开环聚合课件
合集下载
第六章开环聚合(RingOpeningPolymerization)

(kcal/mole)
27.6 26.4 6.5 0.0 6.4 10.0 12.9 12.0 1.5
(kJ/mole)
115 110 27
0 27 42 54 50 6
Ring Size 3,4
Ring Strain
5,6,7
8
Free Energy G= H -TS
# atoms H S
• 内酯可发生阳离子开环聚合,阴离子开环聚合和配位聚合.
• 内酰胺中已内酰胺研究得最多,工业上生产的尼龙-6即由己
内酰胺单体开环聚合制备的。
• 内酰胺除能够发生阳离子开环聚合和阴离子开环聚合外,还 可进行水解聚合.
• 工业上用水解聚合来生产尼龙-6合成纤维。 • ⒌ 含膦和氮的环状化合物 • 由PCl5和NH3制得的环状二氯化氮化膦的三聚体(NPCl2)3 加热到230℃以上发生开环聚合而形成线型高分子。
撑亚胺。四元环亚胺称为吖丁啶或氮杂环丁烷。
•
环亚胺一般只能发生阳离子开环聚合。
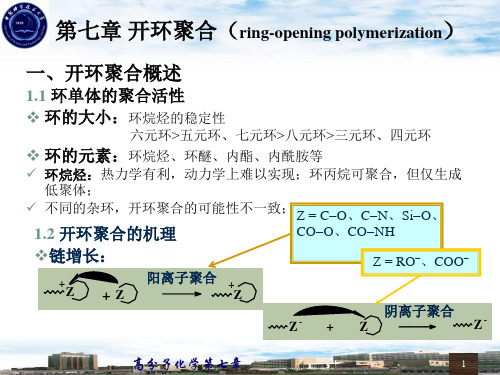
6.1 开环聚合概述
环亚胺要紧有以下两种:
CH2 CH2 N H
吖丙啶
CH2 N H CH2 CH2
吖丁啶
• 环硫化合物中,三元环环硫化合物称为硫化乙烯或噻丙环, 四元环环硫化合物称为噻丁环。
• 三元环硫化合物可发生阳离子开环聚合, 阴离子开环聚合 和配位聚合。按配位聚合可得到立构规整性聚合物。 • 重要的环硫化合物要紧有以下几种。
in Ring
G
[M]e
ext. of pzn @ Equil
3,4 5,6,7
large, neg.
small or zero
small, neg. large, neg. v. low
第七章开环聚合(ring-openingpolymerization)
SiR2
水解-缩合聚合难以生成高分子线形聚硅氧烷;需通过环单体聚合得到;
D4:八烃基环四硅氧烷;D3:六烃基环三硅氧烷
高分子化学第七章
14
聚硅氧烷(polysiloxane)
开环聚合 可进行阴离子、阳离子聚合;
RR R O Si
以丁基锂或萘钠引发的
R
阴离子聚合具有“活性”聚合特征;
Si O
Si
O
O Si
R R
RR
物理特性:低的表面张力和界面张力、 低的玻璃化转变温度;生物相容性
烯烃的开环聚合
R
O Si n R
n CH CH n
高分子化学第七章
n CH CH n n
15
水解-缩合聚合:单体为氯硅氧烷(SiRnCln-4)
CH3
CH3
CH3
Cl Si Cl H2O HO Si Cl + Cl Si OH
CH3 Si O n
CH3
CH3
CH3
CH3
CH3
SiR2 OH + CH3Si(OH)3
SiR2 O Si O SiR2
O
水解-缩合聚合过程中 生成环单体
R = 烷基、苯基、 乙烯基、氢原子
聚磷酸酯(polyphosphoester) biodegradable
高分子化学第七章
13
聚硅氧烷(polysiloxane)
R
O Si
类型:
R
硅 油(低分子量线形聚硅氧烷); SiR2Cl2水解、缩合; 硅树脂(高度交联的聚硅氧烷); SiR2Cl2、SiRCl3与SiCl4水解、缩合; 硅橡胶(轻度交联的聚硅氧烷); 环单体(D4或D3)聚合;
开环聚合

1) prepolymer of polyurethane
2) nonionic surfactants OP-10 C8H17octylphenol -(-EO-)-H EO-adduct
10
(hydrophobic group)
(hydrophilic group)
hydrophobic compound with active H (initiator or starter) KOH R-X-H + EO (catalyst) RX-[-EO-]-H n hydrophobic connecting active H
exp. cal.
115.6 115.8 113.1 69
(25℃)
92.5 88.8 9.2
3 4
60 90
24o44 697.6 9o44 0o44 686.7 664.5
38.6 27.7 5.5
109.7 110.8 105.1 55.3 26.4 27.5 21.8 42.7
5 108
measure: distortion, strain per CH2, strain energy, H, G
1) distortion of bond angles
= 1 (normal valency angle - actual angle between bonds) 2 for 4-member ring, = 1 (109o28 - 90o)=9o44 2 2) strain energy per CH2
60.4 4.2
725.5 21.0
840.6 25.0
- H = -92.1 - H = 63.7 1.2.2 S and G see table of p.6 for S : more double bond
第六章开环聚合

OCH2CCH2 OC+OC OCH2CCH2 OCH2CCH2 OC
R
R
CH2Cl A R CH2Cl CH2Cl A
R=CH2Cl
1)丁氧环(四元环醚)
在0℃或较低温度下,丁氧环经Lewis酸引发,易开环聚合成聚氧化三亚
甲基。但有应用价值的单体却是3,3’-二氯亚甲基丁氧环(丁氧环的衍生
物,聚合产物俗称氯化聚醚),是结晶性成膜材料,熔点为177oC,机械
反应。可与缩聚、加聚并列。
第2页,共50页。
引言
开环聚合的推动力:
环张力的释放
❖ 开环聚合的机理:
大部分属离子聚合(连锁),小部分属逐步聚合
❖ 开环聚合的单体:
环醚、环缩醛、环酯、环酰胺、环硅氧烷等。
环氧乙烷、环氧丙烷、己内酰胺、三聚甲醛等的开环聚
合都是重要的工业化开环聚合反应
第3页,共50页。
8.1 环烷烃开环聚合热力学
能否开环及聚合能力的大小取决于单体环和聚合物线性
结构的相对稳定性
环的元数、构成环的元素(碳环或杂环)、环上的取代
基等对开环的难易都有影响。
有的环状化合物难以开环,如六元环醚等(四氢吡喃、
二氧六环);
有的聚合过程中环状单体和聚合物之间存在平衡,如己内酰
胺;
二官能度单体线性缩聚还有环化倾向。
第4页,共50页。
开环。
-+
AB CH2 CH2
-+
ACH2 CH2OB
O
无取代的和有取代的环烷烃,随着取代程度的增加, (- △H
)依次递减,聚合难度递增。
第7页,共50页。
8.2 杂环开环聚合热力学和动力学
1)热力学因素
环酯、环醚、环酰胺等杂环化合物通常比环烷烃易
R
R
CH2Cl A R CH2Cl CH2Cl A
R=CH2Cl
1)丁氧环(四元环醚)
在0℃或较低温度下,丁氧环经Lewis酸引发,易开环聚合成聚氧化三亚
甲基。但有应用价值的单体却是3,3’-二氯亚甲基丁氧环(丁氧环的衍生
物,聚合产物俗称氯化聚醚),是结晶性成膜材料,熔点为177oC,机械
反应。可与缩聚、加聚并列。
第2页,共50页。
引言
开环聚合的推动力:
环张力的释放
❖ 开环聚合的机理:
大部分属离子聚合(连锁),小部分属逐步聚合
❖ 开环聚合的单体:
环醚、环缩醛、环酯、环酰胺、环硅氧烷等。
环氧乙烷、环氧丙烷、己内酰胺、三聚甲醛等的开环聚
合都是重要的工业化开环聚合反应
第3页,共50页。
8.1 环烷烃开环聚合热力学
能否开环及聚合能力的大小取决于单体环和聚合物线性
结构的相对稳定性
环的元数、构成环的元素(碳环或杂环)、环上的取代
基等对开环的难易都有影响。
有的环状化合物难以开环,如六元环醚等(四氢吡喃、
二氧六环);
有的聚合过程中环状单体和聚合物之间存在平衡,如己内酰
胺;
二官能度单体线性缩聚还有环化倾向。
第4页,共50页。
开环。
-+
AB CH2 CH2
-+
ACH2 CH2OB
O
无取代的和有取代的环烷烃,随着取代程度的增加, (- △H
)依次递减,聚合难度递增。
第7页,共50页。
8.2 杂环开环聚合热力学和动力学
1)热力学因素
环酯、环醚、环酰胺等杂环化合物通常比环烷烃易
高分子化学课件-开环聚合

详细描述
聚碳酸亚丙酯的开环聚合是通过丙二酸和环氧乙烷的反应实现的。在催化剂的作用下,丙二酸和环氧 乙烷发生开环聚合反应,形成聚碳酸亚丙酯。聚碳酸亚丙酯具有优异的耐热性能、阻隔性能和加工性 能,广泛应用于食品包装、电子器件等领域。
聚己内酯的开环聚合
总结词
聚己内酯的开环聚合是一种高效、可控 的聚合方法,可制备出高分子量聚合物 。
能源消耗,实现聚合过程的可持续发展。
循环利用
02
通过循环利用聚合物材料,降低生产成本和资源消耗,同时减
少对环境的污染。
生物降解性
03
研究和发展具有生物降解性的聚合物材料,使其在完成使用寿
命后能够自然降解,减少对环境的长期影响。
05 开环聚合的实例分析
聚乳酸的开环聚合
总结词
聚乳酸的开环聚合是一种环保、可持续的聚合方法,具有广泛的应用前景。
03
02
配位聚合
配位聚合是一种通过过渡金属催化剂将烯烃单体聚合的 方法,具有高活性、高定向性和高立构规整性的特点, 是开环聚合领域的研究热点。
活性聚合技术
活性聚合技术能够实现聚合过程中链自由基的稳定,从 而控制聚合物的分子量和分子量分布,提高聚合物的性 能。
聚合产物的性能改进
功能化聚合物
共聚物
通过在聚合物分子链上引入特定的功 能基团,可以获得具有特殊性能的功 能化聚合物,如导电、发光、磁性等 功能。
合成聚醚类高分子材料
通过开环聚合反应,将环状单体转化为聚醚类高分子材料, 如聚四氟乙烯、聚乙二醇等。这些高分子材料具有优异的耐 化学腐蚀性和生物相容性,广泛应用于制药、电子和化工等 领域。
合成功能性高分子材料
合成导电高分子材料
通过开环聚合反应,将环状单体转化为导电高分子材料,如聚吡咯、聚苯胺等 。这些高分子材料具有良好的导电性能和稳定性,广泛应用于电子器件、传感 器和电池等领域。
聚碳酸亚丙酯的开环聚合是通过丙二酸和环氧乙烷的反应实现的。在催化剂的作用下,丙二酸和环氧 乙烷发生开环聚合反应,形成聚碳酸亚丙酯。聚碳酸亚丙酯具有优异的耐热性能、阻隔性能和加工性 能,广泛应用于食品包装、电子器件等领域。
聚己内酯的开环聚合
总结词
聚己内酯的开环聚合是一种高效、可控 的聚合方法,可制备出高分子量聚合物 。
能源消耗,实现聚合过程的可持续发展。
循环利用
02
通过循环利用聚合物材料,降低生产成本和资源消耗,同时减
少对环境的污染。
生物降解性
03
研究和发展具有生物降解性的聚合物材料,使其在完成使用寿
命后能够自然降解,减少对环境的长期影响。
05 开环聚合的实例分析
聚乳酸的开环聚合
总结词
聚乳酸的开环聚合是一种环保、可持续的聚合方法,具有广泛的应用前景。
03
02
配位聚合
配位聚合是一种通过过渡金属催化剂将烯烃单体聚合的 方法,具有高活性、高定向性和高立构规整性的特点, 是开环聚合领域的研究热点。
活性聚合技术
活性聚合技术能够实现聚合过程中链自由基的稳定,从 而控制聚合物的分子量和分子量分布,提高聚合物的性 能。
聚合产物的性能改进
功能化聚合物
共聚物
通过在聚合物分子链上引入特定的功 能基团,可以获得具有特殊性能的功 能化聚合物,如导电、发光、磁性等 功能。
合成聚醚类高分子材料
通过开环聚合反应,将环状单体转化为聚醚类高分子材料, 如聚四氟乙烯、聚乙二醇等。这些高分子材料具有优异的耐 化学腐蚀性和生物相容性,广泛应用于制药、电子和化工等 领域。
合成功能性高分子材料
合成导电高分子材料
通过开环聚合反应,将环状单体转化为导电高分子材料,如聚吡咯、聚苯胺等 。这些高分子材料具有良好的导电性能和稳定性,广泛应用于电子器件、传感 器和电池等领域。
第八章开环聚合

➢ 可在高分子主链构造中引入多种功能基:酯、醚、酰胺等
➢ 聚合反应前后旳体积收缩比乙烯基单体聚合小
8.2 环状单体开环聚合旳难易取决于两方面原因: ➢ 热力学原因
某些环烷烃转变为线形高分子时旳ΔG
单体
环丙烷 环丁烷 环戊烷 环己烷 环庚烷
环辛烷
ΔG
-22.1 -21.2 -2.2
1.4
-3.9
-8.2
在四氢呋喃等活性较低环醚单体旳聚合反应中,常加入少许活泼 三元环醚单体如环氧乙烷提升引起速率。此时引起反应首先经过活 泼单体形成二级或三级氧鎓离子活性种,再引起低活性旳单体聚合, 此时活泼单体可看作是引起增进剂。
(2)链增长反应
链增长反应为单体旳 O 对增长链旳三级环氧鎓离子活性中心旳 α- C旳亲核攻打反应。以四氢呋喃旳聚合为例:
以环氧乙烷为例,其阴离子开环聚合过程可示意如下:
链引起反应:
链增长反应 :
链增长活性中心为烷氧阴离子。
环氧乙烷旳阴离子聚合体现出活性聚合旳特征,聚合产物旳数 均聚合度为:
X n C[M]0 [I]0
C:单体转化率
醇旳影响 某些金属烷氧化物和氢氧化物引起旳聚合反应体系中,常加入
适量旳醇: ➢ 溶解引起剂,形成均相聚合体系; ➢ 增进增长链阴离子与抗衡阳离子旳离解,增长自由离子浓度, 加紧聚合反应速度。 在醇旳存在下,增长链可和醇之间发生如下互换反应:
互换反应生成旳醇盐可继续引起聚合反应。
与链转移反应不同,互换反应生成旳端羟基聚合物并不是“死” 旳聚合物,而只是休眠种,可和增长链之间发生类似旳互换反应再 引起聚合反应:
经过互换反应,醇也可引起单体聚合,所以若无其他链转移反 应时,聚合产物旳数均聚合度为:
X n C[M]0 [I]0 [ROH]
➢ 聚合反应前后旳体积收缩比乙烯基单体聚合小
8.2 环状单体开环聚合旳难易取决于两方面原因: ➢ 热力学原因
某些环烷烃转变为线形高分子时旳ΔG
单体
环丙烷 环丁烷 环戊烷 环己烷 环庚烷
环辛烷
ΔG
-22.1 -21.2 -2.2
1.4
-3.9
-8.2
在四氢呋喃等活性较低环醚单体旳聚合反应中,常加入少许活泼 三元环醚单体如环氧乙烷提升引起速率。此时引起反应首先经过活 泼单体形成二级或三级氧鎓离子活性种,再引起低活性旳单体聚合, 此时活泼单体可看作是引起增进剂。
(2)链增长反应
链增长反应为单体旳 O 对增长链旳三级环氧鎓离子活性中心旳 α- C旳亲核攻打反应。以四氢呋喃旳聚合为例:
以环氧乙烷为例,其阴离子开环聚合过程可示意如下:
链引起反应:
链增长反应 :
链增长活性中心为烷氧阴离子。
环氧乙烷旳阴离子聚合体现出活性聚合旳特征,聚合产物旳数 均聚合度为:
X n C[M]0 [I]0
C:单体转化率
醇旳影响 某些金属烷氧化物和氢氧化物引起旳聚合反应体系中,常加入
适量旳醇: ➢ 溶解引起剂,形成均相聚合体系; ➢ 增进增长链阴离子与抗衡阳离子旳离解,增长自由离子浓度, 加紧聚合反应速度。 在醇旳存在下,增长链可和醇之间发生如下互换反应:
互换反应生成旳醇盐可继续引起聚合反应。
与链转移反应不同,互换反应生成旳端羟基聚合物并不是“死” 旳聚合物,而只是休眠种,可和增长链之间发生类似旳互换反应再 引起聚合反应:
经过互换反应,醇也可引起单体聚合,所以若无其他链转移反 应时,聚合产物旳数均聚合度为:
X n C[M]0 [I]0 [ROH]
【优】高等有机化学第八章开环聚合PPT资料

但环氧化物由于其三元环张力大,能进行阴离子开环聚合。
然后内酰胺阴离子与内酰胺单体的羰基发生亲核加成反应,使单体开环生成二聚体伯胺阴离子:
可见除环己烷外,其余环烷烃的开环聚合的ΔG 均小于0,反应 以四氢呋喃的聚合为例:
产物聚硅氧烷是一类以Si-O为主链结构的有机硅聚合物,分子量较低时呈液态,称硅油,而高分子量时呈橡胶类的高弹性,称硅橡胶
绝大多数六元环单体不能开环聚合
8.3 阴离子开环聚合反应 8.3.1 环氧化物
环醚是Lewis碱,一般只能进行阳离子开环聚合。但环氧化物由 于其三元环张力大,能进行阴离子开环聚合。引发剂包括金属氢氧 化物、金属烷氧化合物、金属氧化物烷 属化合物以及电子转移 阴离子引发剂等。
以环氧乙烷为例,其阴离子开环聚合过程可示意如下:
。
在热力学上都是可行的,其热力学可行性顺序为: 阳离子开环聚合的链终止反应主要为增长链氧鎓离子与抗衡阴离子结合,如:
二聚体伯胺阴离子不与羰基共轭,不稳定,很快从单体的酰胺基上夺取H形成含环酰胺结构的二聚物,同时生成己内酰胺阴离子:
三元环, 四元环> 八元环 > 五元环,七元环
Ø 动力学因素:动力学上可行的开环方式和反应。
R
O
OO
O OO
环氧化物 环丙醚 四氢呋喃 三聚甲醛
(1)链引发反应
许多用于乙烯基单体阳离子聚合反应的引发剂也可用于环醚
的阳离子开环聚合,包括强质子酸、Lewis酸、碳阳离子源
Ø 可在高分子主链结构中引入多种功能基:酯、醚、酰胺等
Ø 聚合反应前后的体积收缩比乙烯基单体聚合小
8.2 环状单体开环聚合的难易取决于两方面因素:
Ø 热力学因素 以环氧乙烷为例,其阴离子开环聚合过程可示意如下:
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1、自由基开环活性的机理
以DTBP作引发剂,TEMPO作增长链稳定剂,2-亚 甲基-1,3-二氧环庚烷可进行活性开环聚合,具 体反应过程如下:
以α,α’-二溴代二甲苯为引发剂, 溴化亚铜 /2,2’-联吡啶为催化剂, 首次实现了5, 6-苯并-2亚甲基-1, 3-二氧环庚烷的活性自由基开环聚合反 应[40]。
第七节
开环歧化聚合
在催化剂存在下,环烯烃分子中双键开 裂,并以头尾相接的方式连接成大分子的 过程称为开环歧化聚合。环烯烃按结构可 分为单环和多环烯烃。前者以环戊烯为代 表,后者则以降冰片烯为代表。
降冰片烯的开环聚合如下:
对环烯烃的开环歧化聚合的机理,目前普遍接受的 观点是金属卡宾配位化合物引发、增长机理。
CH2CH2O
n
O Na + ROH
R
CH2CH2O
n
+ RO-Na+ OH
交换反应生成的醇盐可继续引发聚合反应。从形 式上看,交换反应与链转移反应相似,但与链转移 反应不同,交换反应生成的端羟基聚合物并不是 “死”的聚合物,而只是休眠种,可和增长链之间 发生类似的交换反应再引发聚合反应:
R CH2CH2O n OH + R CH2CH2O m O-Na+ + R R CH2CH2O CH2CH2O m OH n O-Na+
2、聚合方法比较
方法 因素 推动力 结构单元 特殊基团 开环聚合 连锁聚合 逐步聚合 官能团性质的改 变 聚合过程中有小 分子放出 单体分子间官能 团的相互作用 形成的 苛刻 单体的环张力 化学键键型的 改变 与单体的化学 与单体的化学 组成相同 组成相同 单体分子固有 的 温和 一般 无
聚合条件
引发反应首先从环烯烃中的双键与金属卡宾M=CHR配 位后生成金属杂环丁烷过渡态开始。该过渡态以易 位方式裂解,形成新的增长金属卡宾配位化合物。 新形成的的金属卡宾络合物可继续与环烯烃单体上 的双键形成金属杂环丁烷过渡态,断裂后又形成新 的增长金属卡宾配位化合物,如此反复进行,即得 到高分子聚合物。
开环歧化聚合的应用[30]
• • • • • • • • • • ① 恒比共聚物的合成 ② 理想交替共聚物的合成 ③ 全顺式聚合物和全反式聚合物的合成 ④ 全同立构全顺式共聚物和间同立构全反式聚合 物的合成 ⑤ 嵌段共聚物和接枝共聚物的合成 ⑥ 梳状共聚物和星状共聚物的合成 ⑦ 遥爪聚合物的合成 ⑧ 导电高分子的制备 ⑨ 离子交换树脂的制备 ⑩ 特种性能聚合物的合成
A CH2CH2O n CH2CH2O-M+ + CH2 O CH2 A CH2CH2O CH2CH2O-M+ n+1
CH2
A
CH2CH2OCH2CH2O-M+
环氧化物的阴离子开环聚合具有活性聚合的特点。如不 加入终止剂,则不发生终止反应。
② 交换反应 在醇存在下,增长链与醇之间可发生交换反应:
+
R
文献报道,在发现稀土配位催化剂对环硫丙烷开环 聚合成功地制得高分子量聚合物的基础上,研究稀土 配位催化剂对 CMT的均聚和CMT与环氧氯丙烷(ECH)共 聚的同时,又研究了稀土配位催化剂对CMT与PO的共 聚合。结果表明:稀土催化体系是CMT—PO共聚的优 良催化剂,最大催化效率达6000 g/mol以上,共聚 物的最大特性粘数达1.0dL/g[42]。
第九节
微波开环聚合[46]
以 2,2一二甲基一1,3一丙二醇和氯甲酸乙酯 为原料合成了环状碳酸酯5,5一二甲基一1,3一 二口恶烷一2一酮(DTC),并对DTC在食品添加剂乳 酸锌引发下的微波开环聚合进行了研究。结果表 明:在170 W的微波辐照下,辐照时间对聚合物的 分子量有较大影响,当辐照时间为 17 min时,聚 合物的分子量达到最大,Mw、Mn分别达到208 000 和127000。
第六节 配位开环聚合
烷基金属催化剂主要有A1Et3、A1(i-Bu)3、 ZnEt2、MgEt2等,当它们与其它物质结合形成配 合物或螯合物时,具有很高的催化活性[27]。 环醚单体的阴离子或阳离子聚合很难获取高分 子质量的聚醚,而配位聚合能弥补这方面的不足。 一直以来,配位聚合是环醚开环聚合中研究最多 的[38]。
合成路线如下:
在微波功率为170W、乳酸锌用量为单体DTC的10(摩尔比) 的情况下,辐照时间对单体的转化率、聚合物的分子量及 多分散性的影响见下表:
参考文献
[27] 周立宏,唐辉.环氧化物开环聚合催化体系的研究进 展.精细与专用化学品,2004,12(13):3-12. [28]徐耀良,鲁波,张治国.酸催化下四氢呋喃阳离子均聚 机理的量子化学研究.石油学报(石油加 工),2007,23(6):56-60. [29] 李弘,仝浩,王春伟等.活性开环歧化聚合法合成带羧 酸酯基功能高分子. 离子交换与吸附,2005,21(4):358364.
3、环状单体的聚合活性
• 聚合反应的平衡常数既取决于热力学因素,也 取决于动力学因素: • 若要开环聚合能自发进行,需要ΔG<0
单体
ΔG(kal/ mol)
环丙烷 环丁烷
-22.1 -21.2
环戊烷 环己烷
-2.2 1.4
环庚烷
-3.9
环辛烷
-8.2
九元环以上开环聚合的可能性很小,因此环烷烃开 环聚合的能力顺序为:3、4 > 8 > 5、7
从图中可看出,随着时间的延长,聚合物数均分子量呈 线性增长,分子量分布几乎保持不变。
由上图可看出,聚合物的数均分子量随着聚合物收率的增 加呈线性增长,分子量分布几乎保持不变,符合活性聚合 的标准。
影响开环歧化聚合立体结构的主要是电 子和立体因素:
• • • • • • • • • • • ① 反应中心的立体拥挤程度 ② 过渡金属周围配体的数目和性质 ③ 过渡金属的性质 ④ 反应前线轨道的对称性 ⑤ 过渡金属的氧化态 ⑥ 助催化剂的类型 ⑦ 增殖金属卡宾的性质和电子结构 ⑧ 金属环丁烷中间体的几何结构 ⑨ 反应环烯烃的性质 ⑩ 所得的聚烯烃的结构 还有反应温度和溶剂的性质等
烷氧基铝是一类阴离子配位催化剂,在环氧乙 烷开环聚合中的作用机理如下:
RO Al RO OR + O CH2 RO CH2 RO Al CH2 O CH2 RO 单体 RO Al CH2 O CH2 RO OCH2CH2OR RO OR RO Al O CH2CH2OR
RO Al OCH2CH2OCH2CH2OR 链增长
① 链引发
② 链增长
引发反应生成的三级氧鎓离子活性种后,单体分 子上的氧易亲核进攻活性种中的碳原子而加成增长。
③ 链终止
阳离子开环聚合的链终止反应主要为增长链氧鎓 离子与抗衡阴离子结合,如:
四氢呋喃阳离子均聚反应的实质是四氢呋喃 分子中的o原子按SN2反应机理进攻五元环氧鎓离 子增长物种的α位C原子,从而不断生成新的叔 氧鎓离子增长物种。四氢呋喃与仲氧鎓离子之间 的反应路径由两步组成,首先四氢呋喃与仲氧鎓 离子生成中间体,此步为无能垒的放热反应,放 出的能量为128.438kJ/mol,然后中间体经过渡 态生成叔氧鎓离子,该步反应的能垒为182.012 kJ/mol[28]。
第二节 逐步开环聚合
己内酰胺的水解聚合反应比较复杂,包括己内酰 胺水解开环反应、通过加成聚合和缩合聚合使分子 链增长的反应、通过酰胺链交换改变分子量分布的 反应、环状低聚物的生成及其它副反应。 己内酰胺的水解聚合反应: 开环聚合:在有水存在和一定温度条件下,己内 酰胺水解,在酰胺键处开环,形成氨基己酸
加成聚合:
缩合聚合:
是分子链末端基之间发生的反应,可以使聚合度增加, 并产生小分子水。
酰胺交换反应:
分子链与端氨基交换:
分子链间酰胺交换:
分子链与端羧基的酰胺交换:
环状齐聚物的生成与平衡:
第三节 自由基开环聚合
• 自由基开环聚合有两个显著的特点:一是聚合过 程中体积收缩较小,甚至发生体积膨胀;二是可 合成出主链上带有各种杂原子和官能团的聚合物。
2、自由基开环活性的影响因素
•① 环上取代基的类型
当R1和R2为氢和烷基 取代基时,由于引发生成 的链自由基有较大的位阻, 因而难与烯丙基阻聚反应 相竞争,易与自由基偶合 终止。如果R1和R2中有 一个为苯取代基,则由于 开环生成了稳定的苄基自 由基,单体能进行开环聚 合反应。
②环的大小
环大小对自由基开环聚合的影响是由环张力的大 小和环的位阻综合决定的。 环张力: 六元环(很稳定,开环活性小,聚合物 内总会有一定量的未开环结构单元)<四元环(张 力大,开环活性较大)<七元环单体(任何条件下 自由基开环聚合反应完全)。
2、三聚甲醛的阳离子开环聚合
以三聚甲醛为单体,BF3-H2O为催化剂体系,聚 合过程: 链引发
链增长
链终止
第五节 阴离子开环聚合
在阴离子开环聚合中,任何有活泼氢的化合物 (如水、醇)均可作为引发剂。引发反应被认为是 环氧化物与碱金属氢氧化物或其醇盐作用产生了 醇盐阴离子,而这种阴离子在增长阶段通过若干 单体分子的连续开环反应生成长链聚合物,一般 认为此反应是链式反应[27]。
从以上可见,聚降冰片烯的开环聚合本质 是双键不断异位,分子链逐渐扩大的过程,聚 合后,单体中的双键、单键和环节构在所得聚 合物的重复单元中依然保持不变,只是连接方 式发生了变化。因此是一种完全不同于传统聚 合的新的聚合方法,所得的聚合物具有十分特 殊的性能。
活性开环歧化聚合[29]
以DCPD为原料,经解聚制成CPD,再与丙烯酸叔 丁酯 (tBA)经Diels—Alder反应合成了带羧酸酯基 的降冰片烯单体即:2一叔丁氧羰基一5一降冰片烯。
开 环 聚 合
许静 2008环聚合 自由基开环聚合 阳离子开环聚合 阴离子开环聚合
配位开环聚合 开环歧化聚合 开环共聚合 微波开环聚合