高等有机化学 第五章 芳环讲解
09.09有机化学二脂环、芳环
1
CH3
3
螺[2.4]庚烷
2
5-甲基螺[2.4]庚烷
桥环——共用两个C 7 CH2–CH–CH2 1 2 CH2 CH2 8 3 5 4 CH2–CH–CH2 6 共用C称为——桥头碳
编号从一个桥头C开始,先编最长桥,经另 一桥头C,再编次长桥,最后编短桥。
桥环化合物的命名 • • • • 组成环的碳原子总数称为某烷 词头为“双环” 编号:从某一桥头C起,从大环 小环 各“桥”的碳原子数,按由大到小的次序写在 “双环”和“某烷”之间的方括号里,数字用 圆点分开。 • 环上有取代基时,取代基有最小序号(和)。
三、环己烷衍生物的构象 甲基环己烷
CH3 |
CH3
CH3 5% 大基团位于e键稳定 95%
H3C
CH2CH3
CH3
CH2CH3
本章要点
1. 环烃的命名 2.脂环烃的化学性质
3.环己烷及衍生物的构象
本章习题:p109
1,
6(2、3)
回顾4 环烷烃的命名 常规命名 • 以碳环为母体,侧链为取代基 • 母体的名称同直链烷烃,不同在于前加一“环”
多官能团取代苯命名原则
1. 主官能团有最小序号。
2. 多取代时,序号和最小。
OH | OH | CH3 | NO2
|
|
COOH
| CH3
4 - 甲基苯酚
3 – 羟基苯甲酸
2 – 硝基甲苯
OCH3
NO2
p-硝基苯甲醚 (4-硝基苯甲醚)
常见芳基
CH2—
苯基
苄基
phenyl
benzyl
6 − 2 苯的结构
2.多环芳烃
分子中含有两个和两个以上的苯环
《有机化学》(第四版)第五章 芳烃(习题答案)

第五章 芳烃 芳香性思考题P152 习题5.1 写出四甲(基)苯的构造异构体并命名。
解:CH 3CH 3CH 33CH 3CH 3CH 3H 3CCH 3CH 33H 3C1,2,3,5-四甲(基)苯1,2,3,4-四甲(基)苯1,2,4,5-四甲(基)苯P152 习题5.2 命名下列各化合物或基:解:CH 3C 2H 5CH(CH 3)2CH 2CH 2C=C3HH 3C1-甲基-2-乙基-4-异丙基苯1,2-二苯乙烷顺-2-苯基-2-丁烯(E)-2-苯基-2-丁烯CH 3CH 3CHC(CH 3)3(CH 3)3C 2,6-二甲基苯基β-苯乙基2,2,4,4-四甲基-3-苯基戊烷CH 2CH 22-苯乙基P153 习题5.3写出下列各化合物或基的结构式:(1) 异丁苯CH 2CHCH 3CH 3(2) 间甲苯基环戊烷CH 3(3) (E)-1,2-二苯乙烯C=CH PhPhH(4) 顺-5-甲基-1-苯基-2-庚烯HHCH 2CHCH 2CH 3CH 2CH 3(5) 二苯甲基C 6H 5C 6H 5 (6) 3-苯基-2-丙烯基CH 2CH=CH C 6H 5P156 习题5.4 甲苯的沸点比苯高30.5℃,而熔点低~100℃,为什么?解:甲苯的相对分子质量大于苯,分子间色散力大于苯,因比甲苯的沸点也高于苯;但苯分子的对称性好,晶格能大于甲苯,因此苯的熔点高于苯。
P161 习题5.5 写出乙苯与下列试剂作用的反应式(括号内是催化剂):(1) Cl 2(FeCl 3) (2) 混酸 (3) 正丁醇(BF 3) (4) 丙烯(无水AlCl 3) (5) 丙酸酐(CH 3CH 2CO)2O(无水AlCl 3) (6) 丙酰氯CH 3CH 2COCl(无水AlCl 3) 解:(1)CH 3CH 2CH 3CH 2ClClCH 2CH 3Cl FeCl 3+(2) 混酸CH 3CH 2CH 3CH 2NO 2+NO 2CH 2CH 3(3)3323C 2H 5CHCH 2CH 33CH 2CH 3+CHCH 2CH 3CH 2CH 33(4)233+CH(CH 3)2CH 2CH 3CH 2CH 3CH 2CH 3CH(CH 3)2(5)3223+COCH 2CH 3CH 2CH 3CH 2CH 3CH 2CH 3CCH 2CH 3O(6)323CH 2CH 3+COCH 2CH 3CH 2CH 3CH 2CH 3CCH 2CH 3OP161习题5.6 由苯和必要的原料合成下列化合物:(1)解:HO 24+(或环己烯)(2) 叔丁苯解:CCH 3CH 3CH 3+ CH 2CH 3CH 3H 2SO 4(3) CH 2(CH 2)5CH 3解:HCl+2)5CH 3OCH 3(CH 2)5C OCH 2(CH 2)5CH 3(4)CH 2CH 2OOH O解:OOO+3C CH 2CH 2C OH OOP164 习题5.7 试以苯和必要的原料合成正丙苯。
5第五章芳环上的取代反应

H H C H
C C
C C
H C H H
芳环上离域的π电子的作用,易于发生亲电取代反 应,进攻试剂为正离子或偶极及诱导偶极的正的一端, 而离去的原子团不带成键电子对。 只有当芳环上引入了强吸电子基团,才能发生亲核 取代反应。在亲核取代反应中,进攻试剂是负离子或具 有未共用电子对的原子或基团,离去基团以弱碱负离子 或分子的形式离去,其实用价值不如亲电取代反应。
% %p %m o 相 对 速 率 3 7 3 0 1 7 6 6 9 2 2 9
2) 空间效应越大,对位产物越多:
C(CH3)3 H2SO4 C(CH3)3
100%
SO3H
极化效应:
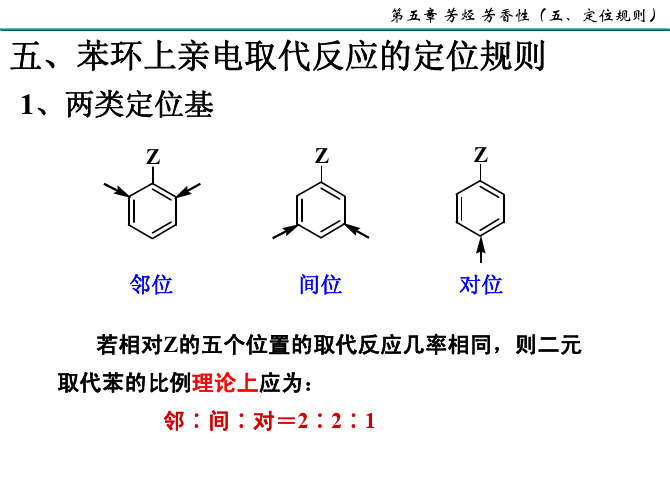
X
X o% p% F 12 88 Cl 30 69 I 32 60
m% 0 1 8
X 具有-I效应,使邻位的电子云密度降低。 F Cl Br I
一. 亲电取代反应
(一) 加成-消除机理
HE
+ E N u
E
σ-络合物 芳正离子
H N O 3
H S O 2 4
N O 2
+ + 2 H S O N O H N O O H S O 2 2 4 3 H 3 4 2
+ N O 2
N O 2 芳正离子生成的 H 一步是决定反应
速率的一步
卤代反应:
e B r r B rF B r + F e B r 3 2 3 B
溴分子在FeBr3的作用下发生极化
R e B r B rB rF R B r + + F e B r B r 3 H
δ
δ
生成芳正离子
R
B r H
R
B r + H
有机化学第五章1讲解
室温 AgCl 立即
AgCl(立即)
加热
AgNO3 AgCl (稍慢) AgCl 稍慢 EtOH
AgCl AgCl
2019/5/10
1.4.2 消除反应
β
α
2019/5/10
2019/5/10
查依采夫规则
2019/5/10
2019/5/10
1.4.3 与金属反应
① 与镁反应
RX + Mg
无水乙醚
④ 与氨作用
卤代烷与氨作用,卤原子被氨基取代生成伯胺
RX + 2 NH3 RNH2 RX RNH2 + NH4X R2NH RX R3N RX R4N X
因为生成的伯胺仍是一个亲核试剂,它可以继 续与卤代烷作用,生成仲胺或叔胺的混合物,故反 应要在过量氨(胺)的存在下进行
2019/5/10
⑤ 与硝酸银作用 (用于鉴别卤代烷烃)
0.194
C—I 218
0.214
故C—X 键比C—H键容易断裂而发生各种化学 反应。
2019/5/10
1.3 卤代烷烃的物理性质
① 沸点:M↑,b.p↑
碳原子数相同的卤代烷:RI>RBr >RCl 支链↑, b.p↓
② 相对密度:一氯代烷<1;一溴代烷和一碘
代烷>1,同系列中,卤代烷的相对密度随 碳原子数的↑而↓ ③ 可燃性:随X原子数目的↑而↓ ④ 不少卤烷带有香味,但卤烷蒸汽有毒,特别 是碘烷,应防止吸入
Corey-House 反应
2019/5/10
反应机理的实验证据:
1.5.1 亲核取代反应历程
以溴甲烷和叔丁基溴水解为例
在80%乙醇 溶液中水解速度: 加入OH- 后: 固定[RX],改变[OH-]: 反应速率方程:
华南理工大学有机化学第5章-2
30%
69%
1%
第五章 芳烃 芳香性(五、定位规则)
(2) 第二类定位基(间位定位基)
对苯环有吸电子作用,使苯环钝化,对后引入基团进入苯环 起着间位定位作用的原子或基团,称为间位定位基。 第二类定位基和定位能力 : -CF3 ―N (CH3)3+ > ―NO2 > ―CN > ―SO3H > ―CHO > -COCH3 > ―COOH > ―COOR > ―CONH2 >―NH3+等. 符合下列条件之一的为间位定位基: 1、与苯环直接相连的原子不饱和的基团或原子(-CX3例外)。 2、与苯环直接相连的原子带有正电荷的基团。 3、对苯环有吸电子性能,使苯环钝化的原子或基团。
+
σ
.... 配合 物 . ..+ ..... I
H E
[
H E
NO2
+
Ib
---Ic
+
H E
]
NO2
NO2
NO2
+
NO2
σ
配合物
....... . .+. H E
Ⅱ
[
+
]
H E
Ⅱa
H E
b
---Ⅱ
H E
H E
Ⅱc
NO2
NO2 H E
NO2 H E
+
NO2
+
σ
配合 物
...... . ...+ ..
NHCOCH3
δ
δ
Cl , Br , I δ 烷基C-Hσ键与苯环存在 σ,π-共轭。
H δ H C H δ
δ
第五章 芳烃 芳香性(五、定位规则)
有机化学第五章芳烃PPT课件

甲苯
乙苯
异丙苯
(2)当苯与烯、炔相连时,习惯上把苯作取代基,不饱和
烃作母体。
-CH=CH2
-C≡CH
苯乙烯(或乙烯第苯1)1页/共114页
苯乙炔
(3) 若烃基的碳链较长或烃链上含有多个苯环时,一般把苯 作取 代基,烃作母体。
CH3 CH3
CH3CH2CH2-CH—CH-CH2
C
2,3-二甲基-1-苯基己烷
的分子,它的真实结构可以用几个经典结构式共同来表示。 这些参与了结构组成的经典结构叫做共振结构式,也叫极
限结构式。
第19页/共114页
+ - ……
O OC
O
O OC
O
OC O O
(二) 共振论的规则 共振结构式的本质是用价键结构式来反映共轭体
系中P电子离域的范围,即用多个合理的价键结构 式来描述电子的瞬间运动状态。
D. 在满足八电子体的共振结构式中,有电荷分离, 负电荷应在电负性较大的原子上。
O
CH3—C—CH2
O-
CH3—C CH2
第24页/共114页
稳定
E. 两个带有相同电荷的原子相隔越近、两个带有相 反电荷的原子 相隔越远,共振式能量高,不稳定, 对杂化体的贡献小
H E
+
+O N
不稳定
O-
三、共振论的优点和缺点 总结:共振论认为共振是一种稳定因素,参与共振 的共振式越多,杂化体越稳定;2)通常以能量最低, 稳定性最高的共振结构为标准,其真实分子即共振 杂化体所降低的能量叫共振能。
芳烃—— 芳香族碳氢化合物
(1)碳氢比高 如:
C : H = 1 : 1 C : H = 10 : 8
沈阳药科大学高等有机化学胡春—— 芳香性

芳香性的定义
1825年法拉第从鲸油裂解产生的气体冷凝液中 发现了苯,接着测定了苯的组成、蒸汽压、熔点、 密度等物理性质,1845年霍夫曼从煤焦油中也提取 出苯这种物质,并发现它非常稳定。1865年德国化 学家凯库勒从苯的分子式C6H6出发,根据苯的一元 取代物只有一种,推断出苯的环状结构式。实验证 明苯类化合物具有特殊的热稳定性,难发生加成和 氧化反应,易发生亲电取代反应,且保持碳环的结 构不变。人们把这种特性称为芳香性。
关于芳香性定义的讨论
(3)从化学性质角度。这是最原始也是最 直观的表现形式,但也是现在用的最少的一 个定义方式,因为化学反应需要涉及许多动 力性质,即与分子的非基态有关,则情况变 得更为复杂。而且在最新制的或理论上推出 的许多芳香化合物,很多都不能进行经典的 芳香族化合物的反应,许多甚至化学性质非 常不稳定。故而,化学性质上的定义已经逐 步被舍弃。
关于芳香性定义的讨论
(4)从分子的磁学形式上比较。这也是现在应用比 较普遍的,被认为最有前景的一个方法。众所周知, 传统芳香性化合物(如苯等)由于π电子的环形离域会 产生抗磁环流,并且可以很方便地从1H NMR 及 13C NMR 谱上得出结论。相似的抗磁环流也被证 明存在于其他芳香化合物中。Schleyer 等人甚至提 出将抗磁系数的上升(diamagnetic susceptibility exaltation)作为唯一可量化的芳香性的标准 。1996 年,Schleyer 又提出将与核无关的化学位移(NICS, nucleus-independent chemical shifts)作为芳香性的 标准。关于NICS 的讨论也正在不断深人中,仍然 有一些问题有待解决。
芳香性的定义
随着量子力学在化学中的应用,进一步从结构上揭开了芳 香性的秘密。在芳香烃分子的芳环上,每个碳原子都以SP2杂 化轨道相互交盖,构成环状平面,处在同一个环平面上的每个 碳原子还剩下一个未杂化的P轨道,这些P轨道轴相互平行,于 是相互间发生交盖重叠,形成“芳香大π键”,所有的π电子 成为环绕整个环平面运动的电子流,完全失去了它们的定域性 。这种芳香大π键又称为非定域键(离域键)。由于π电子离 域的结果,导致体系能量降低,热稳定性增加,键长趋于平均 化,芳环上所有碳碳键都完全相同,键长也完全相等( 0.139nm),它们既不是一般的碳碳单键(0.154nm),也不是 一般的碳碳双键(0.133nm),而是每个碳碳键都具有这种闭 合大π键的特殊性质,在核磁共振光谱上表现出较大的化学位 移(δ值)。目前人们将芳香性跟分子能量较低这个性质联系 起来。人们一直将芳香性化合物所具有特殊的结构稳定性归结 于电子的离域作用,但这种离域作用并不能使共轭多烯有明显 的芳香性。
精品课件!《高等有机化学》_芳环上的亲电和亲核取代反应
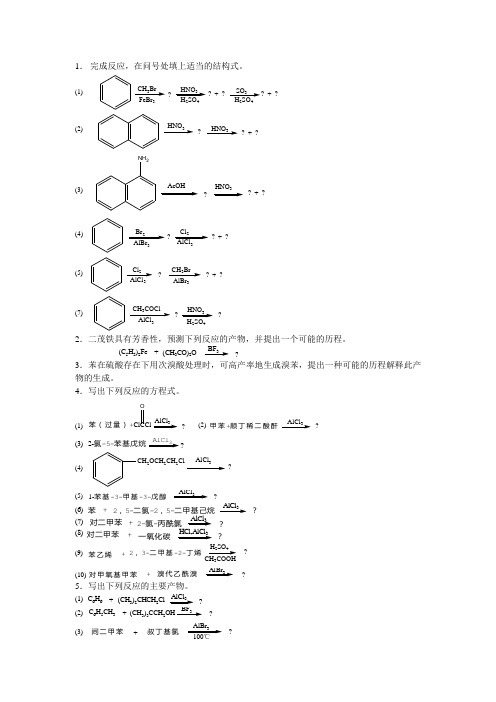
1. 完成反应,在问号处填上适当的结构式。
(1)CH 3Br 3?HNO 324? + ?SO 324HNO 3?? + ?HNO 3(2)NH 2?HNO ? + ?(3)Br 23Cl 23?? + ?(4)(5)Cl 23CH 3Br 3?? + ?(7)CH 3COCl3HNO 324??2.二茂铁具有芳香性,预测下列反应的产物,并提出一个可能的历程。
(C 5H 5)2Fe +(CH 3CO)2OBF 3?3.苯在硫酸存在下用次溴酸处理时,可高产率地生成溴苯,提出一种可能的历程解释此产物的生成。
4.写出下列反应的方程式。
苯(过量)+ClCClOAlCl 3?(1)(2)甲苯+顺丁稀二酸酐AlCl 3?(3) 2-氯-5-苯基戊烷AlCl 3?(4)CH 2OCH 2CH 2ClAlCl ?(5)1-苯基-3-甲基-3-戊醇AlCl 3?(6)苯+2,5-二氯-2,5-二甲基己烷AlCl 3?+AlCl 32-氯-丙酰氯对二甲苯?(7) +HCl,AlCl 3对二甲苯一氧化碳?(8)?+243苯乙烯2,3-二甲基-2-丁烯(9)?+AlBr 3对甲氧基甲苯溴代乙酰溴(10)5.写出下列反应的主要产物。
(1)C 6H 6(CH 3)2CHCH 2Cl AlCl 3?+ (2)C 6H 5CH 3(CH 3)3CCH 2BF 3?+ (3)间二甲苯叔丁基氯AlBr 3?+6.把下列各组化合物按指定性能排序,并解释原因。
酸性:(1)OHCN OHNO2OHNH2OH(2)COOH NO2COOH BrCOOHSO3H(3)COCH2COCH3COCH2COCF3(4)OH OHCH3OHCH3H3CNO2OHNO2O2N碱性:(1)NH2NHCOCH3NH2NO2OONH(2)NH2CH3CH2NH2NH2NO2NO2NH2NO2(3)HNHN H NOO7.完成下列反应。
(1)CH 3+(CH 3)2CHCH 2ClAlCl 3H 2SO 4??(2)??H 2SO 4HNO 324(3)???HNO 324KMnO 4Br 2Fe(4)+??AlCl 3HNO 3H 2SO 4COClO 2N(5)??ClCH 2ClMg PhCH 2Cl乙醚(6)+???H3CCl 2H 2光照(7)+???CH 3CHCHCH 2ClHBrBr 2HO∆NaO(8)+?O 2V 2O 53稀H 2SO 4??(9)????OOONH3OH -Cl 2,NaOHNaNO 2Cu 2Cl 2(10)??°H 3CNH 2NaNO 2,HCl 2(11)?+CH 3ONaCH 3OHClClNO 28.由所给原料合成产物。
高等有机化学 周环反应

对旋:围绕着两个键分别向不同的方向旋转
• 许多类似的反应结果都显示出如下的规律:
• 根据环化时参与反应的电子数是4n或4n+2(n=1 , 2,… 正整数),以及反应的条件是加热或光照,决定该 线性共轭多烯的环合或其逆过程是按顺旋还是对旋方式发 生,这些反应的立体化学结果是一定的,而且可以预测。 电环化反应的立体选择规律见表。
异构体稳定,但在最终产物中常常是内型(endo)占优势。
开链二烯和环状的亲二烯的加成反应经常是遵循 endo 规则的。Alder 内向规则也有例外。
例如,马来酸酐和环 戊二烯加成,产物几 乎全部是endo 异构 体,而热力学更稳定 的exo 产物产率还不 到2%。
(2)顺式规则 顺式规则:Diels 一Alder 反应具有立体专一性,反应产物 保留了双烯和亲双烯的相对立体化学。也就是说,含反式取 代基的亲二烯会得到取代基呈反式构型的加成产物,而含顺 式取代基的亲二烯会得到取代基呈顺式构型的产物.
苯酚的烯丙基醚的重排是Claisen 重排最重要的例子。 • 当烯丙基芳基醚的两个邻位未被取代基占满时,重排主要
得到邻位产物。若两个邻位都被占据,那么烯丙基可以迁 移到对位上。反应是分步进行的,烯丙基先通过Claisen 重排迁移至邻位 ,尔后再经过一次 [3 , 3 ]-迁移 (Cope 重排)到对位,然后经互变异构得到对位烯丙基 酚。对位、邻位均被占满时不发生此类重排反应。
• Claisen 重排是个协同反应,中间经过一个环状过渡态, 所以芳环上取代基的电子效应对重排无影响。二烯酮中间 体可用亲二烯体顺丁烯二酸酐进行Diels 一Alder 反应捕 获。交叉实验证明这一重排是分子内反应而不是分子间反 应。
• 取代的烯丙基芳基醚重排时,无论原来的烯丙基双键是E构型还是Z -构型,重排后新形成的双键构型都是E ,这 是因为重排反应所经过的六元环状过渡态具有稳定椅式构 象。
高等有机—复习总结

立体选择性反应(Stereoselective reactions):
对于某一反应,产物可能是几种立体异构体,但凡 只产生其中一种为主的反应,叫立体选择反应。
立体专一反应(Stereospecitive reaction):
凡互为立体异构体的反应生成不同的立体异构体产物, 这种反应称为立体专一反应.
7) 卡宾的结构 单线态与三线态
8) 卡宾的形成与反应 立体专一性反应
9) 乃春的结构 单线态与三线态
11) 苯炔的结构
10)乃春的形成与反应 立体专一性反应
12)苯炔的形成与反应
非经典正碳离子
写出下面反应的转变历程
第4章 饱和碳原子上的亲核取代反应
➢ SN1反应机理 ➢ 离子对理论 ➢ SN2反应机理 ➢ 邻基参与历程 ➢ SNi反应机理 ➢ 影响亲核取代反应的因素
Question: To which pair will nitronium-salt nitration show greater intermolecular selectivity? (JACS, 1974, 96, 549)
CH3
vs
or
vs NO2
CH3 NO2
Question: 试比较下列底物氯原子的反应活性
第1章 化学成键与分子结构
➢ 共振理论 ➢ 影响分子中电荷分布的因数 ➢ 分子轨道理论(MO) ➢ 芳香性、非芳香性、反芳香性和同芳香性
1.1 共振理论(resonance theory 鲍林提出)
1) 书写共振式时,只允许电子移动,而原子核的位置不动;
2) 共振结构必须符合价键的规则
3) 共价键数目越多的共振式越稳定,而电荷分离的共 振式稳定性降低 ; 4) 在共振式中,负电荷落在电负性较强的原子上较在电 负性较弱的原子上稳定性高;
高等有机化学

如今: 一个高年级本科生大概1天
★ 高效低毒农药、动植物生长调节剂和昆虫信 息物质的研究和开发,为农业的发展提供了重 要的保证。
➢性信息素具有强烈的生理作用。一只雌蚕蛾交配前在 其尾部每秒钟释放出毫微克量的信息素,顺风扩散可 引诱几个km外的雄蚕蛾逆风飞翔到雌蚕蛾。由于检测 仪器的进步,50年代需几十万只、60年代需几万只, 而80年代后则需10只或更少就能准确确定性信息素的 结构。即便样品量很少(< 100g)也能获得确切的结 构信息。
药学 医学
有机化学对于社会进步以及其它学科的发展的贡献也 是巨大的:
例如:
★ 在对重要的天然产物和生命基础物质的研究中,有机化 学取得了丰硕成果。维生素、抗生素、甾体和萜类化合物、 生物碱、碳水化合物、肽、核苷等的发现、结构测定和合 成,为学科本身的发展增添了丰富的内容,为人类的医药 卫生事业提供了有效的武器。
高等有机化学的研究内容与目的
高等有机化学是有机化学的核心部分(core)
高等 有机化学
分子结构的 基本概念
含碳化合物的 反应性
化合物 中间体
结构
反应过程中的结构变化 反应机理
揭示反应的本质、内在规律,把有机反应有机地 联系起来。
第一章 第二章 第三章 第四章 第五章 第六章 第七章 第八章
目录
化学键和分子结构理论
9. 魏荣宝主编 高等有机化学 高等教育出版社
第一章 绪论
一、有机化学
来源:
☆1784: T. Bergman 首次明确定义有机化学 Organic chemistry is the chemistry of carbon compounds
☆ 1808: 瑞典Berzelius首次使用organic chemistry
高等有机化学

• 目前对于反应历程的研究,虽然发展很快, 但绝大部分是属于均相反应,而非均相反应 历程的研究,无论是从广度或深度看,其理 论远远落后于实际的需要。因此,这方面的 研究是目前极待加强的工作。
杂化
2s2 2px12py12pz0
2s1 2px12py12pz1
sp3
基态
激发态
109.5o
H
H CH H
碳原子的sp2杂化轨道
乙烯 CH2=CH2 的结构
激发
杂化
2s2 2px12py12pz0 基态
2s1 2px12py12pz1 激发态
sp2
2pz1
sp2
p
HC H
CH H
HC H
CH H
HC H
CH H
碳原子的sp杂化轨道
激发
杂化
2s2 2px12py12pz0
2s1 2px12py12pz1
sp
基态
激发态
2py12pz1
sp
HCCH
苯的结构:
杂化轨道理论的解释:
苯分子中12个原子共面,其中六个碳原子均采取sp2 杂化,每个碳原子上还剩下一个与σ平面⊥的p轨道, 相互之间以肩并肩重叠形成π66大π键。
• 元素的电负性在同周期中随族数的增大而 递增,在同族中随周期数增大而递减,即愈 是周期表右上角的元素电负性愈大,-I效 应也愈强。
例如: -I效应:-F>-OH>-NH2>-CH3 -F>-Cl>-Br>-I -I效应: -N+R3>-NR2 +I效应: -O->-OR
《高等有机化学》课件-第五章 周环反应

H2C CHCH2CH2CH O
Chairlike TS
联苯芳胺重排反应
烯(ene)反应
The Fischer indole synthesis (in 1883 by Emil Fischer)
完成以下反应:
OH
OCH2CH CHCH3
H Me
Me
D Et
OCH2CH CHCH2CH3
H3C
同面与异面
同面与异面
同面与异面
Diels-Alder Reaction [4π + 2π]
D
D D
D diene
HD HD dienophile
DD D
D DD
DD D
D DD
not observed
• The diene must adopt S-cis conformation
• Syn addition (同位素标记实验)
• Face to face orientation
同面与异面
环加成立体选择规则
环加成立体选择规则
Alder rule
The endo product is usually favored by kinetic control due to secondary orbital interactions.
5.1 电 环 化 反 应
顺旋与对旋
顺旋与对旋
顺旋与对旋
电环化立体选择规则
从天然植物中分离得到的光甾醇,放置一段时间后 发现纯度下降,分析后确定里面出现了麦角甾醇、 焦钙化甾醇、异焦钙化甾醇。解释原因。
CH3
H h CH3 对
H
CH3 H
H H3C
H
H3C CH3
大学有机化学重点知识总结第五章 芳烃 芳香性汇总

COOR > OH > Cl >
COCl > C C
CONH2 > >
NH2 > NO2
COOH
C
C >
1
对-甲酰基苯甲酸
SO 3H
CHO
NH2
COOH
对氨基苯磺酸 1
HO NO2
3-硝基-5-羟基苯甲酸
COOH > C N C C
NH2 Cl OCH3
SO3H > CHO > C C >
COOR > C O > OR >
命名原则: 1. 简单的芳烃以苯环为母体, 称“某苯”
CH3
甲苯 异丙苯
Cl
氯苯
CH3 CHCH3
NO2
硝基苯
2. 当苯环上连有两个以上取代基时,需标明取 代基位次或取代基间的相对位置
二取代苯的3个异构体:
CH3 CH3
CH3 CH3
CH3
CH3
1,2–二甲苯 邻二甲苯 o–
1,3–二甲苯 间二甲苯 m–
CH3 CH CH2 CH3 CHCH2 Cl CH3
H2 SO4 Δ AlCl3 Δ
如何得到直链烷基苯?
*④ 苯环上连有强吸电子基(如–NO2、 -SO3H、-COOH、-COR等取代基)时, 烷基化反应不发生。
NO 2 + CH3CH2Cl
X
不反应 (因为N和O的孤对电子 与AlCl3配合使AlCl3失效)
+ RX AlCl3 R + HX
① 常用催化剂:AlCl3、FeCl3、ZnCl2、 (HF、BF3、H2SO4)
② 常用烷基化试剂:卤代烃、烯烃、 醇、 环醚
有机化学第5章 芳烃

CHO
OCH3 OH
官能团优先次序:
CHO > OH >
4–羟基–3–甲氧基苯甲醛
OCH3
CH3CH2CCH2CHCHO O Cl
4–氧代–2–氯己醛
CH3CHCH CH2 OH
3–丁烯–2–醇
二、苯的结构
第五章 芳烃 芳香性(二、苯的结构)
由元素分析,分子量测定,苯的分子式为:C6H6 易取代,不易加成,不能使溴水和高锰酸钾溶液褪色, 不易氧化。 一取代物只有一种 邻二取代物只有一种,说明具有环状对称结构
H + Br2
Fe
△
(75%)
反应活性:F2 > Cl2 > Br2 > I2
鉴别芳烃。
Br + HBr
苯的溴化反应机理:
第五章 芳烃 芳香性(四、单环芳烃化学性质)
Br Br + FeBr3 + Br 慢
Br Br FeBr3 H Br
Br + Br FeBr3
H Br
H Br
π电子的离域产生共振杂化体:
酰基化反应
CH2Cl
氯甲基化反应
第五章 芳烃 芳香性(四、单环芳烃化学性质)
(1) 卤化(halogenation)
在卤化铁等路易斯酸作用下,苯与卤素作用生成卤化苯的 反应称作卤代反应或卤化反应。
+ X2 Fe or FeX3
X ( X = Cl, Br)
催化剂通常使用的是Lewis 酸: FeCl3, FeBr3 和 AlCl3、Fe
H3C
CH3
1,3,5–三甲苯 均三甲苯
symtrimethylbenzene
CH2CH3 1 2 CH2CH2CH3
【优】高等有机化学芳环亲核取代PPT资料

典型加成-消除历程反应:酚醛树脂的制备
NaOH
当温度超过400C,主要 发生: a.羟甲基相互反应脱 水,生成亚甲基醚。
b.羟甲基基团与苯酚 或多羟基苯酚的活泼H 发生脱水反应,生成 亚甲基基团连接酚基 的结构。
2、消除一加成历程
亲核试剂条件:a.很强的碱性 b.具有亲核性
反应物在亲核试剂作用下消除
二、影响芳环上亲核取代反应的因素
1、反应物的结构对活性 的影响
2、离去基团的影响 3、亲核试剂的影响
1、反应物的结构对活性的影响
芳
环
上
的
亲 核
卤苯主要是消除-加
取
成反应,而它是加
代
成-消除反应。
(1)按加成-消除反应历程进行的亲核取代反应
a、离去基的邻对位有吸电子基时,加速亲核取代反应; 反之反应受阻。
(率2减)小按。消除-加成反应历程进行的亲核取代反应
代基与离去基团的相互位置 试结剂构进 有入关苯。环的位置取决于非离去基的性质以及取
a按、这离种去历基程的进邻行对的位很有少吸,电即子使基是时非,常加活速泼亲的核芳取基代卤反化应物; ,也尚未观察到肯定是SNAr1历程进行 a、对于一取代苯,取代基是离去基时,亲核试剂进入 典型加成-消除历程反应:酚醛树脂的制备 反之反应受阻。
b、对于二取代苯,其中一个取代基是离去基时,亲核 典试型剂加 进成入-苯消环除的历位程置反取应决:于酚非醛离树去脂基的的制性备质以及取
按这种历程进行的很少,即使是非常活泼的芳基卤化物,也尚未观察到肯定是SNAr1历程进行
试剂进入苯环的位置取决于非离去基的性质以及取 进(攻2)试按剂消和除反-加应成物反形应成历一程个进中行间的体亲核取代反离应去基团离去形成稳定产物
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
2019/6/4
26
• 取代基中与苯环相连接的原子带有孤电子
对。
Z
由于Z有未公用电子对,通过共轭作用可以 把电子部分地转移到环中,使得环中的电子
云密度增大,有利于亲电试剂的进攻,生成
σ络合物后,正电荷可以转到Z上。
2019/6/4
27
上面结论还可以用共轭效应来分析定位问题: Z
Óë Y+ ¿É ÒÔ ÐÎ ³É ÒÔ Ï s Âç ºÏ Îï
2019/6/4
28
2019/6/4
12
13
29
☛ o-,p-比m-多一个Lewis 结构式(12或13), 且12和13对共振体的贡献又最大(除H外的 每一个原子都是8隅体),因为o-和p-比m中间体要稳定得多。
☛ 因此这样的Z是邻位、对位定位基。 ☛ 结论:在与环相连的原子上有未公用电子
对的基团是致活基团,且是邻、对位定位基。
2019/6/4
34
这是因为: ① 量子力学计算σ络合物的正电荷在各个C点
上的分布如下:
HH
0.25
0.25
0.1
0.1
0.3
2019/6/4
35
② 邻位有位阻效应,特别是对于大的基团这 个效应更加明显。如:
Br HNO3
Br +
Br NO2
NO2
90% 以上
2019/6/4
36
(3)Orientation in Benzene Rings with More than One Substituent
L
slowly
Y
L
L
Y
Y
1
L Y
fast 2
Y + L+ 3
2019/6/4
9
反应过程生成芳离子中间体,这一正离子可 以写1的共振杂化体形式,也可以用2的离域 式来描述。2称做σ络合物。
对于进攻试剂是偶极离子,中间体按下式 生成:
2019/6/4
10
L
δ+ δ-
+Y Z
L Y + Z-
生成的正离子非常活泼,它可以失去L或 失去Y,变成稳定的芳香结构。若失Y,则 变回原来的原料,实际上未发生反应;若 失去X,发生了Y对X的取代反应。关于此 正离子机理有以下的证据:
Me
H Me
H Me SbF6-
Me
σ络合物
2019/6/4
13
关于σ络合物的生成步骤,有人认为首先 生成π络合物,然后在转换σ络合物。
+ Y+
Y+
p Âç ºÏ Îï Ò² ³Æ ´« ºÉ Âç ºÏ Îï
H +Y
s Âç ºÏ Îï
2019/6/4
Y + H+
²ú Îï
14
已经发现了稳定的π络合物溶液:
第五章 芳环上的取代反应
2019/6/4
1
HC HC
C
H
H C
CH C
H
2019/6/4
2
脂肪族碳上的取代反应大多数是亲核的。
芳香环上的取代反应,则相反。
因为芳环上电子云密度大,它吸引正离子 或偶极分子的正端。
因此进攻试剂是带正电的,而离去基也是 不带走电子对的正离子,即离去基是酸。
以两个基团为例,分以下几种情况讨论
① 两个基团互相加强的,很好预测:
CH3
COOH
CH3 Cl
2019/6/4
37
② 彼此相反,预言困难: 两个基团的定位能大致相等。
NHCOCH3 OCH3
ËÄ Ö ²ú Îï ´ó Ö Ïà µÈ
2019/6/4
38
强活化基 + 较弱活化基(or 钝化基)前 者是控制产物。 间位定位基和o-,p-定位基成间位关系时, 主要进入间位基的邻位(简称邻位效应) 原因尚不太清楚。
fm=
kPhZ/2 × kPhH/6
m
异构体 100
fp= kkPPhhHZ//16×
p 异构体
100
当 f > 1 时,该位置的活泼性比苯大,否
则比苯小。
2019/6/4
46
例如: ☛ 在硝酸与乙酸酐的体系中甲苯的硝化速 度是苯进行硝化反应的23倍。 取代产物的百分比为:
邻
对
间
63%
34%
3%
2019/6/4
21
ZH
邻位
Y
+
3
Z
¶Ô λ
+
YH
6
Z
+
¼ä λ
H
Y
9
2019/6/4
ZH +Y
4
Z
+
YH 7 Z
H +Y 10
ZH Y
+
5
Z
+
YH 8 Z
+
H Y 11
ZH +Y
Z
+
YH
Z
+
H
Y
22
• 若Z是+I基团,中间的电荷可以分配到Z 上去,因此它使得环上的正电荷减少,中 间稳定性增大,即Z是致活基团。
脂肪族亲电取代反应中的离去基是弱碱。
2019/6/4
3
芳环上离域的π电子的作用,易于发生 亲电取代反应,只有当芳环上引入了强吸 电子基团,才能发生亲核取代反应。
2019/6/4
4
芳环 亲电取代反应
2019/6/4
5
1. Mechanism
(1)反应机理
芳香族亲电取代反应,大多数以1种机理 进行,这种机理称为芳正离子机理,在 这个机理中,反应分2步进行:
2019/6/4
17
但是当环上以经有一个取代基Z (e.g. CH3)。
Z
那么就产生了两个问题:
2019/6/4
18
① 此化合物是比苯活泼,还是比苯不活泼? 若是比苯活泼,则Z使苯环活化,若不活 泼则Z使苯环钝化。
② 新进入的基团,可以进入Z的o-,p-,m-, 究竟哪一种为主?这个问题取决于Z的性 质。
邻位σ络合物的Lewis结构式4,和对位σ 络合物的Lewis结构式8的带正电荷原子 直接与Z相连结论:+I基团是致活基团, 且是邻对位定位基。
2019/6/4
23
间位σ络合物则没有这种情况,因此邻o-、 p-对σ络合物比m- 位稳定,因此主要生成o-, p- 取代产物。 结论:+I基团是致活基团,是邻对位定位 基。
CH3 e.g
2019/6/4
41
(2)杂环化合物
2019/6/4
42
N
N
☛ 比苯环的活性差
☛ 喹啉反应一般发生在苯环,而不是吡啶 环,反应比苯苯慢,比吡啶快。
2019/6/4
43
(5)取代基的定量关系
取代基效应与化学活性之间存在一定的 定量关系。 ① 分速度因数与选择性
从定量关系上考虑邻、对、间位取代 难易程度。
2019/6/4
24
• 如果芳环相连的原子为不带孤电子对的I基团。它使整个苯环的电子云密度降低, 形成σ络合物后,Z的-I效应使得环上的正 电荷更加集中,不稳定,反应速度减慢, Z是致钝基团。
2019/6/4
25
o-和p-中间体都有一个正则式其带正电 荷核与Z相邻,而m-则没有,因此生成 m-中间体比o-对- 更稳定,因此取代主要 发生在间位上。 结论:不带孤电子对的-I基团,使苯环钝 化,且是间位定位基。
2019/6/4
11
① 同位素效应,若按SE1机理,则有同位素 效应,即KD小于KH。但实际上,大多数情 况下,没有同位素效应,说明反应速度决 定步骤不是C—H的断裂。
② 正离子中间体的分离,下面中间体已经分 离出来了:
2019/6/4
12
H
Me
Me
SO2(liquid)
+ HF + SbF5
-78℃
2019/6/4
53
3. Some Examples of Reaction.
(1)Nitrogen Electrophiles • 硝化反应
通式
Ar H HNO3 / H2SO4 Ar NO2
ρ 表示了取代基对反应速率的影响。
2019/6/4
52
σ>0, 取代基为吸电子基团; σ< 0, 取代基为供电子基团。
当ρ> 0时, 吸电子基团加速反应; 当ρ <0时,供电子基团加速反应; 当ρ= 0时,取代基对反应影响不大。
根据σ值,可以预测取代基性质; 根据ρ值,可以预测反应机理。
2019/6/4
44
分速度因数 (f) = (6) (k取代)(z产物的百分比) y (k苯)
y-取代位置的数目
通过每一个位置取代苯的活性与苯比较, 把总的速率乘以邻位、间位或对位产物的百 分比,再除以苯的取代速率的结果。
2019/6/4
45
fo=
kPhZ/2 kPhH/6
×
o
异构体 100
2019/6/4
32
• 卤素。卤素的诱导效应和共轭效应相反, 前者使苯环致钝,后者致活,总效应是钝 化。但是卤素是邻对位定位基,这是因为 生成中间体后,C效应是起主导作用的。
2019/6/4
33
(2)Ortho/Pare Ratio
在前面的讨论中,已经提到邻对位定位基, 现在的问题是:若取代基是邻对位定位基, 那么o,p的活性是否相等呢?如果是相等, 那么 o/p=2,但事实上o/p <2。为什么?