抗真菌药物的作用机制及耐药性
抗真菌药物ppt课件

2024/1/30
1
2
3
如念珠菌属感染可选用氟康唑、伊曲康唑等。
根据真菌种类选择药物
如深部真菌感染可选用两性霉素B、氟胞嘧啶等。
根据感染部位选择药物
根据患者年龄、体重、病情等因素调整剂量,确保安全有效。
剂量调整
23
2024/1/30
对于严重或难治性真菌感染,可采用两种或多种抗真菌药物联合治疗。
影响蛋白质合成酶
如特比萘芬、氟康唑等。
代表药物
15
2024/1/30
04
CHAPTER
常用抗真菌药物介绍
16
2024/1/30
通过与真菌细胞膜上的麦角固醇结合,导致细胞膜通透性改变,细胞内重要物质漏失而发挥抗真菌作用。
通过与真菌细胞膜上的麦角固醇结合,形成微孔,导致细胞内物质外泄而死亡。
两性霉素B
抗真菌药物ppt课件
1
2024/1/30
目录
引言真菌的生物学特性抗真菌药物的作用机制常用抗真菌药物介绍抗真菌药物的合理应用抗真菌药物的耐药性及防治策略
2
2024/1/30
01
CHAPTER
引言
3
2024/1/30
真菌感染的定义和分类
由真菌引起的疾病,包括浅表性、皮下组织和系统性感染等。
针对真菌代谢途径的新型药物研究
基于基因组学和蛋白质组学的新药筛选研究
03
38
2024/1/30
THANKS
感谢您的观看。
39
2024/1/30
传播途径
28
2024/1/30
院内感染
社区感染
动物源性感染
29
2024/1/30
抗真菌耐药性的全球问题:流行、机制与管理

抗真菌耐药性的全球问题:流行、机制与管理黄郑雨【摘要】所有严重的真菌感染都需要适当的抗真菌治疗才能取得成功.目前只有少数几类抗真菌药物可用.单一药物耐药性和多药耐药性严重妨碍了对病人的治疗.念珠菌和曲霉的唑类药物耐药是临床上最大的挑战之一,其次是念珠菌属(尤其是光滑念珠菌)的棘白菌素耐药和多药耐药.农业衍生的耐唑烟曲霉以及多重耐药耳道白色念珠菌的出现和传播给人类健康带来了新的威胁.耐药的分子机制在不太敏感菌株中自然发生,并在敏感菌株中快速获得.耐药机制包括药物与靶标相互作用的改变,药物外排泵介导降低细胞内药物浓度,以及与生物膜相关的通透性屏障.虽然耳道念珠菌天生多药耐药,但其它菌株通常通过逐步选择耐药机制而产生多药耐药性.药物处理引起的细胞应激反应促进了细胞的适应,从而导致细胞耐药性的出现.控制耐药性的举措包括有效的抗真菌药物管理计划,快速真菌诊断,治疗药物监测和临床干预.开发更好的诊断工具和有针对性地使用抗真菌药物的策略,对于保持药物效力至关重要.【期刊名称】《国外医药(抗生素分册)》【年(卷),期】2018(039)005【总页数】8页(P前插1-前插2,357-362)【关键词】耐药性;流行;机制;管理【作者】黄郑雨【作者单位】西南大学药学院,重庆400715【正文语种】中文【中图分类】R978.51 前言真菌病原体可引起危及生命的侵袭性疾病(如真菌血症、脑膜炎、肺炎)、严重的慢性疾病(如慢性肺曲霉菌病、过敏性支气管肺曲霉菌病)和复杂的慢性呼吸道疾病(如哮喘、慢性阻塞性肺病)。
这些病原体还会引起反复感染,如口腔和阴道念珠菌病。
病人的免疫抑制是罹患侵袭性真菌感染的重要原因。
这些感染常伴随高死亡率,成功的临床治疗效果需要早期诊断和有效的抗真菌治疗。
然而,抗真菌药物的选择很少,治疗侵袭性疾病的药物仅限于唑类化合物、棘白菌素、多烯类和氟胞嘧啶。
过去的十年,毒性较小药物的开发为抗真菌预防使用和疾病治疗做出了贡献,然而这反过来又导致了耐药性的增加。
抗真菌药物专业版

1.3 伏立康唑
voriconazole
Slide: Malcolm Richardson, 2002
Adapted from JP Donnelly
药代动力学特点
口服给药后迅速吸收,生物利用度达96% 。 食物可影响药物的吸收(生物利用度约下 降20%),因此应在进食后 1~ 2 小时服药。 血浆蛋白结合率约58%;药物在脑脊液中的 浓度接近血浆。 药物通过肝脏代谢(80%)为无活性代谢产 物,由肾脏排出,尿中原形药物不到 5%。 轻到中度肝硬化患者需调整剂量。肾功能 不全患者口服伏立康唑无需调整剂量。
为最重要的广谱抗真菌药物之一,几乎对 所有真菌都有较强的抗菌活性。目前仍是 治疗多种深部真菌病的首选药物。
Slide: Malcolm Richardson, 2002
Adapted from JP Donnelly
药代动力学特点
两性霉素B口服吸收不良且不稳定(<5%),仅能 静脉滴注。 蛋白结合率90-95%,主要在肝脏内代谢,缓慢以 原形自肾排泄,每日约2-5%的给药量由尿派出, 可维持到停药7w后。 吸收后仅有微量药物进入脑脊液,因此,用两性 霉素 B 治疗隐球菌脑膜炎时需鞘内注射(与 5-Fc 合用)。 不易透析除去
5-氟胞嘧啶属于窄谱抗真菌药物,对念珠菌 属、新生隐球菌、申克孢子丝菌、卡氏枝 孢霉、疣状瓶霉等有抗菌作用
药代动力学特点
5-氟胞嘧啶口服吸收良好。 血浆蛋白结合率很低,血循环中游离的药物水平 较高;且分子量小,故容易通过血脑屏障,脑脊 液浓度可达血药浓度的60%-100%。 此药毒性低。5-氟胞嘧啶体内不被分解代谢,主 要通过肾脏清除,口服后90%的药物在24小时内 以原形形式从尿中排出。肾功能衰竭的患者药物 半衰期明显延长,使用该药时要调整剂量。 可经血或腹膜透析排出体外
利奈唑胺耐药标准-概述说明以及解释

利奈唑胺耐药标准-概述说明以及解释1.引言1.1 概述利奈唑胺是一种广谱抗真菌药物,常用于治疗念珠菌感染等真菌性疾病。
然而,随着利奈唑胺的广泛应用,利奈唑胺耐药现象逐渐显现,严重影响了该药物的疗效和临床治疗效果。
为了更好地规范利奈唑胺的使用和防止耐药性的进一步扩散,制定利奈唑胺耐药标准显得尤为重要。
本文将就利奈唑胺耐药标准的重要性、制定原则以及未来的发展方向进行深入探讨,旨在为利奈唑胺的合理应用和管理提供参考。
1.2 文章结构本文主要分为引言、正文和结论三个部分。
在引言部分,将针对利奈唑胺耐药标准这一课题进行概述,介绍利奈唑胺的作用机制和应用范围,以及利奈唑胺耐药现象的发展和影响。
同时,阐述本文的目的和意义,引导读者对该主题有一个整体的了解。
在正文部分,将进一步探讨利奈唑胺的作用机制和应用范围,分析利奈唑胺耐药现象的发展和影响,重点讨论利奈唑胺耐药标准的重要性和制定原则。
通过各种文献资料和研究成果,全面深入地分析利奈唑胺耐药的现状和问题,提出解决问题的思路和方法。
在结论部分,对本文进行总结,强调利奈唑胺耐药标准的必要性和意义,并展望未来利奈唑胺耐药研究的方向和挑战。
同时,提出未来利奈唑胺耐药标准的改进建议,为进一步完善利奈唑胺耐药标准提供参考和指导,促进相关领域的进一步发展和进步。
1.3 目的本文旨在系统地探讨利奈唑胺耐药标准的重要性和制定原则,旨在加深对利奈唑胺耐药现象及其对临床应用的影响的理解。
通过对利奈唑胺耐药标准的重要性进行分析和探讨,我们可以更好地指导临床医生在使用利奈唑胺时的决策,减少耐药性发展的风险,提高治疗效果。
同时,在制定利奈唑胺耐药标准的原则方面,本文旨在为相关研究人员提供指导,确保标准的科学性、严谨性和可操作性,从而为今后的利奈唑胺耐药研究和临床应用提供有益的参考依据。
2.正文2.1 利奈唑胺的作用机制和应用范围利奈唑胺是一种广谱抗真菌药物,其作用机制主要是通过抑制真菌细胞壁的合成而发挥抗真菌活性。
抗真菌药物.

谱抗真菌药物。
抗菌谱
酮康唑具有广谱抗真菌作用。对皮肤癣
菌、糠秕孢子菌、念珠菌属、酵母菌、 皮炎芽生菌、粗球孢子菌、荚膜组织胞 浆菌、巴西副球孢子菌有抗菌作用。对 曲霉、毛霉无抗菌作用。
药代动力学特点
该药在胃肠道容易吸收,口服 400mg 2小 时后,血药浓度达 5 ~ 6mg/l,但是个体 差异较大。胃酸可促进药物吸收,当胃 酸分泌减少或与抑制胃酸分泌的药物同 时服用时药物吸收减少。口服酮康唑后, 药物可分布在皮脂,尿液、乳汁中;但 在脑脊液中含量甚微。
分类
目前常膜,干 扰真菌细胞膜麦角固醇的合成(如唑类、 丙烯胺类和吗啉类)以及损害细胞膜脂 质结构及其功能的药物(如多烯类); ②影响真菌细胞壁合成的药物(如卡泊 芬净);③干扰真菌核酸合成的药物 (如 5- 氟胞嘧啶,灰黄霉素);④作用 机制尚未明确的药物(如碘化钾等)。
这类药物有共同的 N-碳置换的咪唑或三唑环,
包括三唑类和咪唑类两大类。唑类药物是近20 年来发展较快的一类抗真菌药物,在近年来防 治真菌感染中发挥了重要作用。 唑类药物的作用部位在真菌的细胞膜。这类药 物通过抑制细胞色素 P450 依赖酶,使麦角固醇 合成受阻从而破坏了真菌细胞的完整性;同时 使甲基化的固醇堆积,这样则改变了真菌细胞 膜的化学成分,使其通透性发生变化,从而阻 止了真菌细胞的生长繁殖。有些唑类药物在高 浓度时可直接破坏真菌细胞膜,导致细胞内容 物外露,发挥杀菌作用。
药物相互作用
与抗组胺药物特非那定、阿斯咪唑同时服用,
可延长这些药物的半衰期,导致严重心率失常 发生。 酮康唑能使环孢霉素血药浓度升高。与环孢霉 素合用时,应注意监测其血药浓度。 华法林与酮康唑同时服用时可使其抗凝作用增 强,二者同时使用时要注意监测凝血酶原时间。
抗真菌药AntifungalDrugs

13
克霉唑(clotrimazole)
• 克霉唑为广谱抗真菌药。 • 口服不易吸收,血药峰浓度较低,代谢产物大部
分由胆汁排出,1%由肾脏排泄。t1/2均为3.5~5.5h。 • 局部用药治疗各种浅部真菌感染。
14
联苯苄唑(bifonazole)
• 联苯苄唑抑制24-甲烯二氢羊毛固醇转化为脱甲基 固醇,也抑制羟甲基戊二酰辅酶A转化为甲羟戊酸, 从而双重阻断麦角固醇的合成,使抗菌活性明显 强于其他咪唑类抗真菌药,具有广谱高效抗真菌 活性。
• 不良反应轻微,常见胃肠道反应,较少发生肝炎 和皮疹。
20
嘧啶类抗真菌药
氟胞嘧啶(flucytosine)
• 氟胞嘧啶在敏感真菌的细胞内,脱去氨基而形成抗 代谢物5-氟尿嘧啶,再由尿苷-5-磷酸焦磷酸化酶 转变为5-氟尿嘧啶脱氧核苷,抑制胸腺嘧啶核苷合 成酶,阻断尿嘧啶脱氧核苷转变为胸腺嘧啶核苷, 影响DNA合成。
③ 复合物(ABLC),是脂质体与两性霉素B交织 而成
6
制霉菌素 (nystatin)
•抗真菌作用和机制与两性霉素B相似,对念珠菌属 的抗菌活性较高,且不易产生耐药性。 •制霉素主要局部外用治疗皮肤、黏膜浅表真菌感染。 口服吸收很少,仅适于肠道白色念珠菌感染。注射 给药时制霉素毒性大,故不宜用做注射。 •局部应用时不良反应少见。口服后可引起暂时性恶 心、呕吐、食欲不振、腹泻等胃肠道反应。
• 不良反应主要为胃肠道反应,其发生率较氟康唑 低,患者更易耐受。
18
丙烯胺类抗真菌药
• 抑制角鲨烯环氧化酶(squalene epoxidase), 使角鲨烯经此关键酶催化生成麦角固醇受抑制,真 菌细胞壁成分麦角固醇合成障碍,而产生抑菌或杀 菌效应。
抗真菌药物知识点总结

抗真菌药物知识点总结真菌是一类微生物,在自然界与人类生活中广泛存在。
在某些情况下,真菌可以引起感染和疾病,需要使用抗真菌药物进行治疗。
抗真菌药物是一类针对真菌感染的药物,可以有效抑制真菌的生长和繁殖。
本文将对抗真菌药物的知识点进行总结,包括抗真菌药物的分类、药理学、治疗原则以及常见的抗真菌药物等内容。
一、抗真菌药物的分类抗真菌药物可以按照其作用机制、化学结构和临床应用等方面进行分类。
按照作用机制的不同,抗真菌药物可以分为以下几类:1. 抑制真菌壁合成的药物:这类药物可以抑制真菌细胞壁的合成,从而导致真菌细胞的死亡。
常见的抑制真菌壁合成的药物有伊曲康唑、咪康唑等。
2. 抑制真菌细胞膜合成的药物:这类药物可以抑制真菌细胞膜的合成,影响真菌的细胞结构和功能,导致真菌细胞的死亡。
常见的抑制真菌细胞膜合成的药物有酮康唑、酮康唑等。
3. 抑制真菌核酸和蛋白质合成的药物:这类药物可以抑制真菌的核酸和蛋白质合成,从而影响真菌的生物活性,导致真菌细胞的死亡。
常见的抑制真菌核酸和蛋白质合成的药物有氟胞嘧啶、利巴韦林等。
4. 其他抗真菌药物:除了上述几类抗真菌药物外,还有一些其他种类的抗真菌药物,包括拟对生物药物、抗真菌多肽药物等。
二、抗真菌药物的药理学抗真菌药物的药理学是研究抗真菌药物在机体内的吸收、分布、代谢和排泄等过程,以及抗真菌药物的药效学和毒性学等方面的知识。
抗真菌药物的药理学知识对于临床应用和用药安全至关重要。
1. 药物的吸收:抗真菌药物可以经口、静脉、肌肉等途径给药,吸收机制多种多样。
一般来说,抗真菌药物在胃肠道内有一定的吸收,但不同药物的吸收率和速度有所不同。
2. 药物的分布:抗真菌药物在机体内的分布受多种因素的影响,包括蛋白结合率、组织分布率、脂溶率等。
抗真菌药物的分布特点直接影响其在机体内的药效及安全性。
3. 药物的代谢:抗真菌药物在机体内可以发生代谢作用,其代谢产物可能会影响药物的药效和毒性。
抗真菌药物的代谢途径和代谢产物对于药物的研究和临床应用有重要意义。
抗真菌类药物tdm及临床应用

03
临床试验与疗效评 估
新型抗真菌药物在临床试验中显 示出较好的疗效,但仍需进一步 评估。
未来抗真菌药物的发展趋势
联合治疗
未来抗真菌药物的发展可 能会采用联合治疗策略, 以提高治疗效果并降低耐 药性的产生。
靶向治疗
针对真菌特定的生物学特 性或致病机制,开发靶向 药物是未来的一个重要方 向。
免疫治疗
两性霉素B和制霉菌素主要通过抑制 真菌核酸合成,干扰真菌的繁殖和生 长。
干扰真菌细胞膜功能
丙烯胺类抗真菌药物主要通过干扰细 胞膜的通透性,影响营养物质的吸收 和代谢产物的排出,导致真菌死亡。
抗真菌药物的研发历程
20世纪50年代
20世纪70年代
制霉菌素和两性霉素B的发现和应用,开启 了抗真菌药物研发的历程。
开展多中心、大样本的临床研究
通过多中心、大样本的临床研究,可以进一步评估抗真菌药物TDM的临床效果和经济效益,为TDM的推 广应用提供更为充分的证据支持。
THANKS
感谢观看
1 2 3
深部真菌感染
对于一些严重的深部真菌感染,如败血症、脑膜 炎等,需要使用强效的抗真菌药物进行治疗。
免疫缺陷患者的真菌感染
对于免疫缺陷或免疫抑制的患者,如艾滋病、器 官移植受者等,需要特别关注并采取积极的治疗 措施。
长期使用抗生素导致的真菌感染
长期使用抗生素的患者容易发生真菌感染,需要 密切监测并及时治疗。
唑类抗真菌药物的研发和应用,如氟康唑 、伊曲康唑等,为临床治疗深部和浅部真 菌感染提供了有效手段。
20世纪90年代
21世纪初
丙烯胺类抗真菌药物的研发和应用,如特 比萘芬,为治疗皮肤癣菌等浅部真菌感染 提供了新的选择。
嘧啶类抗真菌药物的研发和应用,如氟胞 嘧啶,进一步丰富了抗真菌药物的治疗手 段。
抗真菌药物的作用机制

抗真菌药物的作用机制目前应用于临床的抗真菌药物,就其作用机制分类,大致可以分为3种:1作用于真菌细胞膜中甾醇合成的抗真菌药物,包括:酮康唑等咪唑类药物、多烯类抗生素、两性霉素等,以及烯丙胺类药物特比萘芬等;2作用于真菌细胞壁合成的抗真菌药物,如:棘白菌素类药物卡泊芬净,其抑制真菌细胞壁主要成分1,3-β-D-葡聚糖的合成,以及抑制几丁质合成的日光霉素和多氧霉素等; 3作用于核酸合成的抗真菌药物,如:5-氟胞嘧啶等能抑制或杀灭真菌的药物。
除一些古老的抗真菌外用药如水杨酸、雷琐辛、碘剂、硫黄等外,抗真菌作用显著的新药有抗生素和合成药两大类。
①抗生素。
主要有灰黄霉素、制霉菌素和两性霉素B等。
灰黄霉素只对皮肤癣菌病有效,主要是头癣、体癣、股癣、手足甲癣等,口服时,20~30天为一个疗程,需合并外用治癣药物。
长期使用有少数浅部真菌产生耐药菌株,可换用酮康唑。
制霉菌素治疗胃肠道念珠菌病,外用治疗皮肤粘膜念珠菌感染,也可制成坐药。
两性霉素B主要治疗深部真菌病,如系统性念珠菌病、隐球菌病、曲霉病、结合菌病、芽生菌病、巴西副球孢子菌病、球孢子菌病和组织胞浆菌病等。
将此药加入5%葡萄糖溶液中,缓慢静脉滴注。
②合成药。
包括:咪唑类药物如克霉唑、益康唑、咪康唑和酮康唑等、氟胞嘧啶、丙烯胺衍生物。
5-氟胞嘧啶治疗念珠菌病、隐球菌病和着色芽生菌病。
克霉唑、益康唑和咪康唑基本供外用。
咪康唑也可静脉滴注。
酮康唑也可口服。
外用时主要治疗皮肤真菌病和皮肤念珠菌病。
口服和静脉滴注主要治疗深部和浅部的真菌病。
抗真菌药容易影响白细胞及肝功能,长期使用造成一过性GPT上升或白细胞下降,停药可愈。
5-氟胞嘧啶从尿中排泄,肾功能不良者可在血中聚集,引起中毒,故肾功能差者应禁用或慎用。
两性霉素B可损伤肾脏,并引起血钾降低,有人有发冷、发热反应,少数人可引起血栓性静脉炎。
酮康唑应特别注意肝脏受损问题。
长期使用可引起血中雄激素水平降低和肾上腺皮脂功能受到抑制。
抗真菌类药物与临床耐药性

随着免 疫缺 陷 、 官 移植 的病 人增 多 , 器 以及 大量 抗 生 素的使 用 . 真菌感 染 的发生 率逐 年上 升 。 同时伴
感 染治 疗无 效 .并从 患 者体 内分 离 出与 最初 感染 相
同的真 菌 ( 即具 有 相 同的生 物 型 、基 因型 或基 因亚
随抗真 菌药 物 的广泛 使用 .造 成抗 真菌 药物 的耐药
菌药 物治疗 时 。 患者 临床 症状 和体 征 消失 . 感 染 真 但 菌却未 能清 除 : 时治疗 失败 而分 离 出的致 病菌不伴 有 最小抑菌浓度 ( nm ln ii r n ert nMI 升 mii a ihbt y 0 ct i , C) o c ao 高 。这 是 由于还 有多 种 因素 可能 导致 抗真 菌 治疗 失
真菌耐 药时 需要 慎重 ,只有排 除 了其 它 可能 造成 失 败 的因素 .确认 这种 抗真 菌 药物 对某 一 患者 的 真菌
的耐 药性 发 生率 较低 。只有 少数 较 罕见 的真 菌如 部
大 差异 。
[ () 2 1患者 体 内定 植 或感 染 的真 菌发 生 基 因 突变 . 】 : 从
而产 生耐药 ;2 由于化疗 药物 的选择压 力作用 . 患 () 使 者 体 内定 植或 感染 的固有 耐药 的非 优 势菌 成为 优 势 菌 ;3 患 者一 开始 就被 耐药 的真菌感 染 。 () 但 在 临床上 。 断 耐药性 比较 困难 . 时用 抗真 判 有
4真菌 耐 药性 的发 展史 【 5 J
早 期 的抗 真菌 药物 的使 用 .由于早 期 真菌感 染
发生 率较 低 , 上早 期抗 真菌 药 物 的品种 不 多 . 患 加 故
者使 用抗 真 菌类 药 物也 不多 .因此这 些 抗真 菌药 物
抗真菌药物的种类与作用机制综述

Chakraborty等[25]研究粪壳菌素sordarin是蛋白质翻译过程中延长因子EF抑制剂,阻止真菌蛋白质的进行。
Zhao等[26]研究N肉豆蔻酰基转移酶抑制剂,可抑制白色念珠菌等真菌蛋白质的生物合成。
2.4作用于真菌的电子呼吸链Ma等[27]研究抗霉素A和于链霉菌的UK2A,对真菌呼吸链的电子传递具有抑制作用,引起真菌的死亡。Fudou等[28]发现于粘细菌的Haliangicin,能够专一性的抑制呼吸链中蛋白复合物III的电子传递。另外,从粘细菌的代谢产物中发现了多种具有抑制电子传递的抗真菌化合物。
Mandala等[22]发现两个结构相类似的IPC合成酶抑制剂khafrefungin和rustmicin.
rustmicin是IPC合成酶的可逆性抑制剂,特异性作用IPC合成酶;khafrefugin对白色念珠菌和新型隐球菌IPC合成酶的强的抑制作用。
2.3抑制真菌蛋白质的生物合成Kinsman等[23]从Graphimumputredinis菌液中分离出GR,能抑制真菌的蛋白合成,具有抗真菌活性。Okada等[24]从Penicillium minioluteum发酵液中分离出BE31405,对白色念珠菌、新型隐球菌均具有抑制活性,主要抑制真的蛋白合成。
结语
抗真菌药物种类繁多,其作用机制各不相同,随着唑类药物广泛应用,真菌耐药现象不断出现,已出现药物间交叉耐药,对临床治疗带来严重威胁。本研究对近年来真菌药物的种类、结构、作用机制等方面进行了阐述。近年来,又相继发现许多新型抗真菌药物,并提出了抗真菌药物作用的新靶点,成功开发出一些活性强大,疗效确切的抗真菌药。
抗微生物药名词解释

抗微生物药名词解释抗微生物药是用于治疗细菌、真菌、病毒和寄生虫等微生物感染的药物。
以下是关于抗微生物药的一些常见名词解释。
1. 抗生素:抗生素是治疗细菌感染的药物,可以通过杀死细菌或抑制其生长来治疗感染。
抗生素可以根据它们的作用机制分为多个类别,包括青霉素、四环素、氨基糖苷类等。
2. 抗真菌药物:抗真菌药物用于治疗真菌感染,包括念珠菌、白色念珠菌、肺曲霉病等。
抗真菌药物可以通过杀死真菌或阻止其生长来治疗感染。
3. 抗病毒药物:抗病毒药物用于治疗病毒感染,包括流感病毒、艾滋病毒等。
抗病毒药物可以通过干扰病毒的复制和传播来减轻感染的严重程度。
4. 抗寄生虫药物:抗寄生虫药物用于治疗由原虫、线虫、蠕虫和虱子等寄生虫引起的感染,如疟疾、血吸虫病、衣原体感染等。
抗寄生虫药物可以通过杀死寄生虫或阻止其生长来治疗感染。
5. 抗菌谱:抗菌谱指抗微生物药物对不同微生物的抗菌活性范围。
广谱抗菌药物可以同时对多种细菌起作用,而窄谱抗菌药物则只对特定菌株有效。
6. 耐药性:耐药性是指微生物对抗微生物药物产生的抵抗性。
当微生物对抗微生物药物产生耐药性时,原本有效的药物可能无法继续治疗感染。
7. 二重感染:二重感染是指一个人同时感染两种或更多不同的微生物,如细菌和真菌。
二重感染可能导致治疗困难,因为不同的微生物可能对抗微生物药物具有不同的敏感性。
8. 阻断剂:阻断剂用于抑制微生物对抗微生物药物的抗性机制,以提高药物的疗效。
阻断剂可以与微生物产生的酶结合,从而抑制其对抗微生物药物的降解能力。
总之,抗微生物药是一类用于治疗细菌、真菌、病毒和寄生虫感染的药物。
它们可以通过不同的作用机制来杀死或抑制微生物的生长,从而达到治疗感染的目的。
然而,随着微生物对药物的耐药性增加,研发新的抗微生物药物和阻断剂变得愈发重要。
抗真菌药物耐药监测及预警制度

抗真菌药物耐药监测及预警制度简介抗真菌药物耐药性是指微生物对抗真菌药物的治疗效果降低或丧失的现象。
随着抗真菌药物的广泛应用,耐药性问题日益突出。
为了有效监测和预警抗真菌药物的耐药情况,制定抗真菌药物耐药监测及预警制度是非常重要的。
监测和评估- 监测对象:对抗真菌药物耐药性进行监测的对象包括临床病原真菌和环境中的真菌。
- 监测方法:采用标准化的实验方法进行真菌耐药性的监测和评估,包括最小抑菌浓度(MIC)实验、亲和力试验等。
- 监测指标:监测指标主要包括真菌对抗真菌药物的敏感性和耐药性水平,以及对不同抗真菌药物的耐药性谱。
数据收集和分析- 数据收集:建立真菌耐药性数据库,收集和整理全国范围内的监测数据,并与国际数据库进行比对和分析。
- 数据分析:通过统计分析真菌耐药性的时空分布规律,识别耐药性的变化趋势和高耐药区域,为制定针对性的预防和治疗策略提供科学依据。
预警机制和应对策略- 预警指标:根据监测和分析结果,确定抗真菌药物的耐药性预警指标,如超过一定耐药率、出现多药耐药等。
- 预警机制:建立抗真菌药物耐药性的预警机制,包括监测结果的实时更新、预警信息的发布和信息共享等。
- 应对策略:根据预警信息,制定相应的应对策略,包括加强感染控制措施、优化抗真菌药物使用策略、促进研发创新等,以减少抗真菌药物耐药性的发生和传播。
宣传和培训- 宣传意义:通过宣传抗真菌药物耐药监测及预警制度的重要性,增强社会对该问题的关注度。
- 培训措施:开展相关培训,提高医务人员和实验人员对抗真菌药物耐药性监测和评估的能力。
结论抗真菌药物耐药监测及预警制度的建立和实施,对于预防和控制抗真菌药物耐药性具有重要意义。
通过规范化的监测、数据分析、预警和应对策略,有效应对抗真菌药物耐药性问题,提高真菌感染的防治水平。
抗真菌药物艾莎康唑的临床应用

➢艾莎康唑对毛霉菌的抗菌活 性与两性霉素B相似,对曲 霉菌的活性与伏立康唑相似。
➢耐药取决于CYP51A突变的类 型,伴有TR34/L9H突变的菌 株对三唑类呈现泛耐药。
LOG五、O临床应用
侵袭性曲霉
01
侵袭性毛霉菌
02
临床 应用
中枢性神经系统感染
艾莎康唑主要经过肝脏中CYP3A4和3A5的代谢;且治疗过程中无 需常规新型药物浓度监测。
艾莎康唑主要经过粪便(46.1%)和尿液(45.5%)排出体外。
LOG三O、抗菌谱
真菌种类
烟曲霉 土曲霉 黄曲霉 白色念珠菌 耳念珠菌 都柏林念珠菌 光滑念珠菌 季也蒙念珠菌
抗菌活性
++ ++ ++ + ± + ± +
艾莎康唑 的临床应用
时间:XXXXX 汇报人:XXXXX
目录
1
作用机制
2
药代动力学特性
3
抗菌谱
4
交叉耐药性
5
临床应用
6
常见不良反应
一、作用机制
➢ 艾莎康唑为一种三唑类抗真菌药物,抗菌谱广,对常见霉菌、酵母菌和双向真菌 均有活性。
➢ 艾莎康唑通过抑制羊毛甾醇14-α-脱甲基酶来阻断麦角固醇合成,促使真菌细胞凋 亡。
真菌种类
克柔念珠菌 葡萄牙念珠菌 近Байду номын сангаас滑念珠菌
热带念珠菌 隐球菌 暗色真菌 镰刀霉菌 毛霉菌
抗菌活性 真菌种类
+
尖端赛多孢菌
+
多育赛多孢菌
+
毛孢子菌
+
氟康唑、伏立康唑等临床抗真菌药物分类及作用机制、抗菌谱、临床应用及注意事项
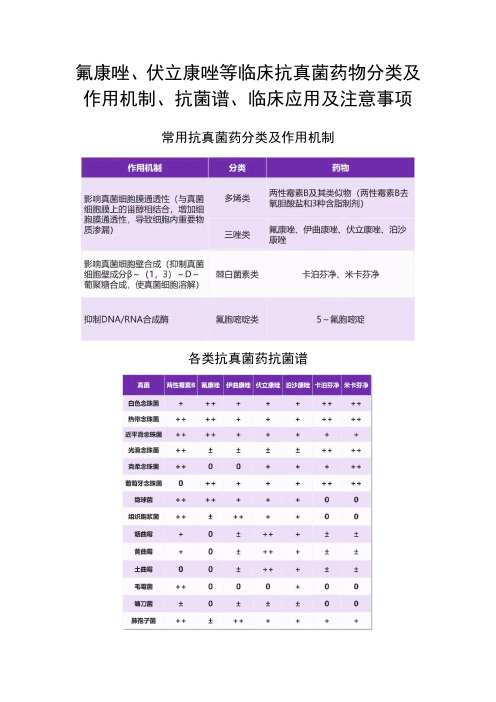
氟康唑、伏立康唑等临床抗真菌药物分类及作用机制、抗菌谱、临床应用及注意事项常用抗真菌药分类及作用机制各类抗真菌药抗菌谱0 不推荐;+ 有活性;++ 推荐;±不确定各类抗真菌药临床应用及注意事项三唑类三唑类抗真菌药物具体用药方案见下表 3。
注意事项:1、不良反应中胃肠道反应最常见,如恶心、呕吐、腹痛、腹泻等。
其次为皮肤反应,包括皮疹瘙痒和荨麻疹等。
2、因可导致严重心律紊乱,本类药物禁止与西沙必利、阿司咪唑、特非那定和三唑仑合用。
3、伊曲康唑不可用于充血性心力衰竭以及有充血性心力衰竭史的患者。
4、因通过细胞色素P450 同工酶代谢,与华法林、环孢素A、他克莫司、奥美拉唑、非核苷类逆转录酶抑制剂、苯二氮䓬类、他汀类、双氢吡啶钙通道阻滞剂、磺脲类口服降糖药和长春碱等药物存在相互作用。
棘白菌素类用于侵袭性念珠菌病、对其他抗真菌药物治疗无效或不耐受的曲霉病。
该类药物是伴有中性粒细胞减少的念珠菌血症,且近期有唑类使用史患者的首选用药。
卡泊芬净成人剂量第 1 日 70 mg,以后每日 50 mg,静脉滴注 1 小时(疗效不佳者可每日 70 mg)。
米卡芬净成人剂量 50—100 mg/d,静脉滴注,曲霉感染可增至每日 50—300 mg。
氟胞嘧啶类口服是首选方法。
不能口服的患者,可以静脉滴注或腹腔内灌注,治疗系统性念珠菌感染,为避免耐药性出现,开始即宜以大剂量。
肾功能正常的成人 150 mg/(kg.d) 或 5—8 g/d,分 4 次服用,间隔 6 小时。
如果肾功能受损,首剂应减至 25 mg/kg,之后调整剂量和给药间隔。
注意事项:因极易产生耐药,一般不单独用药,常合并两性霉素 B 或三唑类抗真菌药物同时使用。
多烯类(两性霉素 B 及其含脂制剂)药物不经黏膜吸收,胃肠道只吸收少量,故只有静脉制剂,其半衰期长达 24 h,可每日 1 次给药。
组织穿透力弱,不易通过血脑屏障进人脑脊液,经单用静滴该药治疗真菌性脑膜炎无效者,可同时鞘内给药。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
由于药物首先与外源加入的甾醇物质发生生物化 学作用,而使细胞膜上的甾醇免遭药物的作用。
1、多烯类抗真菌抗生素的作用机制
抗生素发挥作用时首先与膜结合,其结合程度与膜 内甾醇含量成正比。 结合后生成的膜——抗生素复合物,使细胞质膜结 构发生改变,在膜脂质双层中形成由多烯大环内酯 抗生素与胆固醇结合的环状化合物,构成亲水通道, 致使细胞内容物向胞外泄漏。 所泄漏的物质种类与抗生素的性质、浓度及作用 时间有关,如钾离子、无机磷、有机磷、氨基酸、 磷酸酯直至核酸、蛋白等,从而产生杀菌作用。
一、作用于真菌细胞膜的抗真菌抗生素 和合成药物
目前在临床上广泛使用的这类抗真菌药物有 三种结构类型:
唑类; 多烯类; 烯丙胺硫代氨基甲酸酯类 (allylaminethiocarbamates)。 这类药物的作用靶位为真菌细胞膜的主要组成— —麦角甾醇。
不同抗真菌药物的作用靶位
(一)、作用于真菌细胞膜中甾醇合成 的抗真菌合成药物
靶酶合成量增加
靶酶拷贝数增加
2、烯丙胺类抗真菌药物 1)、烯丙胺类抗真菌药物的作用机制
虽然与其他麦角甾醇生物合成抑制剂类抗真菌药物的结构 不同,烯丙胺类的特比萘芬和萘替芬的作用机制也是抑制 麦角甾醇的生物合成。 特比萘芬在体内外对皮肤真菌具有很强的抗菌活性,并对 某些唑类抗菌药物产生耐药性的菌株也有效。
不同抗菌药物的协同作用
单独使用抗细菌药物RNA聚合酶抑制剂利福 平时无抗真菌活性,但当与两性霉素B合并用 药时,对多种真菌具有活性。
产生这一协同作用的原因是由于两性霉素B对 真菌细胞膜的作用而增加了细胞对利福霉素 的吸收。
不同抗菌药物的协同作用
合并使用两性霉素B和核苷类抗真菌药物5-氟 胞嘧啶(5FC)于念珠菌感染的老鼠模型,同 样能够产生协同效应。
1、多烯类抗真菌抗生素的作用机制
利用这一特性,结合使用一些原先不能通过真菌胞膜的药 物,使其发挥作用,菲律宾菌素与甾体结合后形成的复合物 在膜内发生重排,以至膜结构破坏成为碎片,从而使真菌被 杀死。 这类抗生素的毒副作用是由于其对细胞质膜脂质双层中的 固醇类结合专一性不强而损伤正常人体细胞所引起的。
1:靶酶过量产生;2:药物结构被改变;3:药物被外排蛋白泵出; 4:药物在细胞壁水平/细胞膜水平被阻止;5:细胞由于药物的作用而产 生的补偿途径以使细胞保持活性;6:某些能够将钝化的药物转化为活性 药物的酶被抑制;7:细胞产生某些能够降解药物的酶并分泌至胞外。
真菌对唑类抗菌药物产生耐药性的生物化学机制
两性霉素B的应用
尽管两性霉素B是一个最为重要的抗真菌药物,但由 于其较窄的治疗指数(therapeutic index),并且肾 毒性较强,使其在临床应用上受到了一定的限制。为 了改变这一不足,近年来,对两性霉素B脂质体的研 究取得了很大的成功。
目前已经有多种两性霉素B脂质体剂型被用于临床, 从而有效地改善了高剂量使用时对哺乳细胞的毒性。
作用机制 药物作用靶位改变 (14α-甾醇去甲基酶 ) 原因 备注
靶位发生改变致使药物不 靶位有活性,但对药物 能与之结合,但不改变与 的亲和力降低 外源底物的结合能力
Байду номын сангаас
甾醇生物合成改变
缺乏△5,6-去饱和酶
导致14α-甲基 fecosterol的积累, 而不是麦角甾醇的积累
胞内靶酶合成量减低
细胞膜中脂和甾醇改变, 透过细胞膜的能力降低; 特异性药物外排泵过量表 主动外排泵 达(CDR1、PDR5和BEN) 麦角甾醇过量合成,导 致氟康唑和依曲康唑的 交叉耐药性
FLU:氟康唑; ITRA:依曲康唑; VOR: voriconazole; TERB:特比萘芬
2)、真菌对唑类抗真菌药物的耐药性机制
真菌对唑类药物产生耐药性的主要作用机制是由 于药物的作用靶酶结构发生修饰,或是降低了这 类药物与靶酶接触的机会,或是两者同时兼之。 还没有发现由于药物结构被修饰而造成耐药性的 作用机制。
第三节 抗真菌药物的作用机制与真菌耐药性 机制
抗真菌药物的作用机制
抗真菌药物的作用靶位集中在细胞表面:干扰细 胞膜的合成如唑类药物氟糠唑等和多烯大环内酯 类如两性霉素B等;
干扰细胞壁中几丁质的合成如日光霉素和多氧霉 素等;
干扰细胞壁中1,3-β-葡聚糖的合成如卡帕芬净等; 干扰细胞表面甘露糖蛋白复合物的合成如 pradimicin等 .
A:由两性霉素B在细胞膜上产生的孔道; B: 两性霉素B与细胞膜上的胆甾醇以氢键的形式结合, 从而破坏细胞膜的结构产生孔道
两性霉素B(AMB)的应用
两性霉素B适用于治疗大多数深部真菌病,如 隐球菌、假丝菌、曲霉菌、毛霉菌、球孢子 菌、膜组织胞浆菌和芽生菌等引起的各种脏 器和全身感染;也可用于治疗皮肤和粘膜真 菌病,以及使用广谱抗生素时预防并发真菌病。 两性霉素B可作为全身或深部真菌感染的首选 药物,如隐球菌脑膜炎、真菌性菌血症、真菌 性心内膜炎、肺部真菌感染、真菌性肠炎、 真菌性角膜溃病及阴道炎等。
1969年咪康唑和克霉唑(clotrimazole,局部)被 用于临床;1974年依康唑被用于临床;
1978年描述了阿莫罗芬(amorolfine);1979年 咪康唑parenreral制剂在英国上市; 1981年酮康唑口服制剂在美国得到批准上市;同 年第一个烯丙胺类药物萘替芬(naftifine)进入 临床试验; 1987年开始研究开发多烯类药物的脂质体制剂;
耐药机制的不同点
到目前为止,细菌通过改变抗菌药物的分子结构, 以使药物难以到达作用靶位并与之结合而发挥抗 菌作用的耐药性机制(这是细菌对ß-内酰胺类抗 生素、氨基糖苷类抗生素和糖肽类抗生素等药物 产生耐药性的主要作用机制),以及改变抗菌药 物作用靶位的作用机制等,对于真菌耐药性来说, 还没有被确证。 尽管有一篇报道皮肤寄生的真菌能够降解制霉菌 素,但没有足够的理由证明这是造成真菌耐药性 的主要原因。
第二节 真菌耐药性与细菌耐药性的异同点
主要不同点
一是真菌的细胞结构和生活史与细菌的具有较大 的差别,如大多数真菌具有二倍体性质和其生活 周期较长;
二是药物的作用靶位不同,如大多数抗细菌药物 的作用靶位是抑制细菌细胞壁重要组分肽聚糖的 合成,而大多数抗真菌药物的作用靶位是抑制真 菌细胞膜重要组分麦角甾醇的合成或抑制其功能 的发挥。
1)、唑类抗真菌药物的作用机制
早期的咪康唑、依康唑和酮康唑等除了抑制14α-去甲基 酶的活性外,对存在于膜上的其他一些有关的酶也具有抑 制活性:如用新近开发的voriconazole处理白念珠菌时发 现有酵母甾醇和角鲨烯(squalene)的累积,但还不清楚 造成这一结果的原因是由于voriconazole与细胞膜上的一 些与麦角甾醇合成有关的酶发生交互作用,还是由于 14α-去甲基酶的活性被抑制后产生的次级效应所致。 另外,这类药物的作用机制与作用对象有关如,氟康唑和 依曲康唑除了抑制新型隐球酵母的14α-去甲基酶的活性 外,还能够抑制将钝叶鼠曲草素酮(obtusifolione)还 原成为相应的醇(obtusifoliol),从而导致甲基化的甾 醇前体累积。
抗真菌药物的作用机制及耐药性
第一节
抗真菌药物发展简介
第一个发现并被用于临床的为上世纪30年代 末,从微生物发酵代谢产物中分离得到的灰 黄霉素;
1944年报道了唑类化合物的抗真菌作用;
1949年从微生物代谢产物中分离得到了制霉 菌素;1956年报道了两性霉素B的抗真菌活性; 1958年灰黄霉素被用于临床;同年,上市了 第一个唑类抗真菌药物;1960年两性霉素B被 用于临床; 1962年报道了氟胞嘧啶(flucytosine)的抗 真菌活性;
N N N O
S
H3C
O
Cl
H3C
N
N
O H
O Cl Cl
Cl
特康唑
噻康唑
1)、唑类抗真菌药物的作用机制
麦角甾醇作为构成真菌细胞膜的重要成分,对于维持细胞 膜的流动性、生物调节以及立体结构等起着重要的作用, 而构成细胞膜的甾醇应该是C-14位去甲基的。 已有的研究表明:唑类抗真菌药物的主要作用靶位是血红 素蛋白,该蛋白共催化抑制依赖于细胞色素P450的羊毛甾 醇(lanosterol)的14α-去甲基;而当14α-去甲基酶的活性 受到抑制,则不能合成麦角甾醇而只能累积诸如羊毛甾醇、 4,14-二甲基酵母甾醇(4,14-dimethylzymosterol)、 24-亚甲基二氢羊毛甾醇(24-methylenedihydrolanosterol) 等14α-甲基化的甾醇前体,由这样的甾醇构成的真菌细胞 膜的结构和功能都发生了变化。
Cl
N N O O N H 3C N O H O Cl Cl
N
N
酮康唑
克霉唑
N N N O O N N O H O Cl Cl
CH 3 H 3C N
N N
依曲康唑
N N N N N N OH F F
N Cl N Cl O Cl
氟康唑
依康唑
N Cl N Cl O Cl Cl
咪康唑
N Cl N
奥昔康唑
耐药机制的不同点
而已有的研究表明:抗真菌药物的作用靶位的改 变,降低药物与作用靶位的接触是真菌产生耐药 性的主要机制。 再则,从基因水平的研究来看,细菌产生高度耐 药的一个主要原因是由于带有耐药性基因的载体 的相互传递所致,如质粒、转座子和噬菌体等;
而对真菌耐药性的研究,目前只发现是由于广泛 与药物接触而产生的选择压力所致。
产生这一协同效应的原因推测为由于两性霉 素B与细胞膜上麦角甾醇的交互作用导致细胞 膜结构的改变,从而促进了5FC的吸收。
不同抗菌药物的协同作用
合并使用细菌细胞壁抑制剂ß -内酰胺类抗生素和 蛋白质抑制剂氨基糖苷类抗生素,对肠球菌也能 够产生作用机制相似的协同作用如,体外合并使 用青霉素和链霉素于粪肠球菌时,在胞内测得的 链霉素的浓度比单独使用链霉素时的要高。 但是,两性霉素B与5FC的协同作用与上面的机制 有所不同,细菌细胞壁抑制剂ß -内酰胺类抗生素 和蛋白质抑制剂氨基糖苷类抗生素的协同作用是 相继发挥的而不是同时发挥的。