构与功能。
此外,通过比较不同物种之间的基因序列差异,我们可以推断出基因的进化关系和功能的变化。
其次,基因序列分析与挖掘可以帮助我们发现新的基因和功能元件。
通过对已知基因序列的比对和数据库的搜索,我们可以发现新的基因以及与其相关的调控元件。
例如,通过在不同物种基因组中搜索高度保守的序列,我们可以推断出这些序列在基因调控中起到重要的作用,进而挖掘出新的调控元件。
此外,基因序列分析与挖掘还可以帮助我们研究基因组的组织和调控。
基因组是一个生物体内所有基因的集合,通过对基因组序列的分析,我们可以了解基因组的结构和特点。
例如,通过比较基因组中基因的分布情况,我们可以揭示基因的聚集规律和染色体的结构。
同时,基因组的调控是维持生物体正常功能和发育的重要因素,通过分析基因组序列中的启动子、增强子等调控序列,我们可以揭示基因的表达调控机制。
为了实现基因序列分析与挖掘的目标,生物信息学研究中有许多常用的工具和算法。
比对算法(如Smith-Waterman算法和BLAST算法)可以用来比较不同序列之间的相似性和差异性。
序列标注工具(如GFF和GTF格式)可以帮助我们对基因序列进行标注和注释,以便于后续的功能分析。
生物信息学中的基因序列分析与挖掘

生物信息学中的基因序列分析与挖掘在生物信息学领域中,基因序列分析和挖掘是两个重要的研究方向。
基因序列是生物体内部编码基因信息的一系列碱基序列,通过对基因序列的分析和挖掘,可以深入了解生物体的遗传特征和功能。
本文将探讨基因序列分析和挖掘的主要方法和应用,以及其在生命科学研究中的重要性。
基因序列分析是从生物信息学角度来研究基因序列的结构、功能和进化的过程。
通过分析基因序列中的碱基组成、编码蛋白质的开放阅读框、剪切位点等特征,可以推测基因的功能和表达模式。
其中一项重要的分析工具是比对算法。
比对算法可以将待分析序列与已知序列进行比较,从而找到相似的区域,推断它们的共同起源和功能。
常用的比对算法包括BLAST和Smith-Waterman算法等。
基因序列挖掘是利用计算方法从基因组中挖掘出具有重要生物学功能的基因序列。
通过挖掘基因组中的保守序列、启动子区域、调控序列等关键元素,可以发现潜在的基因和功能。
序列挖掘的关键工具是基于机器学习和数据挖掘的算法。
例如,通过训练一个分类器,可以预测一个序列是否为启动子区域,从而鉴定潜在的基因。
基因序列分析和挖掘在生命科学研究中具有重要的应用价值。
首先,它们可以帮助科学家理解基因的结构和功能,为研究生物体的遗传特征和调控机制提供基础。
其次,基因序列分析和挖掘可以辅助药物研发和疾病诊断。
通过分析疾病相关基因的序列特征,可以发现导致疾病的突变和变异,为精准医学的发展提供依据。
另外,基因序列分析和挖掘还可以帮助揭示物种进化的过程。
通过比较不同物种的基因序列,可以推测它们的共同祖先和进化关系。
近年来,基因组学技术的快速发展使得大规模基因序列数据的获得变得更加容易。
然而,这也给基因序列分析和挖掘带来了挑战。
首先,如何高效地处理和分析大规模的序列数据是一个问题。
随着技术的发展,分析算法和计算工具也需要不断更新和优化。
其次,如何准确地解释序列分析的结果也是一个难题。
只有合理地解读分析结果,才能得出准确的结论和预测。
生物信息学作业-序列查找与分析

一、序列(1)从NCBI网站中查找人类钙网蛋白的基因序列,登录号为AY047586.1,序列长度为1402 bp,CDS区为54..1307bp。
序列如图3。
图1. NCBI网站中查找人类钙网蛋白序列图2. 人类钙网蛋白序列的相关信息图3 人类钙网蛋白的FASTA格式序列(2)通过blast比对获得相似性前6条的序列:白犀牛钙网蛋白(XM_004442548.1 )、野猪胸腺克隆(AK398467.1)、鼠的钙网蛋白(X53363.1)、小家鼠钙网蛋白( NM_007591.3)、褐家鼠钙网蛋白( NM_022399.2)、现代人互补DNA克隆( BC107102.2),对7条序列的CDS区进行比对分析,并构建系统进化树。
图4 进行BLAST的界面图5 BLAST之后的结果图6 BLAST之后的结果图7 MAGA的运行结果图8 MEGA的运行结果图9 系统进化树二、对人类钙网蛋白的蛋白质进行一级结构的预测从NCBI中搜索人类钙网蛋白的蛋白序列,其登录号为AAL13126.1,序列如图所示:图10 人类钙网蛋白的蛋白序列通过protparam(/tools/protparam.html)对人类钙网蛋白的蛋白质的基本理化性质进行预测,结果显示该蛋白编码氨基酸数目为417,相对分子质量为48141.5 Da,理论pI值4.29。
图11 protparam的首页图12 蛋白质的氨基酸数目.相对分子质量.理论pI值.氨基酸组成图13 蛋白质的正/负电荷残基数.分子式.总原子数图14 蛋白质的消光系数.半衰期.不稳定系数.脂肪系数.总平均亲水性三、对该蛋白质二级结构进行预测(亲疏水性、跨膜区、结构域等)(1)通过protscale (/tools/protscale.html)网站进行亲疏水性预测。
图15 亲疏水性工具protscale首页图16 亲疏水性预测的结果图17 亲疏水性预测的结果(2)通过http://www.cbs.dtu.dk/services/TMHMM-2.0这个网站,对该蛋白质进行跨膜区预测图19 跨膜区工具TMHMM首页图19 跨膜区预测结果(3)通过(http://smart.embl-heidelberg.de/)这个网站,对该蛋白质进行结构域预测图19 结构域工具smart页面图20 结构域预测结果四、通过(/)这个网站,对该蛋白质三级结构进行预测图21 三维工具swiss-modle页面图22 三级结构预测结果个人收获生物信息学通俗的说法就是利用数学和计算机知识来处理生物数据,在这一个学期内的学习中,不仅学习到了有关生物信息学的一些理论知识,而且也使我们接触到了怎样用现代技术来处理得到的数据,每一步做出来的东西都让我们很惊讶也很惊喜,原来生物世界是这么美妙。
生物信息学中的DNA序列分析方法与工具介绍

生物信息学中的DNA序列分析方法与工具介绍DNA序列分析是生物信息学领域中的重要研究内容,通过对DNA序列进行分析可以揭示生物基因组的组成、结构和功能,为进一步的生物学研究提供了重要的信息。
本文将介绍DNA序列分析的一些常用方法和工具。
首先要介绍的是DNA序列比对方法。
DNA序列比对是将一个DNA序列与另一个DNA序列进行对比,以确定两个序列之间的相似性和差异性。
在DNA序列比对中有两种常见的方法,即全局比对和局部比对。
全局比对是将整个序列进行比对,适用于两个相似的序列。
而局部比对则是找出序列中的一个片段,与另一个序列进行比对,适用于两个不太相似的序列。
常用的DNA序列比对工具有BLAST(Basic Local Alignment Search Tool)和BWA (Burrows-Wheeler Aligner)。
其次是DNA序列组装方法。
DNA序列组装是将大量的DNA 片段拼接起来,以重建原始DNA序列。
DNA序列组装是一项复杂的任务,需要解决重复片段的问题和利用辅助信息进行拼接。
目前,在DNA序列组装中常用的方法有重叠组装方法和重建图方法。
重叠组装是通过比对DNA序列片段之间的重叠区域来进行拼接,常用的重叠组装工具有SOAPdenovo和Velvet。
而重建图方法则是通过构建一张图,将DNA序列的片段作为节点,辅助信息作为边,来进行拼接,常用的重建图工具有SPAdes和ABySS。
DNA序列分析中还有一个重要的方法是序列标识和注释方法。
序列标识是将DNA序列进行标记,以便于后续的分析和注释。
常用的序列标识方法有基因预测和开放阅读框(ORF)预测。
基因预测是通过寻找DNA序列中具有编码蛋白质的基因,以确定基因的位置和功能。
而ORF预测则是通过寻找DNA序列中具有编码蛋白质的开放阅读框,以确定蛋白质编码区域。
常用的序列标识工具有GeneMark和Glimmer。
此外,DNA序列分析中还有一些其他的方法和工具。
生物信息学序列分析

• 由于密码子偏性的研究近年来一直是一个热点,因此 研究的指标也出现得很多,如可以衡量特定基因偏性 大小的密码子偏爱指CBI(Morton1993)和最优密码子 使用频率FOp(Lavnerand Kotlar2005) 等。多种多样 的技术和方法促进了密码子偏性的研究,但是也产生 了一些的研究结果之间存在了的不一致,特别是有些 方法仅仅能运用于局限的物种或某些特定的基因中。 因此在使用这些新开发的方法时,必须了解每一种方 法背后的假设和推论,才能确保结果的正确性。
表4 64种可能的碱基三联体密码子及相应的氨基酸数(据图1序列)
相邻碱基之间的关联将导致更远碱基 之间的关联,这些关联延伸距离的估计 可以从马尔科夫链(Markov chain)理论 得到(Javare和Giddings,1989)
什么是HMM? Hidden Markov Models (HMMs, 隐马尔可夫模型) 最早是在上个世纪60年代末70年代初提出来的一种 概率论模型。进入80年代以后,逐渐被利用在各个领 域。主要的应用领域: 语音识别系统。 生物学中的DNA/protein序列的分析。 机器人的控制。 文本文件的信息提取。
第7章 序列分析
一、初级序列分析
序列的组成/分子量/等电点分析
2
碱基组成
DNA序列一个显而易见的特征是四种碱基类 型的分布。尽管四种碱基的频率相等时对数学 模型的建立可能是方便的,但几乎所有的研究 都证明碱基是以不同频率分布的。
生物信息学的生物序列分析

生物信息学的生物序列分析生物信息学是应用计算机科学和统计学的原理与方法,对生物学数据进行分析的学科。
在生物学研究中,生物序列分析是生物信息学的一个重要研究方向。
生物序列是DNA、RNA或蛋白质的线性排列,通过对生物序列进行分析,可以揭示其结构、功能、进化及与疾病之间的关系,对于生物学的研究和应用具有重要意义。
一、序列比对序列比对是生物序列分析的常见任务之一,它用于将两个或多个生物序列进行比较,并找到它们之间的相似性和差异。
在序列比对中,一种常见的方法是使用动态规划算法,比如Smith-Waterman算法和Needleman-Wunsch算法。
这些算法通过对序列中的字符进行匹配、替代、插入和删除等操作,计算出两个序列之间的最佳匹配程度。
二、基因预测基因预测是通过分析DNA序列,确定其中的基因以及它们的起始点、终止点和剪切位点等信息。
基因预测的方法包括基于序列比对的方法和基于统计学模型的方法。
基于序列比对的方法将已知的基因序列与待预测序列进行比对,从中找出相似片段,并据此预测新的基因。
基于统计学模型的方法则通过建立统计学模型,综合考虑启动子、终止子、剪切位点等特征,对序列进行分析和预测。
三、蛋白质结构预测蛋白质结构预测是根据给定的氨基酸序列预测其对应的三维结构。
蛋白质的结构与其功能密切相关,因此对蛋白质结构的预测具有重要的科学价值和实际应用。
蛋白质结构预测的方法包括基于比对的方法、基于进化信息的方法和基于物理化学原理的方法。
这些方法通过模拟蛋白质的折叠过程,寻找最稳定的结构,并预测出相应的结构信息。
四、进化分析进化分析是通过比较不同物种的序列,揭示它们之间的进化关系和演化历史的方法。
进化分析可以通过构建系统发育树或计算序列之间的相似性矩阵等手段来实现。
系统发育树是描述物种间亲缘关系的图表,通过对多个序列进行比对和计算,可以推断出物种的进化关系及其相对的亲缘程度。
相似性矩阵则用于表示不同序列之间的相似性程度,从而揭示序列的进化关系。
生物信息学 实验 核酸序列分析

核酸序列分析【实验目的】1、掌握核酸序列检索的基本步骤;2、熟悉基于核酸序列比对分析的真核基因结构分析(内含子/外显子分析);3、掌握使用DNAclub软件进行核酸序列的基本分析;【实验内容】1、使用Entrez信息查询系统检索人瘦素 (leptin) 的mRNA、基因组DNA、外显子等核酸序列,连接提取该序列内容,阅读序列格式的解释,理解其含义;2、使用DNAclub对上述核酸序列进行分析’3、使用DNAclub软件对人瘦素 (leptin) 的mRNA序列进行可读框架分析;4、使用NCBI查询系统进行人瘦素 (leptin) 的基因组序列分析【实验方法】1、调用Internet浏览器,并在其地址栏输入Entrez网址:/Entrez ;2、在Search后的选择栏中选择nucleotide;3、在输入栏输入homo sapiens leptin;4、点击go后显示与LEP相关的序列信息,5、查找人leptin 的mRNA或基因,点击序列接受号后显示序列详细信息;6、将序列转为FASTA格式保存7、将上述核酸序列输入DNAClub软件进行序列基本分析(反向或互补序列转换,开放阅读框寻找,序列翻译,酶切位点查找);8、根据基因定位信息查找人瘦素的基因组DNA (Contig) 的序列接受号及序列识别号,点击序列接受号显示序列详细信息;9、分析人瘦素 (leptin) 的基因组序列;查找外显子与内含子序列。
【作业】1、归纳对人瘦素 (leptin) 的核酸序列分析的结果,列出主要的分析结果;2、写出人leptin mRNA序列酶切位点3个。
ORIGIN1 GTAGGAATCG CAGCGCCAGC GGTTGCAAG g taaggccccg gcgcgctcct tcctccttct 61 ctgctggtct ttcttggcag gccacagggc cccacacaac tctggatccc ggggaaactg 121 agtcaggagg gatgcagggc ggatggctta gttctggact atgatagctt tgtaccgagt ......10681 ctccttgcag tgtgtggttc cttctgtttt cag GCCCAAG AAGCCCATCC TGGGAAGGAA 10741 A ATG CATTGG GGAACCCTGT GCGGATTCTT GTGGCTTTGG CCCTATCTTT TCTATGTCCA 10801 AGCTGTGCCC ATCCAAAAAG TCCAAGATGA CACCAAAACC CTCATCAAGA CAATTGTCAC 10861 CAGGATCAAT GACATTTCAC ACACG gtaag gagagtatgc ggggacaaag tagaactgca 10921 gccagcccag cactggctcc tagtggcact ggacccagat agtccaagaa acatttattg ......13021 aggcagccca gagaatgacc ctccatgccc acggggaagg cagagggctc tgagagcgat 13081 tcctcccaca tgctgagcac ttgttctccc tcttcctcct gcatag CAGT CAGTCTCCTC 13141 CAAACAGAAA GTCACCGGTT TGGACTTCAT TCCTGGGCTC CACCCCATCC TGACCTTATC 13201 CAAGATGGAC CAGACACTGG CAGTCTACCA ACAGATCCTC ACCAGTATGC CTTCCAGAAA 13261 CGTGATCCAA ATATCCAACG ACCTGGAGAA CCTCCGGGAT CTTCTTCACG TGCTGGCCTT13321 CTCTAAGAGC TGCCACTTGC CCTGGGCCAG TGGCCTGGAG ACCTTGGACA GCCTGGGGGG13381 TGTCCTGGAA GCTTCAGGCT ACTCCACAGA GGTGGTGGCC CTGAGCAGGC TGCAGGGGTC13441 TCTGCAGGAC ATGCTGTGGC AGCTGGACCT CAGCCCTGGG TGC TGA GGCC TTGAAGGTCA13501 CTCTTCCTGC AAGGACTACG TTAAGGGAAG GAACTCTGGC TTCCAGGTAT CTCCAGGATT......16081 CACTAGATGG CGAGCATCCT GGCCAACATG GTGAAACCCC GTCTCTACTA AAAACACAAA16141 AGTTAGCTGA GCGTGGTGGC GGGCGCCTGT AGTCCCAGCC ACTCGGGAGG CTGAGACAGG16201 AGAATCGCTT AAACCTGGGA GGCGGAGAGT ACAGTGAGCC AAGATCGCGC CACTGCACTC16261 CGGCCTGATG ACAGAGCGAG ATTCCGTCTT AAAAAAAAAA AAAAAAAAGT TTGTTTTTAA16321 AAAAATCTAA ATAAAATAAC TTTGCCCCCT GC在genbank查询到有关leptin基因的资料,阅读资料回答以下问题:在genbank的登录号是哪个?属于leptin 的哪一种分子类型?来源于什么物种?该基因在染色体上的定位情况?Leptin基因有几个外显子,几个内含子?哪一段是ORF区域,其编码的蛋白质检索号是哪个,编码的蛋白质包含多少氨基酸,信号肽、成熟肽序列分别为哪一段,LOCUS NM_000230 3444 bp mRNA linear PRI 13-DEC-2009 DEFINITION Homo sapiens leptin (LEP), mRNA.SOURCE Homo sapiens (human)source 1..3444/organism="Homo sapiens"/mol_type="mRNA"/chromosome="7"/map="7q31.3"gene 1..3444/gene="LEP"/db_xref="GeneID:3952"exon 1..29/number=1exon 30..201/number=2CDS 58..561/product="leptin precursor"/protein_id="NP_000221.1"sig_peptide 58..120mat_peptide 121..558exon 202..3427/number=3LOCUS NC_000007 16352 bp DNA linear CON 10-JUN-2009 DEFINITION Homo sapiens chromosome 7, GRCh37 primary reference assembly.ACCESSION NC_000007 REGION: 127881331..127897682 GPC_000000031SOURCE Homo sapiens (human)FEATURES Location/Qualifierssource 1..16352/organism="Homo sapiens"/mol_type="genomic DNA"/db_xref="taxon:9606"/chromosome="7"gene 1..16352/gene="LEP"/note="Derived by automated computational analysis usinggene prediction method: BestRefseq."/db_xref="GeneID:3952"mRNA join(1..29,10714..10885,13127..16352)/product="leptin"/note="Derived by automated computational analysis usinggene prediction method: BestRefseq."/transcript_id="NM_000230.2"/db_xref="GeneID:3952"CDS join(10742..10885,13127..13486)/note="Derived by automated computational analysis usinggene prediction method: BestRefseq."/codon_start=1/product="leptin precursor"/protein_id="NP_000221.1"/db_xref="GeneID:3952"。
生物信息学中的序列分析方法

生物信息学中的序列分析方法生物信息学是一门应用生物学和计算机科学最为紧密结合的学科,其研究内容主要涉及到生物体内分子生物学、基因组学、蛋白质组学等多个方面。
其中,序列分析是生物信息学中非常重要的一个研究领域,它主要涉及到DNA、RNA、蛋白质等生物分子序列的比较、识别、预测等一系列方法。
本文将着重介绍生物信息学中的序列分析方法,探讨其应用和局限性。
一、序列比对序列比对是序列分析中最基本的方法之一,它通过比对两个或多个生物分子的序列,揭示这些序列之间的相似性或差异性,帮助我们理解生物分子在进化和功能上的关系。
序列比对的方法包括全局比对、局部比对和多序列比对等。
其中,全局比对旨在比较整个序列,常用的算法有Needleman-Wunsch算法和Smith-Waterman算法,而局部比对则着眼于区域相似性,常用的方法有BLAST(基本局部比对搜索工具)。
多序列比对相对较为复杂,可以通过多种方法比对多个序列,如CLUSTAL、T-COFFEE等。
序列比对在分析基因家族、确定物种来源、鉴定疾病基因等方面应用广泛。
此外,序列比对还可以用于辨别序列中的重复部分、鉴定非编码序列、识别重要保守位点等方面。
二、基因预测基因预测,即通过生物分子的DNA序列,推测其可能的基因位置和长度,是生物信息学中的重要研究方向。
目前常用的预测方法包括基于启动子区域的方法、基于开放阅读框(ORF)的方法、基于比对的方法、基于深度学习的方法等。
其中,基于ORF的方法最为简单和常用,它通过找到所有可能的ORF,再从中确定具有编码序列的部分作为基因。
而基于比对的方法则是利用已知的基因序列比对待预测序列,识别编码区域的方法,此方法主要局限在于需要已有相关物种的基因序列。
基于深度学习的方法则是近年来的热点方向之一,它通过构建神经网络模型,从海量数据中提取特征,实现对基因的高效预测。
基因预测在基因组注释方面很有用,还可以应用于生物多样性研究、疾病诊断、新药发现等方面。
生物信息学9序列分析

表1 九种完整DNA序列的碱基组成
表2 人类胎儿球蛋白基因不同区段的碱基组成
二.碱基相邻频率
分析DNA序列的主要困难之一是碱基相邻的频率 不是独立的。碱基相邻的频率一般不等于单个碱基 频率的乘积
例: 鸡血红蛋白β链的mRNA编码区的438个碱基
图1 鸡β球蛋白基因编码区的DNA序列 (GenBank:CHKHBBM,记录号J00860)
拟南芥phyA 部份RNA
五、从序列中寻找基因
1.基因及基因区域预测
基因按其功能可分为结构基因和调控基因:结构基因可 被转录形成mRNA,并进而转译成多肽链;调控基因是 指某些可调节控制结构基因表达的基因。在DNA链上, 由蛋白质合成的起始密码开始,到终止密码子为止的一 个连续编码序列称为一个开放阅读框(Open Reading Frame,ORF)。结构基因多含有插入序列,除了细菌和病 毒的DNA中ORF是连续的,包括人类在内的真核生物的 大部分结构基因为断裂基因,即其编码序列在DNA分子 上是不连续的,或被插入序列隔开。断裂基因被转录成 前体mRNA,经过剪切过程,切除其中非编码序列(即内 含子),再将编码序列(即外显子)连接形成成熟mRNA, 并翻译成蛋白质。假基因是与功能性基因密切相关的 DNA序列,但由于缺失、插入和无义突变失去阅读框而 不能编码蛋白质产物。
三.同向重复序列分析
除了分析整个序列碱基关联程度的特征外,我们常对寻找 同向重复序列(direct repeats)之类的问题感兴趣。Karlin等 (1983)给出了完成这一分析的有效算法。该法采用由特定的几 组碱基字母组成的不同亚序列或称为字码(word)。只需要对整 个序列搜索一次。给一碱基赋以值α,例如A、C、G、T的值为 0、1、2、3。由X1、X2、…、Xk 共k个字母组成的每一种不同 的字码按:
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
一、序列
(1)从NCBI网站中查找人类钙网蛋白的基因序列,登录号为AY047586.1,序列长度为1402 bp,CDS区为54..1307bp。
序列如图3。
图1. NCBI网站中查找人类钙网蛋白序列
图2. 人类钙网蛋白序列的相关信息
图3 人类钙网蛋白的FASTA格式序列
(2)通过blast比对获得相似性前6条的序列:白犀牛钙网蛋白(XM_004442548.1 )、野猪胸腺克隆(AK398467.1)、鼠的钙网蛋白(X53363.1)、小家鼠钙网蛋白( NM_007591.3)、褐家鼠钙网蛋白( NM_022399.2)、现代人互补DNA克隆( BC107102.2),对7条序列的CDS区进行比对分析,并构建系统进化树。
图4 进行BLAST的界面
图5 BLAST之后的结果
图6 BLAST之后的结果
图7 MAGA的运行结果
图8 MEGA的运行结果
图9 系统进化树
二、对人类钙网蛋白的蛋白质进行一级结构的预测
从NCBI中搜索人类钙网蛋白的蛋白序列,其登录号为AAL13126.1,序列如图所示:
图10 人类钙网蛋白的蛋白序列
通过protparam(/tools/protparam.html)对人类钙网蛋白的蛋白质的基本理化性质进行预测,结果显示该蛋白编码氨基酸数目为417,相对分子质量为48141.5 Da,理论pI值4.29。
图11 protparam的首页
图12 蛋白质的氨基酸数目.相对分子质量.理论pI值.氨基酸组成
图13 蛋白质的正/负电荷残基数.分子式.总原子数
图14 蛋白质的消光系数.半衰期.不稳定系数.脂肪系数.总平均亲水性
三、对该蛋白质二级结构进行预测(亲疏水性、跨膜区、结构域等)
(1)通过protscale (/tools/protscale.html)网站进行亲疏水性预测。
图15 亲疏水性工具protscale首页
图16 亲疏水性预测的结果
图17 亲疏水性预测的结果
(2)通过http://www.cbs.dtu.dk/services/TMHMM-2.0这个网站,对该蛋白质进行跨膜区预测
图19 跨膜区工具TMHMM首页
图19 跨膜区预测结果
(3)通过(http://smart.embl-heidelberg.de/)这个网站,对该蛋白质进行结构域预测
图19 结构域工具smart页面
图20 结构域预测结果
四、通过(/)这个网站,对该蛋白质三级结构进行预测
图21 三维工具swiss-modle页面
图22 三级结构预测结果
个人收获
生物信息学通俗的说法就是利用数学和计算机知识来处理生物数据,在这一个学期内的学习中,不仅学习到了有关生物信息学的一些理论知识,而且也使我们接触到了怎样用现代技术来处理得到的数据,每一步做出来的东西都让我们很惊讶也很惊喜,原来生物世界是这么美妙。
同时,通过它也使人类对生物的研究更提升了一个水平。
在以后的学习生活中,我会一如既往的保持原有的严谨性,不仅学习好我的理论知识,还锻炼好我的动手能力。
11。