已知可沥滤物测定方法验证及确认
已知可沥滤物测定方法验证及确认

已知可沥滤物测定方法验证及确认技术审查指导原则一、前言医疗器械的可沥滤物(Leachables)是指在临床使用过程中,医疗器械与水或使用中有关的液体、气体作用等介质作用时,从该医疗器械中释放出的化学物质的统称。
可沥滤物一般包括灭菌残留剂、工艺残留物、降解产物以及材料中的单体及添加剂(包括稳定剂、抗氧化剂、增塑剂、着色剂等)。
在医疗器械产品与人体接触并发挥作用的过程中,可沥滤物也在或短期或长期地对人体产生安全性方面的危害。
可沥滤物安全性评价首要任务是建立拟研究物质的允许限量(Allowable limit),其次,应在临床模拟最坏使用环境下测定其释放量(Release),并根据其释放量是否超过其在该产品该预期用途下的允许限量,形成完整的可沥滤物安全性研究报告,其中,可沥滤物的释放量测定方法的设计和方法学验证是评价可沥滤物安全性研究报告质量和可靠性的重要依据。
本指导原则是对医疗器械已知可沥滤物测定方法研究的一般要求,申请者应依据具体产品的特性和拟研究可沥滤物性质对注册申报资料的内容进行充实和细化。
申请者还应依据具体可沥滤物的特性和分析方法确定释放量测定方法的设计和方法学验证参数的具体内容是否适用,若不适用,需具体阐述其理由及相应的科学依据。
本指导原则旨在帮助和指导申请者对医疗器械产品注册申报资料进行准备,以满足技术审评的基本要求。
同时有助于审评机构对该类产品进行科学规范的审评,提高审评工作的质量和效率。
本指导原则是对申请者和审查人员的指导性文件, 但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其它方法,也可以采用,但是需要提供详细的研究资料和验证资料。
应在遵循相关法规的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将适时的进行调整。
二、适用范围本指导原则适用于医疗器械注册申报时对已知可沥滤物释放量的研究和产品技术审评的参考。
最新一次性使用血液灌流器注册技术审查指导原则

一次性使用血液灌流器注册技术审查指导原则(征求意见稿)本指导原则旨在指导注册申请人对一次性使用血液灌流器(以下简称灌流器)注册申报资料的准备及撰写,同时也为药品监督管理部门对注册申报资料的技术审评提供技术参考。
本指导原则系对灌流器产品注册申报资料的一般要求,申请人应依据产品的具体特性对注册申报资料的内容进行充分说明和细化,并依据具体产品的特性确定其中的具体内容是否适用,若不适用,需详细阐述理由及相应的科学依据。
本指导原则是供申请人和审查人员使用的指导文件,不涉及注册审批等行政事项,亦不作为法规强制执行,如有能够满足法规要求的其他方法,也可以采用,但应提供详细的研究资料和验证资料。
应在遵循相关法规的前提下使用本指导原则。
本指导原则是在现行法规、标准体系及当前认知水平下制定的,随着法规、标准体系的不断完善和科学技术的不断发展,本指导原则相关内容也将适时进行调整。
一、适用范围本指导原则所涉及的灌流器,是指采用活性炭或吸附树脂等非特异性吸附剂材料,配合血液灌流装置,用于清除病人体内内源性和外源性药物、毒物的产品,但不适用于免疫吸附等其他以特异性吸附方式清除血液中毒性物质的灌流器产品。
1其他非特异性吸附血液灌流器产品可参考使用。
二、注册申报资料要求(一)综述资料1.概述灌流器为Ⅲ类医疗器械,分类编码10-04-02。
产品名称应符合《医疗器械通用名称命名规则》(国家食品药品监督管理总局令第19号)的规定,并解释申报产品名称的确定依据。
2.产品描述说明灌流器适用范围、工作原理、灭菌方式、结构组成、原材料、性能指标、有效期以及区别于其他同类产品的特征等内容。
必要时提供图示。
3.型号规格对于各种型号规格的结构组成、功能、特征等方面加以描述,且应当明确各型号规格的区别。
4.包装说明有关灌流器的包装信息,应当说明与灭菌方法相适应的最初包装材料。
5.适用范围和禁忌症(1)适用范围;(2)预期使用环境;(3)适用人群;(4)禁忌症(如适用)。
医疗器械注册技术审查指导原则目录2023年

11
隐球菌荚膜多糖抗原检测试剂注册技术审查指导原则(2023年第4号)
2023年1月19日
12
遗传性耳聋相关基因突变检测试剂注册技术审查指导
2023年1月19日
原则(2023年第4号)
13
肺炎支原体IgMZIgG抗体检测试剂注册技术审查指导原则(2023年第4号)
2023年1月19日
2023年9月24日
29
3D打印患者匹配下颌骨假体注册审查指导原则(2023年第62号)
2023年9月24日
30
个性化匹配骨植入物及工具医工交互质控审查指导原则(2023年第62号)
2023年9月24日
31
输注产品针刺伤防护装置要求与评价技术审查指导原则(2023年第62号)
2023年9月24日
32
2019年9月4日
111
口腔数字印模仪注册技术审查指导原则(2019年第37号)
2019年7月4日
112
植入式给药装置注册技术审杳指导原则(2019年第25号)
2019年5月22日
113
宫内节育器注册技术审查指导原则(2019年第25号)
79号)
2019年11月15日
91
肌电生物反馈治疗仪注册技术审查指导原则(2019年
2019年11月15日
第79号)
92
医用诊断X射线管组件注册技术审查指导原则(2019年第79号)
2019年11月15日
93
直接检眼镜注册技术审查指导原则(2019年第79号)
2019年11月15日
94
医疗器械产品受益-风险评估注册技术审查指导原则(2019年第79号)
2023年7月6日
滤芯检测项目和流程

滤芯检测项目和流程滤芯检测是在滤芯生产过程中对滤芯进行品质检查和性能测试的一项重要工作。
滤芯的质量和性能直接影响到滤芯的使用效果和寿命,因此,滤芯检测是确保滤芯质量的重要环节。
本文将从滤芯检测项目和流程两个方面来详细介绍滤芯检测的内容和方法。
一、滤芯检测项目1.外观检查:包括滤芯的表面有无划痕、顶帽、端盖是否完整,是否有变形等。
2.直径检测:测量滤芯的外直径和内直径,以确保尺寸的精度和一致性。
3.过滤效率检测:通过在一定条件下,将一定浓度的颗粒物或溶液通过滤芯,通过检测进出口的颗粒物浓度来确定滤芯的过滤效率。
4.破碎强度测试:通过在一定的压力下施加力,检测滤芯的破碎强度,以确保滤芯在使用过程中不容易发生损坏和破裂。
5.渗透率测试:通过测量滤芯材料的渗透率,以确定滤芯的过滤效果和材料的质量。
6.净水流量测试:测量滤芯通过单位时间内的净水流量,以确认滤芯的工作效果和流量。
二、滤芯检测流程1.准备工作:确定滤芯检测的标准和规范,准备检测所需的设备和试剂。
2.外观检查:对滤芯进行外观检查,确保滤芯的表面完整无划痕并符合要求。
3.尺寸检测:使用测量工具测量滤芯的外直径、内直径和长度,确保滤芯尺寸的精度和一致性。
4.过滤效率检测:将一定浓度的颗粒物或溶液通过滤芯,分别测量进出口的颗粒物浓度,并计算滤芯的过滤效率。
5.破碎强度测试:将滤芯固定在测试设备上,施加一定的压力,记录滤芯的破碎强度。
6.渗透率测试:将水或其他液体通过滤芯,测量液体通过滤芯的时间和量,计算滤芯的渗透率。
7.净水流量测试:将净水通过滤芯,测量单位时间内的净水流量。
8.数据分析和评估:根据检测结果,对滤芯的质量和性能进行评估和分析。
9.检测报告:根据检测结果编写检测报告,包括滤芯的各项指标和评估。
10.问题处理和改进:对于检测中发现的问题,及时进行处理和改进,确保滤芯的质量和性能达到要求。
以上是滤芯检测的项目和流程的简要介绍,滤芯的质量和性能对滤芯的使用效果和寿命有着重要的影响,因此,滤芯生产过程中的检测工作非常重要。
医疗器械已知可沥滤物测定方法验证及确认注册技术审查指导原则

附件2医疗器械已知可沥滤物测定方法验证及确认注册技术审查指导原则医疗器械的可沥滤物(Leachables)是指医疗器械或材料在临床使用过程中释放出的物质的统称。
可沥滤物一般包括灭菌残留剂、工艺残留物、降解产物以及材料中的单体及添加剂(包括稳定剂、抗氧化剂、增塑剂、着色剂等)。
在医疗器械产品与人体接触并发挥作用的过程中,可沥滤物也在或短期或长期地对人体产生安全性方面的危害。
可沥滤物安全性评价首要任务是建立拟研究物质的允许限量(Allowable limit),其次,应在模拟临床最坏使用环境下测定其释放量(Released amount),并根据其释放量是否超过其在该产品该预期用途下的允许限量,形成完整的可沥滤物安全性研究报告,其中,可沥滤物的释放量测定方法的设计和方法学验证是评价可沥滤物安全性研究报告质量和结果可靠性的重要依据。
本指导原则是对医疗器械已知可沥滤物测定方法研究的一般要求,申请者应依据具体产品的特性和拟研究可沥滤物性质对注册申报资料的内容进行充实和细化。
注册申请人还应依据具体可沥滤物的特性和分析方法确定释放量测定方法的设计和方法学验证参数的具体内容是否适用,若不适用,需具体阐述其理由及相应的科学依据。
—1 —本指导原则旨在帮助和指导注册申请人对医疗器械产品注册申报资料进行准备,以满足技术审评的基本要求。
同时有助于审评机构对该类产品进行科学规范的审评,提高审评工作的质量和效率。
本指导原则是对注册申请人和审查人员的指导性文件,但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其它方法,也可以采用,但是需要提供详细的研究资料和验证资料。
应在遵循相关法规的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将适时的进行调整。
一、适用范围本指导原则适用于医疗器械注册申报时对已知可沥滤物释放量的研究和产品技术审评的参考。
默克密理博除菌过滤验证

过滤工艺相关法规及验证内容解析时优默克密理博验证实验室认识我们使用的过滤器⏹折叠式⏹膜在支撑材料间⏹热熔⏹2-5种结构材料议题与过滤相关的法规及指南检查缺陷及解决方案如何进行过滤器验证世界各国GMP法规要求进行过滤器验证⏹US GMP 21 CFR Parts 210 & 211美国GMPAppropriate written procedures…shall be established and followed. Suchproceduresshall include validation of any sterilization process必须建立和跟进相应的证明性文件….,,这些文件中包括所有的除菌工艺的验证文件。
⏹EU GMP Annex 1 Sterile Medicinal Products欧盟GMP 无菌药品附录All sterilization processes should be validated.所有除菌工艺必须进行验证。
⏹Australian TGA GMP澳大利亚TGA GMPFiltration processes used as the sterilizing step for products should bevalidated.除菌级的过滤工艺应该验证。
⏹Health Canada GMP加拿大GMPDocumented evidence is available establishing validation and validity ofeachsterilization process.每步除菌工艺必须要有验证其有效性的证明性文件。
FDA法规对过滤器验证的要求FDA Aseptic Processing Guidelines (2004) FDA无菌工艺指南•Filter validation should be conducted using the worst case conditions, (pH, temperature, flow rate, pressures etc.)挑战试验必须模拟最差工艺条件• Challenge fluid should simulate product as closely as in practice 挑战液应该尽可能保持和实际产品相同中国新版GMP附录1第四十一条过滤器应当尽可能不脱落纤维。
委托书医疗器械已知可沥滤物测定方法验证及确认注册技术审查指导原则

委托书医疗器械已知可沥滤物测定方法验证及确认注册技术审查指导原则背景医疗器械是为了预防、诊断、治疗、缓解疾病、损伤或残疾,改善人体结构或功能所用的器材及设备。
在市场上,医疗器械的质量和安全性是至关重要的。
对于一些可沥滤物测定的医疗器械,相关部门出台了验证和确认的政策,以确保这些器械的质量和性能符合适用的标准和法规。
目的此文档的目的是为了帮助委托单位了解医疗器械可沥滤物测定方法的验证和确认步骤,并为注册技术审查提供指导原则。
这些步骤和指导原则旨在确保可沥滤物测定的医疗器械的质量和可靠性。
测定方法验证和确认步骤医疗器械可沥滤物测定方法的验证和确认步骤包括以下内容:1.分析目标和数据要求2.设计验证方案3.执行验证实验4.统计分析结果5.报告结论分析目标和数据要求在分析目标和数据要求阶段,需要明确验证的目标和测量指标。
重要的因素包括医疗器械的性能要求、试验方法、样品数目和数据的可靠性要求等。
在明确目标和数据要求之后,需要确定可沥滤物测定方法的验证方案。
设计验证方案在设计验证方案阶段,需要确定验证方案的具体内容,包括实验设备、试验方法、样品要求、试验步骤等。
验证方案应该要充分考虑到实验的风险,并进行谨慎评估。
执行验证实验在执行验证实验的阶段,需要根据验证方案一步步地执行实验,并采取适当的措施来控制各项因素的影响,如控制温度、湿度、时间等。
要确保实验数据的准确性和一致性,并记录实验结果。
统计分析结果在统计分析结果的阶段,需要根据实验数据对样本的性能和测量指标进行比较和分析。
这需要应用统计学的原则,例如t检验和方差分析等。
报告结论在报告结论的阶段,需要汇总实验结果,撰写详细的验证报告并撰写验证结论。
这个验证结论将用于医疗器械的注册和技术审查的指导。
注册技术审查指导原则注册技术审查的指导原则应包括以下内容:1.首先,应了解本地区的法规和标准,以便明确医疗器械的销售权限和相关要求。
2.其次,需要明确可沥滤物测定方法的测量标准并评估医疗器械可沥滤物测定方法的准确性和可靠性。
净水原材料的检验项目和检验方法

净水原材料的检验项目和检验方法下载提示:该文档是本店铺精心编制而成的,希望大家下载后,能够帮助大家解决实际问题。
文档下载后可定制修改,请根据实际需要进行调整和使用,谢谢!本店铺为大家提供各种类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by this editor. I hope that after you download it, it can help you solve practical problems. The document can be customized and modified after downloading, please adjust and use it according to actual needs, thank you! In addition, this shop provides you with various types of practical materials, such as educational essays, diary appreciation, sentence excerpts, ancient poems, classic articles, topic composition, work summary, word parsing, copy excerpts, other materials and so on, want to know different data formats and writing methods, please pay attention!1. 引言净水处理过程中原材料的质量控制直接影响着水的净化效果和安全性。
医疗器械安全和性能基本原则基本清单编写指导
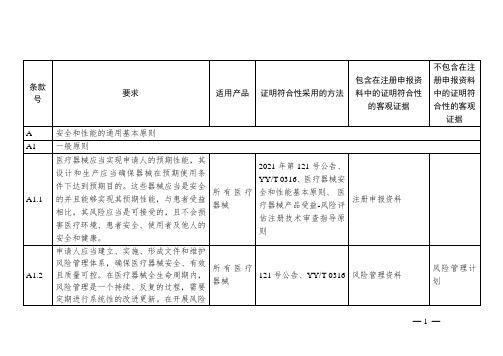
GB 18278.1、GB 18279系列、GB 18280系列、YY/T 1276、YY/T 1464、
GB/T 19974、YY/T 1463、无菌医疗器械灭菌工艺注册技术审查指导原则、其他适用的标准或指导原则
灭菌研究、稳定性研究、包装说明、产品技术要求及检测报告
包装过程确认
A4.4
无菌医疗器械应按照经验证的方法进行加工、生产、包装和灭菌,其货架有效期应按照经验证的方法确定。
无菌医疗器械
GB 18278.1、GB 18279系列、GB 18280系列、YY/T 1276、YY/T 1464、
GB/T 19974、YY/T 1464、无菌医疗器械灭菌工艺注册技术审查指导原则、其他适用的标准或指导原则
所有医疗器械
2021年第121号公告、YY/T 0316、医疗器械安全和性能基本原则、医疗器械产品受益-风险评估注册技术审查指导原则
注册申报资料
A1.2
申请人应当建立、实施、形成文件和维护风险管理体系,确保医疗器械安全、有效且质量可控。在医疗器械全生命周期内,风险管理是一个持续、反复的过程,需要定期进行系统性的改进更新。在开展风险管理时,申请人应当:a)建立涵盖所有医疗器械风险管理计划并形成文件;b)识别并分析涵盖所有医疗器械的相关的已知和可预见的危险(源);c)估计和评价在预期使用和可合理预见的误使用过程中,发生的相关风险;
适用的标准或指导原则
标签
A5
环境和使用条件
A5.1
如医疗器械预期与其他医疗器械或设备整合使用,应确保整合使用后的系统,包括连接系统,整体的安全性,且不影响器械本身的性能。整合使用上的限制应明确标识和/或在使用说明书中明确。对于需要使用者处理的连接,如液体、气体传输、电耦合或机械耦合等,在设计和生产过程中尽可能消除或降低所有可能的风险,包括错误连接或安全危害。
一次性使用无菌闭合夹注册技术审查指导原则

附件4一次性使用无菌闭合夹注册技术审查指导原则本指导原则旨在帮助和指导注册申请人对一次性使用无菌闭合夹注册申报资料进行准备,以满足技术审评的基本要求。
同时有助于审评机构对该类产品进行科学规范的审评,提高审评工作的质量和效率。
本指导原则是对一次性使用无菌闭合夹注册申报资料的一般要求,注册申请人应依据具体产品的特性对注册申报资料的内容进行充实和细化。
注册申请人还应依据具体产品的特性确定其中的具体内容是否适用,若不适用,需具体阐述其理由及相应的科学依据。
本指导原则是对注册申请人和审评人员的指导性文件,但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,应在遵循相关法规的前提下使用本指导原则。
如果有能够满足相关法规要求的其他方法,也可以采用,但是需要提供详细的研究资料和验证资料。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将进行适时的调整。
—1 —一、适用范围本指导原则所适用的产品一次性使用无菌闭合夹(以下简称闭合夹)主要用于外科手术中夹闭血管或闭合管状组织,包括中小动静脉、胆管等,不适用于大动脉和大静脉。
闭合夹应用于人体后不被降解吸收,为永久植入器械。
本指导原则不适用于可吸收性闭合夹和术中临时夹闭组织或血管、术后取出的闭合夹和止血夹,不适用于带有施夹、除夹等功能的产品,但可参考其适用部分内容。
二、注册申报资料要求(一)综述资料1.概述(1)依据医疗器械分类目录,闭合夹管理类别为三类医疗器械,分类编码02-06-01。
(2)产品名称:通常由1个核心词和不超过3个特征词确定产品通用名称,建议使用“闭合夹”、“止血夹”作为核心词,以“一次性使用无菌”或“内窥镜”或“腹腔、血管”等(专用使用部位)作为特征词命名。
产品名称应符合《医疗器械通用名称命名规则》及《无源手术器械通用名称命名指导原则》等医疗器械命名有关指南的规定。
2.产品描述产品描述应全面、详细,至少应包括申报产品名称、结构组成及图示、型号规格及划分依据、工作原理、灭菌方法、预期用途、适用部位、技术性能指标及其制定依据,以及区别于其他同—2 —类产品的特征等内容。
医疗器械包装材料生物相容性 生物相容性化学表征评价终点医疗器械包装风险管理生物学评价

医疗器械包装材料生物相容性生物相容性化学表征评价终点医疗器械包装风险管理生物学评价本文旨在通过解读医疗器械包装的标准和指南,帮助读者更好地进行医疗器械包装的风险管理和生物学评价。
关键词:生物相容性化学表征评价终点医疗器械包装风险管理生物学评价医疗器械作为特殊商品,要求其安全、有效[1]。
医疗器械包装,尤其是无菌屏障系统,作为器械整体的一部分,同样对其包装材料的生物相容性有着特殊的要求。
本文通过对有关法规和指南中相关内容的解读,希望为读者正确理解和进行医疗器械包装的生物相容性评价,提供较全面的参考信息。
医疗器械包装生物相容性评价的意义器械包装材料与医疗器械的相容性是许多监管机构的要求。
由于多数医疗器械都是与人体直接或者间接接触,可能会产生生物相容性的问题。
因此,与医疗器械直接接触的初级(一级)包装材料必须进行,以确保它们不会对医疗器械的物理、化学或生物特性产生负面影响,从而保证人体接触和使用的安全性。
标准“ISO 11607-1(GB/T 19633.1)材料、无菌屏障系统和包装系统的要求”中明确要求:包装中使用的材料对与之接触的医疗器械无不良影响,并且需要进行生物相容性和毒理学特性的项目和考察[2]。
此可能包括对包装材料相关信息的收集,经验和文献的研究,一系列化学测试和生物测试以及毒理学的安全。
这种也可能会得出这样的结论:如果收集的信息充分,能够证实包装与已上市产品的包装生物等同,或者与设计包装具有相同的安全使用历史,则无需进行测试。
因此,包装相容性评价是器械相容性评价有机整体的一部分,对于安全用械有着重要的意义。
医疗器械包装生物相容性评价的标准和指南目前,ASTM F2475-20是直接关于医用器械包装材料生物相容性评价的指南[3],版(2020年版)是根据ISO 10993-1:2018[4]原则和理念,对前一版指南(2011年版)在使用范围、术语定义、评价流程以及信息性附录等内容上,进行了全面更新。
委托书医疗器械已知可沥滤物测定方法验证及确认注册技术审查指导原则

委托书医疗器械已知可沥滤物测定方法验证及确认注册技术审查指导原则1. 引言医疗器械确认注册技术审查是确保医疗器械在市场销售前安全有效的一项重要工作。
委托书医疗器械已知可沥滤物测定方法验证是医疗器械确认注册技术审查的必要环节之一。
因此,制定相应的指导原则具有重要意义。
2. 术语定义2.1. 委托书医疗器械指委托人与申请人签订有关医疗器械注册申请文件的法律文件。
2.2. 已知可沥滤物指采用透析膜或滤器可从液体中分离出的具有生物活性的大分子物质。
2.3. 方法验证指证明特定应用下的新方法满足特定目的的过程。
2.4. 确认注册技术审查指在医疗器械注册申请中进行技术审查,以确认医疗器械的技术要求符合国家和地方规定要求的过程。
3. 委托书医疗器械已知可沥滤物测定方法验证指导原则3.1. 原则性要求委托书医疗器械已知可沥滤物测定方法验证应当符合以下原则:1.确保委托书医疗器械与已验证的方法之间不存在任何差异。
2.根据国家标准或相关技术规范,对委托书医疗器械进行严格的方法验证。
3.对委托书医疗器械的方法验证需要进行完整的建档记录,包括相应的文件、实验记录、数据分析和评估。
3.2. 方法验证方案编制编制方法验证方案应当包括以下内容:1.方法验证所需的仪器、试剂和材料准备及检查。
2.方法验证实验的设计方案和程序,包括实验的目的、方法、实验流程、数据记录和结果分析等。
3.3. 实验方法验证委托书医疗器械的方法验证应当进行合理的实验设计和科学的数据处理。
实验过程中需要注意以下问题:1.样品处理: 通过对委托书医疗器械的样品进行处理,包括加样、提取、稀释等,确保样品中已知可沥滤物的浓度均匀分布。
2.实验方法选择: 根据样品类型、测量范围、检测灵敏度等特点,选择合适的实验方法。
3.常数精度检验: 对实验仪器和试剂的常数进行检验,确保实验精度和准确性。
4.系统精度检验: 测量一组样品多次,计算各次测量值之间的差异。
结果应在规定精度范围内。
有关物质(包括已知杂质)检查方法验证标准操作规程

有关物质(包括已知杂质)检查方法验证标准操作规程目的为保证检测工作的可靠性和可重现性,在未知样品的检测前必须对检测方法进行验证以证明所采用的检测方法适合于相应的检测要求。
范围建立药品质量标准时、药品生产工艺变更时、制剂组分发生变更时、原分析方法修订时均应进行有关物质检测的方法学的验证。
责任人检测员、项目负责人、各级项目经理:要求系统、全面验证含量测定方法并记录整理验证数据。
程序4.1 验证内容:杂质检测方法的建立基于方法学研究,主要包括专属性试验、检测限试验、溶液稳定性试验等内容,如果定量检测杂质则需进行线性、精密度、稳定性、重现性及回收率等试验,从不同的角度、层面验证分析方法的可行,从而保证药品中的杂质能够有效地检测。
4.2 杂质检测方法建立验证及可接受标准1)专属性试验主要通过破坏性实验实现。
破坏性试验,也称为强制降解试验(stressing test),它是在人为设定的特殊条件下,如酸、碱、氧化、高温、光照等,引起药物的降解,通过对降解产物的测定,验证检测方法的可行性,分析药物可能的降解途径和降解机制。
每项破坏性试验通常包括以下内容:酸降解一般采用0.1mol/L-1mol/L盐酸或硫酸;碱降解采用0.1mol/L-1mol/L的氢氧化钠溶液;氧化降解采用合适的过氧化氢溶液。
以上三种试验,为了加快反应或者提高降解强度,必要时可以加热或提高浓度;高温试验通常温度高于加速试验温度的10℃,如50℃、60℃等,对于原料药有时需考虑水溶液或混悬液的降解,或者考虑在不同的pH 值条件下的降解;光照试验条件可采用4500LX。
破坏性试验的具体条件,与具体药物密切相关,需结合具体药物的特点,选择合适的条件,使药物有一定量的降解,并对可能的降解途径和降解机制进行分析,保证实验的意义。
药物经强力破坏产生的降解产物通常采用色谱法测定,需结合药物和可能降解产物的理化性质,选择不同的色谱方法(HPLC、GC、TLC)或检测器,有时可采用不同分离机理的色谱系统。
整形用面部植入假体注册技术审查指导原则

附件4整形用面部植入假体注册技术审查指导原则本指导原则旨在为药品监管部门对注册申报资料的技术审评提供技术指导,同时也为注册申请人进行整形用面部植入假体的产品注册申报提供参考。
本指导原则系对整形用面部植入假体的一般要求,注册申请人应依据具体产品的特性对注册申报资料的内容进行充实和细化,并依据具体产品的特性确定其中的具体内容是否适用。
本指导原则是对注册申请人和审评人员的技术指导性文件,不包括注册审批所涉及的行政事项,亦不作为法规强制执行。
如果有其他科学合理的替代方法,也可以采用,但是需要提供依据及相关资料。
应在遵循相关法规和标准的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制订的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将进行适时的调整。
一、适用范围本指导原则所涉及的整形用面部植入假体是指人工合成不可吸收材料制备的,用于面部填充的,仅以物理占位作用机理获得整形效果的产品。
具体产品的适用范围需根据产品性能特点及临床数据进行确定。
目前境内已上市的该类产品材质主要有硅橡胶、膨体聚四氟乙烯/全氟乙丙烯、高密度聚乙烯等。
—1 —其他材料制备的整形用植入假体可参考本指导原则中适用的部分。
二、技术审查要求注册申报资料按照国家药品监督管理局医疗器械注册相关规章进行提供,尤其注意以下几方面内容:(一)产品名称根据《医疗器械分类规则》(国家食品药品监督管理总局令第15号)、《医疗器械通用名称命名规则》(国家食品药品监督管理总局令第19号)等相关文件确认产品名称并论述其确定依据。
该类产品通用名一般为“整形用面部植入假体、面部整形植入填充材料”。
可添加材料化学名称,如“膨体聚四氟乙烯面部植入整形填充材料”;可反映具体植入部位,如“植入性硅橡胶鼻假体”。
(二)注册单元划分根据《医疗器械注册单元划分指导原则》(国家食品药品监督管理总局通告2017年第187号)对申报产品的注册单元进行确认,原则上材料成分不同的整形用面部植入假体应划分为不同的注册单元,如硅橡胶与膨体聚四氟乙烯产品作为不同的注册单元;硅橡胶成分配比、硫化程度不同时作为不同的注册单元。
已知可沥滤物测定方法验证及确认技术审查指导原则

已知可沥滤物测定方法验证及确认技术审查指导原则一、前言医疗器械的可沥滤物(Leachables)是指在临床使用过程中,医疗器械与水或使用中有关的液体、气体作用等介质作用时,从该医疗器械中释放出的化学物质的统称。
可沥滤物一般包括灭菌残留剂、工艺残留物、降解产物以及材料中的单体及添加剂(包括稳定剂、抗氧化剂、增塑剂、着色剂等)。
在医疗器械产品与人体接触并发挥作用的过程中,可沥滤物也在或短期或长期地对人体产生安全性方面的危害。
可沥滤物安全性评价首要任务是建立拟研究物质的允许限量(Allowable limit),其次,应在临床模拟最坏使用环境下测定其释放量(Release),并根据其释放量是否超过其在该产品该预期用途下的允许限量,形成完整的可沥滤物安全性研究报告,其中,可沥滤物的释放量测定方法的设计和方法学验证是评价可沥滤物安全性研究报告质量和可靠性的重要依据。
本指导原则是对医疗器械已知可沥滤物测定方法研究的一般要求,申请者应依据具体产品的特性和拟研究可沥滤物性质对注册申报资料的内容进行充实和细化。
申请者还应依据具体可沥滤物的特性和分析方法确定释放量测定方法的设计和方法学验证参数的具体内容是否适用,若不适用,需具体阐述其理由及相应的科学依据。
本指导原则旨在帮助和指导申请者对医疗器械产品注册申报资料进行准备,以满足技术审评的基本要求。
同时有助于审评机构对该类产品进行科学规范的审评,提高审评工作的质量和效率。
本指导原则是对申请者和审查人员的指导性文件, 但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其它方法,也可以采用,但是需要提供详细的研究资料和验证资料。
应在遵循相关法规的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将适时的进行调整。
二、适用范围本指导原则适用于医疗器械注册申报时对已知可沥滤物释放量的研究和产品技术审评的参考。
医疗器械注册技术审查指导原则汇编(带下载链接,更新至2020年4月)
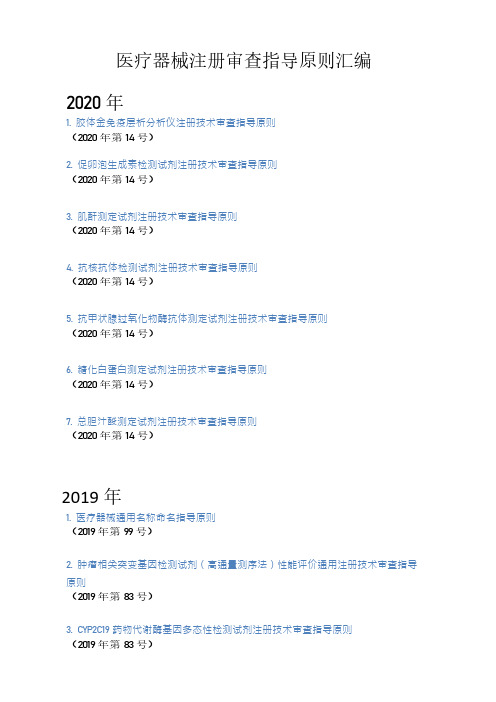
医疗器械注册审查指导原则汇编2020年1. 胶体金免疫层析分析仪注册技术审查指导原则(2020年第14号)2. 促卵泡生成素检测试剂注册技术审查指导原则(2020年第14号)3. 肌酐测定试剂注册技术审查指导原则(2020年第14号)4. 抗核抗体检测试剂注册技术审查指导原则(2020年第14号)5. 抗甲状腺过氧化物酶抗体测定试剂注册技术审查指导原则(2020年第14号)6. 糖化白蛋白测定试剂注册技术审查指导原则(2020年第14号)7. 总胆汁酸测定试剂注册技术审查指导原则(2020年第14号)2019年1. 医疗器械通用名称命名指导原则(2019年第99号)2. 肿瘤相关突变基因检测试剂(高通量测序法)性能评价通用注册技术审查指导原则(2019年第83号)3. CYP2C19药物代谢酶基因多态性检测试剂注册技术审查指导原则4. 基于细胞荧光原位杂交法的人类染色体异常检测试剂注册技术审查指导原则(2019年第80号)5. 呼吸道病毒多重核酸检测试剂注册技术审查指导原则(2019年第80号)6. 基于核酸检测方法的金黄色葡萄球菌和耐甲氧西林金黄色葡萄球菌检测试剂注册技术审查指导原则(2019年第80号)7. 沙眼衣原体和/或淋病奈瑟菌核酸检测试剂注册技术审查指导原则(2019年第80号)8. 氨基酸、肉碱及琥珀酰丙酮检测试剂注册技术审查指导原则(2019年第80号)9. 肢体加压理疗设备注册技术审查指导原则(2019年第79号)10. 医疗器械产品受益-风险评估注册技术审查指导原则(2019年第79号)11. 直接检眼镜注册技术审查指导原则(2019年第79号)12. 医用诊断X射线管组件注册技术审查指导原则(2019年第79号)13. 肌电生物反馈治疗仪注册技术审查指导原则(2019年第79号)14. 牙科种植手术用钻注册技术审查指导原则(2019年第79号)15. 人工复苏器注册技术审查指导原则(2019年第79号)16. 上下肢主被动运动康复训练设备注册技术审查指导原则(2019年第79号)17. 一次性使用内镜用活体取样钳注册技术审查指导原则(2019年第79号)18. 血浆速冻机注册技术审查指导原则(2019年第79号)19. 肠内营养泵注册技术审查指导原则(2019年第79号)20. 牙根尖定位仪注册技术审查指导原则(2019年第79号)21. 尿动力学分析仪注册技术审查指导原则(2019年第79号)22. 辅助生殖用胚胎移植导管注册技术审查指导原则(2019年第78号)23. 医疗器械已知可沥滤物测定方法验证及确认注册技术审查指导原则(2019年第78号)24.心肺转流系统体外循环管道注册申报技术审查指导原则(2019年第78号)25. 无源植入性骨、关节及口腔硬组织个性化增材制造医疗器械注册技术审查指导原则(2019年第70号)26. 晚期非小细胞肺癌临床试验终点技术指导原则(2019年第64号)27. 子宫内膜射频消融设备注册技术审查指导原则(2019年第59号)28. 口腔数字印模仪注册技术审查指导原则(2019年第37号)29. 脑利钠肽/氨基末端脑利钠肽前体检测试剂注册技术审查指导原则(2019年第11号)30. 总甲状腺素检测试剂注册技术审查指导原则(2019年第11号)31. 孕酮检测试剂注册技术审查指导原则(2019年第11号)32. 降钙素原检测试剂注册技术审查指导原则(2019年第11号)2018 年1.医用激光光纤产品注册技术审查指导原则(2018年第130号)2.一次性使用胆红素血浆吸附器注册技术审查指导原则(2018年第126号)3. 一次性使用活检针注册技术审查指导原则(2018年第126号)4. 外科纱布敷料注册技术审查指导原则(2018年修订)(2018年第120号)5.吻(缝)合器产品注册技术审查指导原则(2018年修订)6. 一次性使用吸痰管注册技术审查指导原则(2018年第120号)7. 护脐带注册技术审查指导原则(2018年修订)(2018年第116号)8.全瓷义齿用氧化锆瓷块注册技术审查指导原则(2018年修订)(2018年第116号)9.手动轮椅车注册技术审查指导原则(2018年第116号)10. 骨科外固定支架注册技术审查指导原则(2018年修订)(2018年第107号)11. 一次性使用医用喉罩注册技术审查指导原则(2018年修订)(2018年第107号)12. 骨水泥套管组件注册技术审查指导原则(2018年第107号)13. 用于罕见病防治医疗器械注册审查指导原则(2018年第101号)14. 鼻饲营养导管注册技术审查指导原则(2018年修订)(2018年第80号)15. 一次性使用无菌导尿管注册技术审查指导原则(2018年修订)(2018年第80号)16. 定制式义齿注册技术审查指导原则(2018年修订)(2018年第80号)17. 软性接触镜临床试验指导原则(2018 年第51 号)18. 角膜塑形用硬性透气接触镜临床试验指导原则(2018 年第51 号)19. 无源植入性医疗器械临床试验审批申报资料编写指导原则(2018 年第40 号)20. 麻醉咽喉镜注册技术审查指导原则(2018 年第30 号)21. 内镜清洗消毒机注册技术审查指导原则(2018 年第30 号)22. 睡眠呼吸监测产品注册技术审查指导原则(2018 年第30 号)23. 手术显微镜注册技术审查指导原则(2018 年第25 号)24. 医用洁净工作台注册技术审查指导原则(2018 年第25 号)25. 眼压计注册技术审查指导原则(2018 年第25 号)26 脉搏波速度和踝臂指数检测产品注册技术审查指导原则(2018 年第25 号)27. 冠状动脉药物洗脱支架临床前研究指导原则(2018 年第21 号)28. 冠状动脉药物洗脱支架临床试验指导原则(2018 年第21 号)29. 软性接触镜注册技术审查指导原则(2018 年第18 号)30. 人类体外辅助生殖技术用液注册技术审查指导原则(2018 年第18 号)31 气腹机注册技术审查指导原则(2018 年第15 号)32. 医用低温保存箱注册技术审查指导原则(2018 年第15 号)33. 电子尿量计注册技术审查指导原则(2018 年第15 号)35. 口腔曲面体层X射线机注册技术审查指导原则(2018 年第9 号)36. 硬性光学内窥镜(有创类)注册技术审查指导原则(2018 年第54 号)37. 结核分枝杆菌特异性细胞免疫反应检测试剂注册技术审查指导原则(2018 年第57 号)38. 持续葡萄糖监测系统注册技术审查指导原则(2018 年第56 号)39. 眼科飞秒激光治疗机注册技术审查指导原则(2018 年第53 号)40. 眼科超声诊断设备注册技术审查指导原则(2018 年第55 号)41. 眼科光学相干断层扫描仪注册技术审查指导原则(2018 年第44 号)42. 超声软组织切割止血系统注册技术审查指导原则(2018 年第37 号)43. 肠道病毒核酸检测试剂注册技术审查指导原则(2018 年第36 号)44. 抗人球蛋白检测试剂注册技术审查指导原则(2018 年第36 号)45. 幽门螺杆菌抗原/抗体检测试剂注册技术审查指导原则(2018 年第36 号)46. 人表皮生长因子受体(EGFR)突变基因检测试剂(PCR 法)注册技术审查指导原则(2018 年第36 号)47. X 射线计算机体层摄影设备注册技术审查指导原则(2018 年第26 号)48. 丙氨酸氨基转移酶测定试剂注册技术审查指导原则(2018 年第8 号)50. 同型半胱氨酸测定试剂注册技术审查指导原则(2018 年第8 号)51. 胰岛素测定试剂注册技术审查指导原则(2018 年第8 号)52. C-肽测定试剂注册技术审查指导原则(2018 年第8 号)53. D-二聚体测定试剂(免疫比浊法)注册技术审查指导原则(2018 年第9 号)54. 载脂蛋白B 测定试剂注册技术审查指导原则(2018 年第9 号)55. 载脂蛋白A1 测定试剂注册技术审查指导原则(2018 年第9 号)56. 接受医疗器械境外临床试验数据技术指导原则(2018 年第13 号)57. 质子碳离子治疗系统临床评价技术审查指导原则(2018 年第4号)58. 全血及血液成分贮存袋注册技术审查指导原则(2018 年第3 号)59. 一次性使用输注泵(非电驱动)注册技术审查指导原则(2018 年第3 号)60. 血液浓缩器注册技术审查指导原则(2018 年第3 号)61. 医疗器械临床试验设计指导原则(2018 年第6 号)2017 年1. 移动医疗器械注册技术审查指导原则(2017 年第222 号)2. 动物源性医疗器械注册技术审查指导原则(2017 年修订版)(2017 年第224 号)3. 促黄体生成素检测试剂(胶体金免疫层析法)注册技术审查指导原则(2017 年第213 号)4. 心肌肌钙蛋白I/肌红蛋白/肌酸激酶同工酶MB 检测试剂(胶体金免疫层析法)注册技术审查指导原则(2017 年第213 号)5. 电解质钾、钠、氯、钙测定试剂注册技术审查指导原则(2017 年第213 号)6. 高密度脂蛋白胆固醇测定试剂注册技术审查指导原则(2017 年第213 号)7. 胱抑素C 测定试剂(胶乳透射免疫比浊法)注册技术审查指导原则(2017 年第213 号)8. 全自动血型分析仪注册技术审查指导原则(2017 年第209 号)9. ABO、RhD 血型抗原检测卡(柱凝集法)注册技术审查指导原则(2017 年第209 号)10. 人表皮生长因子受体2 基因扩增检测试剂盒(荧光原位杂交法)注册技术审查指导原则(2017 年第209 号)11. 丙型肝炎病毒核酸基因分型检测试剂盒注册技术审查指导原则(2017 年第209 号)12. 治疗呼吸机临床评价技术审查指导原则(2017 年第212 号)13. 子宫内膜去除(热传导、射频消融)设备临床评价技术审查指导原则(2017 年第212 号)14. 细胞治疗产品研究与评价技术指导原则(试行)(2017 年第216 号)15. 生物显微镜注册技术审查指导原则(2017 年第199 号)16. 紫外治疗设备注册技术审查指导原则(2017 年第199 号)17. 裂隙灯显微镜注册技术审查指导原则(2017 年第199 号)18. 输液泵注册技术审查指导原则(2017 年第199 号)19. 电动洗胃机注册技术审查指导原则(2017 年修订版)(2017 年第199 号)20. 小型蒸汽灭菌器注册技术审查指导原则(2017 年第198 号)21. 动态心电图系统注册技术审查指导原则(2017 年第198 号)22. 血管内球囊扩张导管用球囊充压装置注册技术审查指导原则(2017 年第198 号)23. 验光仪注册技术审查指导原则(2017 年第198 号)24. 中央监护软件注册技术审查指导原则(2017 年第198 号)25. 医疗器械注册单元划分指导原则(2017 年第187 号)26. 红外线治疗设备注册技术审查指导原则(2017 年修订版)(2017 年第177 号)27. 医用控温毯注册技术审查指导原则(2017 年修订版)(2017 年第177 号)28. 中频电疗产品注册技术审查指导原则(2017 年修订版)(2017 年第177 号)29. 脉搏血氧仪注册技术审查指导原则(2017 年修订版)(2017 年第177 号)30. 牙科手机注册技术审查指导原则(2017 年修订版)(2017 年第177 号)32. 电动轮椅车注册技术审查指导原则(2017 年第180 号)33. 耳腔式医用红外体温计注册技术审查指导原则(2017 年第180 号)34. 医用吸引设备注册技术审查指导原则(2017 年修订版)(2017 年第180 号)35. 小型分子筛制氧机注册技术审查指导原则(2017 年修订版)(2017 年第180 号)36. 超声多普勒胎儿监护仪注册技术审查指导原则(2017 年修订版)(2017 年第177 号)37. 超声理疗设备注册技术审查指导原则(2017 年修订版)(2017 年第177 号)38. 超声洁牙设备注册技术审查指导原则(2017 年修订版)(2017 年第177 号)39. 视野计注册技术审查指导原则(2017 年修订版)(2017 年第177 号)40. 防褥疮气床垫注册技术审查指导原则(2017 年修订版)(2017 年第177 号)41. 酶标仪注册技术审查指导原则(2017 年第154 号)42. 一次性使用心电电极注册技术审查指导原则(2017 年第154 号)43. 动态血压测量仪注册技术审查指导原则(2017 年第154 号)44. 心电图机注册技术审查指导原则(2017 年第154 号)45. 病人监护产品(第二类)注册技术审查指导原则(2017 年第154 号)46. 红外乳腺检查仪注册技术审查指导原则(2017 年修订版)47. 医用臭氧妇科治疗仪注册技术审查指导原则(2017 年修订版)(2017 年第146 号)48. 骨组织手术设备注册技术审查指导原则(2017 年修订版)(2017 年第146 号)49. 牙科种植机注册技术审查指导原则(2017 年第124 号)50. 无源植入性医疗器械货架有效期注册申报资料指导原则(2017 年修订版)(2017 年第75 号)51. 超声多普勒胎儿心率仪注册技术审查指导原则(2017 年第60 号)52. 电动牵引装置注册技术审查指导原则(2017 年修订版)(2017 年第60 号)53. 电动手术台注册技术审查指导原则(2017 年修订版)(2017 年第60 号)54. 影像型超声诊断设备(第二类)注册技术审查指导原则(2017 年第60 号)55. 胎儿染色体非整倍体(T21、T18、T13)检测试剂盒(高通量测序法)注册技术审查指导原则(2017 年第52 号)56. 人工颈椎间盘假体注册技术审查指导原则(2017 年第23 号)57. 手术电极注册技术审查指导原则(2017 年修订版)(2017 年第41 号)58. 聚氨酯泡沫敷料产品注册技术审查指导原则(2017 年第44 号)59. 牙科纤维桩产品注册技术审查指导原则(2017 年第44 号)60. 髋关节假体系统注册技术审查指导原则(2017 年第23 号)61. 结核分枝杆菌复合群耐药基因突变检测试剂注册技术审查指导原则(2017 年第25 号)62. 手术无影灯注册技术审查指导原则(2017 年第30 号)63. 腹腔镜手术器械技术审查指导原则(2017 年第30 号)64. 电动病床注册技术审查指导原则(2017 年修订版)(2017 年第30 号)65. 人工耳蜗植入系统注册技术审查指导原则(2017 年第35 号)66. 腔镜用吻合器产品注册技术审查指导原则(2017 年第44 号)67. 医疗器械网络安全注册技术审查指导原则(2017 年第13 号)68. 半导体激光治疗机(第二类)注册技术审查指导原则(2017 年修订版)(2017 年第41 号)69. 医用电子体温计注册技术指导原则(2017 年修订版)(2017 年第41 号)70. 注射泵注册技术审查指导原则(2017 年修订版)(2017 年第41 号)71. 可见光谱治疗仪注册技术审查指导原则(2017 年第40 号)72. 软性纤维内窥镜(第二类)注册技术指导原则(2017 年修订版)(2017 年第40 号)73. 硬管内窥镜(第二类)注册技术审查指导原则(2017 年修订版)(2017 年第40 号)74. 钙磷/硅类骨填充材料注册技术审查指导原则(2017 年第14 号)75. 中心静脉导管产品注册技术审查指导原则(2017 年第14 号)76. 袜型医用压力带注册技术审查指导原则(2017 年第14 号)77. 医用磁共振成像系统临床评价技术审查指导原则(2017 年第6 号)78. 体外除颤产品注册技术指导原则(2017 年第6 号)79. 口腔颌面锥形束计算机体层摄影设备注册技术审查指导原则(2017 年第6 号)80. 光固化机注册技术审查指导原则(2017 年第6 号)81. 人工耳蜗植入系统临床试验指导原则(2017 年第3 号)2016 年1. 眼科超声乳化和眼前节玻璃体切除设备及附件注册技术审查指导原则(2016 年第162 号)2. 一次性使用血液透析管路注册技术审查指导原则(2016 年第146 号)3. 人红细胞反定型试剂注册技术审查指导原则(2016 年第131 号)4. 牙科种植体(系统)注册技术审查指导原则(2016 年第70 号)5. 牙科基托聚合物材料注册技术审查指导原则(2016 年第70 号)6. 一次性使用脑积水分流器注册技术审查指导原则(2016 年第70 号)7. 可吸收性外科缝线注册技术审查指导原则(2016 年第70 号)8. 脊柱后路内固定系统注册技术审查指导原则(2016 年第70 号)9. 椎间融合器注册技术审查指导原则(2016 年第70 号)10. β2-微球蛋白检测试剂盒(胶乳增强免疫比浊法)注册技术审查指导原则(2016 年第29 号)11. 甘油三酯测定试剂盒注册技术审查指导原则(2016 年第29 号)12. 唾液酸检测试剂盒(酶法)注册技术审查指导原则(2016 年第29 号)13. 促甲状腺素检测试剂注册技术审查指导原则(2016 年第29 号)14. 乳酸脱氢酶测定试剂盒注册技术审查指导原则(2016 年第29 号)15. 糖化血红蛋白测定试剂盒(酶法)注册技术审查指导原则(2016 年第29 号)16. 白蛋白测定试剂(盒)注册技术审查指导原则(2016 年第29 号)17. 自动尿液有形成分分析仪注册技术审查指导原则(2016 年修订版)(2016 年第22 号)18. 助听器注册技术审查指导原则(2016 年修订版)(2016 年第22 号)19. 医用雾化器注册技术审查指导原则(2016 年修订版)(2016 年第22 号)20. 牙科综合治疗机注册技术审查指导原则(2016 年修订版)(2016 年第22 号)21. 血液透析用制水设备注册技术审查指导原则(2016 年修订版)(2016 年第22 号)22. 血糖仪注册技术审查指导原则(2016 年修订版)(2016 年第22 号)23. 生化分析仪注册技术审查指导原则(2016 年修订版)(2016 年第22 号)24. 凝血分析仪注册技术审查指导原则(2016 年修订版)(2016 年第22 号)25. 半自动化学发光免疫分析仪注册技术审查指导原则(2016 年修订版)(2016 年第22 号)26. 尿液分析仪注册技术审查指导原则(2016 年修订版)(2016 年第22 号)27. X 射线诊断设备(第二类)注册技术审查指导原则(2016 年修订版)(2016 年第22 号)28. 电子血压计(示波法)注册技术审查指导原则(2016 年修订版)(2016 年第22 号)29. 磁疗产品注册技术审查指导原则(2016 年修订版)(2016 年第22 号)30. 治疗呼吸机注册技术审查指导原则(2016 年第21 号)31. 强脉冲光治疗仪注册技术审查指导原则(2016 年第21 号)32. 脉搏血氧仪设备临床评价技术指导原则(2016 年第21 号)33. 植入式心脏起搏器注册技术审查指导原则(2016 年修订版)(2016 年第21 号)34. 医用X 射线诊断设备(第三类)注册技术审查指导原则(2016 年修订版)(2016 年第21 号)35. 高频手术设备注册技术审查指导原则(2016 年第21 号)36. 透明质酸钠类面部注射填充材料注册技术审查指导原则(2016 年第7 号)37. 腹腔、盆腔外科手术用可吸收防粘连产品注册技术审查指导原则(2016 年第7 号)38. 可吸收止血产品注册技术审查指导原则(2016 年第7 号)39. α-氰基丙烯酸酯类医用粘合剂注册技术审查指导原则(2016 年第6 号)40. 一次性使用膜式氧合器注册技术审查指导原则(2016 年第6 号)41. 医学图像存储传输软件(PACS)注册技术审查指导原则(2016 年第27 号)42. 正压通气治疗机注册技术审查指导原则(2016 年第27 号)43. 大型蒸汽灭菌器注册技术审查指导原则(2016 年第27 号)44. 腹膜透析机注册技术审查指导原则(2016 年第27 号)45. 医用内窥镜冷光源注册技术审查指导原则(2016 年第27 号)46. 振动叩击排痰机注册技术审查指导原则(2016 年第27 号)47. 碱性磷酸酶测定试剂盒注册技术审查指导原则(2016 年修订版)(2016 年第28 号)48. 人绒毛膜促性腺激素检测试剂(胶体金免疫层析法)注册技术审查指导原则(2016年修订版)(2016 年第28 号)49. C反应蛋白测定试剂盒注册技术审查指导原则(2016 年修订版)(2016 年第28 号)50. 大便隐血(FOB)检测试剂盒(胶体金免疫层析法)注册技术审查指导原则(2016年修订版)(2016 年第28 号)51. 缺血修饰白蛋白测定试剂盒注册技术审查指导原则(2016 年修订版)(2016 年第28 号)52. 肌酸激酶测定试剂(盒)注册技术审查指导原则(2016 年修订版)(2016 年第28 号)2015 年1. 全自动化学发光免疫分析仪技术审查指导原则(2015 年第93 号)2. 人乳头瘤病毒(HPV)核酸检测及基因分型试剂技术审查指导原则3. 过敏原特异性IgE 抗体检测试剂技术审查指导原则(2015 年第93 号)4. 丙型肝炎病毒核糖核酸测定试剂技术审查指导原则(2015 年第93 号)5. 结核分枝杆菌复合群核酸检测试剂注册技术审查指导原则(2015 年第65 号)6. 影像型超声诊断设备(第三类)技术审查指导原则(2015 年修订版)(2015 年第112 号)7. 离心式血液成分分离设备技术审查指导原则(2015 年第112 号)8. 质子/碳离子治疗系统技术审查指导原则(2015 年第112 号)9. 乙型肝炎病毒基因分型检测试剂技术审查指导原则(2015 年第32 号)10. 影像型超声诊断设备新技术注册技术审查指导原则(2015 年第33 号)11. 医疗器械软件注册技术审查指导原则(2015 年第50 号)12. 雌激素受体、孕激素受体抗体试剂及检测试剂盒技术审查指导原则(2015 年第11 号)13. 医疗器械临床评价技术指导原则(2015 年第14 号)2014 年1. 体外诊断试剂临床试验技术指导原则(2014 年第16 号)2. 体外诊断试剂说明书编写指导原则(2014 年第17 号)3. 医疗器械产品技术要求编写指导原则4. 硬性角膜接触镜说明书编写指导原则(2014 年第3 号)5. 软性亲水接触镜说明书编写指导原则(2014 年第3 号)6. 植入式心脏电极导线产品注册技术审查指导原则(2014 年第10 号)7. 药物滥用检测试剂技术审查指导原则(2014 年第2 号)8. 肿瘤个体化治疗相关基因突变检测试剂技术审查指导原则(2014 年第2 号)9. 弓形虫、风疹病毒、巨细胞病毒、单纯疱疹病毒抗体及G 型免疫球蛋白抗体亲合力检测试剂技术审查指导原则(2014 年第2 号)10. 医用磁共振成像系统注册技术审查指导原则(2014 年第2 号)11. 金属接骨板内固定系统产品注册技术审查指导原则(2014 年第6 号)12. 血液透析浓缩物产品注册技术审查指导原则(2014 年第6 号)13. 一次性使用避光输液器产品注册技术审查指导原则(2014 年第6 号)14. 一次性使用血液分离器具产品注册技术审查指导原则(2014 年第6 号)15. 牙科树脂类充填材料产品注册技术审查指导原则(2014 年第6 号)16. 心脏射频消融导管产品注册技术审查指导原则(2014 年第5 号)17. 一次性使用皮肤缝合器产品注册技术审查指导原则(2014 年第7 号)18. 一次性医用喉罩产品注册技术审查指导原则(2014 年第7 号)19. 护脐带产品注册技术审查指导原则(2014 年第7 号)20. 一次性使用引流管产品注册技术审查指导原则(2014 年第7 号)21. 医用口罩产品注册技术审查指导原则(2014 年第7 号)22. 碱性磷酸酶检测试剂盒产品注册技术审查指导原则(已废止)(2014 年第7 号)23. 缺血修饰白蛋白测定试剂产品注册技术审查指导原则(已废止)(2014 年第7 号)24. 肌酸激酶测定试剂盒产品注册技术审查指导原则(已废止)(2014 年第7 号)25. C 反应蛋白定量检测试剂盒产品注册技术审查指导原则(已废止)(2014 年第7 号)26. 牙科手机产品注册技术审查指导原则(已废止)(2014 年第7 号)27. 脉搏血氧仪产品注册技术审查指导原则(已废止)(2014 年第7 号)28. 医用电子体温计产品注册技术审查指导原则(已废止)(2014 年第7 号)29. 电动洗胃机产品注册技术审查指导原则(已废止)(2014 年第7 号)30. 医用控温毯产品注册技术审查指导原则(已废止)(2014 年第7 号)2013 年1. 一次性使用鼻氧管产品注册技术审查指导原则(2013 年第8 号)2. 义齿制作用合金产品注册技术审查指导原则(2013 年第8 号)3. 一次性使用配药用注射器产品注册技术审查指导原则(2013 年第8 号)4. 一次性使用无菌手术包类产品注册技术审查指导原则(2013 年第8 号)5. 负压引流装置产品注册技术审查指导原则(2013 年第8 号)6. 流式细胞仪配套用检测试剂注册技术审查指导原则(2013 年第3 号)7. 人类免疫缺陷病毒检测试剂临床研究注册技术审查指导原则(2013 年第3 号)8. 病原体特异性M 型免疫球蛋白定性检测试剂注册技术审查指导原则(2013 年第3 号)9. 乙型肝炎病毒脱氧核糖核酸定量检测试剂注册技术审查指导原则(2013 年第3 号)10. 疝修补补片产品注册技术审查指导原则(2013 年第7 号)11. 一次性使用透析器产品注册技术审查指导原则(2013 年第3 号)12. 生物芯片类检测试剂注册技术审查指导原则(2013 年第3 号)13. 金标类检测试剂注册技术审查指导原则(2013 年第3 号)14. 核酸扩增法检测试剂注册技术审查指导原则(2013 年第3 号)15. 发光免疫类检测试剂注册技术审查指导原则(2013 年第3 号)16. 酶联免疫法检测试剂注册技术审查指导原则(2013 年第3 号)2012 年1. 全瓷义齿用氧化锆瓷块产品注册技术审查指导原则(2012 年第210 号)2. 麻醉机和呼吸机用呼吸管路产品注册技术审查指导原则(2012 年第210 号)3. 吻(缝)合器产品注册技术审查指导原则(2012 年第210 号)4. 手术动力设备产品注册技术审查指导原则(2012 年第210 号)2011 年1. 流行性感冒病毒抗原检测试剂注册申报资料指导原则(2011 年第540 号)2. 流行性感冒病毒核酸检测试剂注册申报资料指导原则(2011 年第540 号)3. 一次性使用手术衣产品注册技术审查指导原则(食药监办械函[2011]187 号)4. 天然胶乳橡胶避孕套产品注册技术审查指导原则(食药监办械函[2011]187 号)5. 定制式义齿产品注册技术审查指导原则(食药监办械函[2011]187 号)6. 一次性使用真空采血管产品注册技术审查指导原则(食药监办械函[2011]187 号)7. 3A 类半导体激光治疗机产品注册技术审查指导原则(食药监办械函[2011]187 号)8. 角膜塑形用硬性透气接触镜说明书编写指导原则(食药监办械函[2011]143 号)9. 乳房植入体产品注册技术审查指导原则(食药监办械函[2011]116 号)10. 接触镜护理产品注册技术审查指导原则。
可沥滤物标准

可沥滤物标准
1.定义:可沥滤物是指通过特定筛网后得到的固体颗粒物,在滤纸上的残留量符合本标准规定的物质。
2. 分类:可沥滤物按来源分为化学和生物两类。
3. 要求:可沥滤物应无毒、无害、无异味、无异色,符合国家环境保护标准。
4. 试验方法:采用干燥筛分法或湿筛分法进行测试,测试结果应符合本标准规定。
5. 标志:可沥滤物应标注生产企业名称、产品名称、规格型号、生产日期、批号等信息,并注明符合本标准。
6. 包装:采用密封包装,防潮、防尘、防震,保证产品质量。
7. 运输:采用无害化运输方式,防止受潮、震动和外力损坏。
8. 贮存:贮存于阴凉、干燥、通风、清洁的仓库内,远离火源、易燃和易爆物品。
本标准适用于化工、医药、冶金、矿山、食品等行业生产的可沥滤物。
- 1 -。
医疗器械已知可沥滤物测定方法验证及确认注册技术审查指导原则精选全文完整版

可编辑修改精选全文完整版附件2医疗器械已知可沥滤物测定方法验证及确认注册技术审查指导原则医疗器械的可沥滤物(Leachables)是指医疗器械或材料在临床使用过程中释放出的物质的统称。
可沥滤物一般包括灭菌残留剂、工艺残留物、降解产物以及材料中的单体及添加剂(包括稳定剂、抗氧化剂、增塑剂、着色剂等)。
在医疗器械产品与人体接触并发挥作用的过程中,可沥滤物也在或短期或长期地对人体产生安全性方面的危害。
可沥滤物安全性评价首要任务是建立拟研究物质的允许限量(Allowable limit),其次,应在模拟临床最坏使用环境下测定其释放量(Released amount),并根据其释放量是否超过其在该产品该预期用途下的允许限量,形成完整的可沥滤物安全性研究报告,其中,可沥滤物的释放量测定方法的设计和方法学验证是评价可沥滤物安全性研究报告质量和结果可靠性的重要依据。
本指导原则是对医疗器械已知可沥滤物测定方法研究的一般要求,申请者应依据具体产品的特性和拟研究可沥滤物性质对注册申报资料的内容进行充实和细化。
注册申请人还应依据具体可沥滤物的特性和分析方法确定释放量测定方法的设计和方法—1 —学验证参数的具体内容是否适用,若不适用,需具体阐述其理由及相应的科学依据。
本指导原则旨在帮助和指导注册申请人对医疗器械产品注册申报资料进行准备,以满足技术审评的基本要求。
同时有助于审评机构对该类产品进行科学规范的审评,提高审评工作的质量和效率。
本指导原则是对注册申请人和审查人员的指导性文件,但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其它方法,也可以采用,但是需要提供详细的研究资料和验证资料。
应在遵循相关法规的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将适时的进行调整。
一、适用范围本指导原则适用于医疗器械注册申报时对已知可沥滤物释放量的研究和产品技术审评的参考。
液体过滤器的验证方案
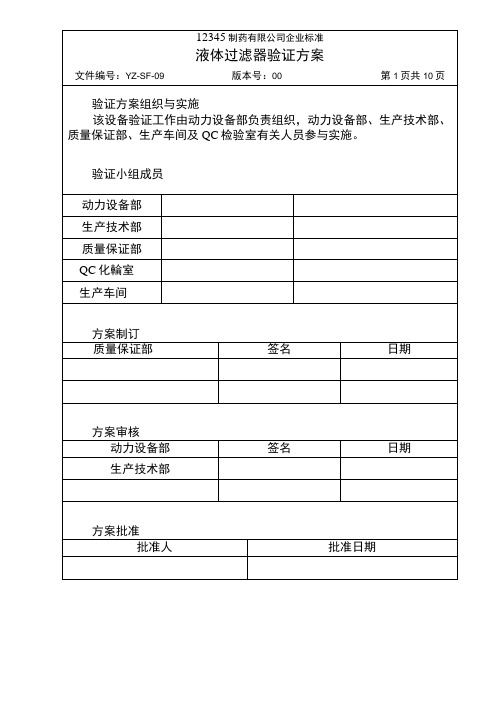
12345制药有限公司企业标准液体过滤器验证方案文件编号:YZ-SF-09版本号:00第色页共_10_页5. 2. 3试验方法将微孔滤膜过滤器用液体充分浸湿后,逐步加大气体的压力至发 泡点的临界压力的80%,将系统密闭,在规定时间内观察并记录压力的 下降情况.继续升压,直至在过滤器下侧浸入水的管中有稳定的气流 发生。
记录气泡第一次出现时压力的读数。
5. 2. 4评价标准:5. 2. 4. 1压力保持实验:0. 26Mpa, lOmin 内压降〈5% 5. 2. 4. 2起泡点压力: 孔径/ U m 最低起泡点压力/ Mpa 压力保持实验值/ Mpa 0. 2 Um 0. 34 0. 24 0. 45 U m 0. 24 0. 17压力表5. 3微生物挑战性试验5. 3.1目的:用来检测无菌过滤器去除溶液中微生物的能力。
5. 3. 2材料和方法5. 3. 2. 1阳性对照:生理盐水。
5. 3. 2. 2生物指示剂:缺陷假单抱菌(ATCC19146)。
5. 3. 2. 3菌液浓度:每平方厘米有效过滤面积应达到10’个菌的 挑战水平。
5. 3. 2.4试验压力:约为020MPa5. 3. 2. 5 试验流量:筒式过滤器为每分钟 2L/0. lm 2-3. 86L/0. Im 3 • min5. 3.3微生物挑战性试验示意图5. 3. 3试验步骤5. 3. 3.1将过滤系统灭菌5. 3. 3. 2用无菌生理盐水润湿过滤器,之后进行过滤器的完好性12345制药有限公司企业标准液体过滤器验证方案文件编号:YZ-SF-09版本号:00第6页共10页试验。
5. 3. 3. 3将此溶液用一阴性对照用无菌过滤器压滤,培养并检查无 菌。
5. 3. 3. 4将事先标定浓度的微生物悬浮液装入适当容器,并对待 试验的过滤器进行挑战性试验,操作同上。
5. 3. 3. 5进行过滤器的完好性检查,确认试验过程中滤膜没有损 坏。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
已知可沥滤物测定方法验证及确认技术审查指导原则一、前言医疗器械的可沥滤物(Leachables)是指在临床使用过程中,医疗器械与水或使用中有关的液体、气体作用等介质作用时,从该医疗器械中释放出的化学物质的统称。
可沥滤物一般包括灭菌残留剂、工艺残留物、降解产物以及材料中的单体及添加剂(包括稳定剂、抗氧化剂、增塑剂、着色剂等)。
在医疗器械产品与人体接触并发挥作用的过程中,可沥滤物也在或短期或长期地对人体产生安全性方面的危害。
可沥滤物安全性评价首要任务是建立拟研究物质的允许限量(Allowable limit),其次,应在临床模拟最坏使用环境下测定其释放量(Release),并根据其释放量是否超过其在该产品该预期用途下的允许限量,形成完整的可沥滤物安全性研究报告,其中,可沥滤物的释放量测定方法的设计和方法学验证是评价可沥滤物安全性研究报告质量和可靠性的重要依据。
本指导原则是对医疗器械已知可沥滤物测定方法研究的一般要求,申请者应依据具体产品的特性和拟研究可沥滤物性质对注册申报资料的内容进行充实和细化。
申请者还应依据具体可沥滤物的特性和分析方法确定释放量测定方法的设计和方法学验证参数的具体内容是否适用,若不适用,需具体阐述其理由及相应的科学依据。
本指导原则旨在帮助和指导申请者对医疗器械产品注册申报资料进行准备,以满足技术审评的基本要求。
同时有助于审评机构对该类产品进行科学规范的审评,提高审评工作的质量和效率。
本指导原则是对申请者和审查人员的指导性文件, 但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其它方法,也可以采用,但是需要提供详细的研究资料和验证资料。
应在遵循相关法规的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平下制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将适时的进行调整。
二、适用范围本指导原则适用于医疗器械注册申报时对已知可沥滤物释放量的研究和产品技术审评的参考。
本指导原则仅适用于已知可沥滤物研究,已知可沥滤物的信息可以通过以下途径获得:1)从原料供应商处获得材料的组成信息并预测潜在的可沥滤物;2)通过生产工艺文件获得额外的加工助剂信息,例如脱模剂、粘合剂;3)通过已有医疗器械用材料的标准、文献等资料查阅获得潜在的可沥滤物信息;4)通过浸提试验(Extraction study)获得信息,预测潜在的可沥滤物。
本指导原则不适用于对未知可沥滤物的测定研究,但部分内容可参考使用。
三、注册申报资料要求1.已知可沥滤物信息企业应提供已知可沥滤物的基本信息,包括:化学名称、化学式及结构式、CAS 号(如有)、来源、最终医疗器械中的使用及加工方式、用途(适用时)、添加量(如适用),如可能,还应提供可沥滤物的理化性能,如酸碱性、密度、熔点、沸点、溶解特性及相应溶解度、极性特性等。
2.已知可沥滤物安全性研究的基本步骤应首先确认该已知可沥滤物是否为原材料或最终医疗器械生产过程中添加的添加剂剂,如抗氧化剂、稳定剂等成分。
如果是,应进一步确认该添加剂在原材料或最终医疗器械中的添加总量是否超过该已知可沥滤物的允许限量,如果未超过,则一般无需对该可沥滤物本身的安全性做进一步的研究,如果超过了允许限量,则应模拟临床实际使用最恶劣环境,通过浸提试验研究,获得其最大溶出量(可参考附录1流程图),并根据其允许限量形成完整的安全性评价报告。
值得注意的是,可沥滤物来源可能不仅是器械原材料及工艺信息中提供的添加剂、单体、加工助剂本身,某些情况下,器械及其原材料在生产、制造、贮存及使用等过程中产生的上述化学物质的水解、降解或反应产物等宜同时纳入可沥滤物风险评估的考虑。
3.浸提液制备的论述3.1 浸提方式选择浸提是通过最大限度的实验室模拟获得医疗器械临床实际使用环境获下可沥滤物释放的进入人体的物质的量,然而大部分医疗器械很难模拟临床使用条件,特别是长期植入类器械,因此某些情况下可通过可浸提物(extractables)研究替代可沥滤物研究,即采用浸提方式制备浸提液进行已知可沥滤物测定,但是务必对浸提方式进行论述,证明浸提条件是严于器械临床使用条件。
常用的浸提方式包括模拟浸提、加严浸提、加速浸提和极限浸提(具体可参考GB/T 16886.12部分定义),一般来说模拟浸提最接近于实际,极限浸提(此处及此后提到的极限浸提均指的是所用溶剂符合下面论述前提下的溶剂)获得结果可能大于或等于患者实际可能接受到的剂量或模拟浸提获得的结果。
但有些特殊情况下,因实际使用条件很难在实验室进行模拟,则优先推荐使用极限浸提法,比如产品属于持久接触(接触时间>30天)等。
需要说明的是,某些器械采用模拟浸提法可能会导致相对较大的浸提体积,这种情况下,则可能需要极大地提高对残留物测定的灵敏度以满足安全性评价的要求。
极限浸提法排除了时间对剂量测定的影响,不能保证所测得的可沥滤物在与器械接触的第一天或第一个月完全放给患者,但如果通过极限浸提法测定出产品上存在的全部残留量符合安全性评价的要求,则一般不需要再进行模拟法研究。
除采用前述浸提的方式外,有时还可以采用其他方式获得拟研究物质。
比如分析某些医疗器械产品上易挥发性有机物时可以采用顶空进样分析,这种分析方法一般更加适用于挥发性可沥滤物,并同样能够达到预期的研究目的。
对于浸提方法,可采用加严方式进行浸提,如:循环、剪碎浸泡、超声浸提等,如果通过加严方式溶出量超过毒理学评估阈值时,可考虑临床实际使用情况。
制备浸提液需要考虑典型样品的选择、浸提介质、浸提比例、浸提时间、浸提温度、浸提方式等。
但所选浸提条件均不应引起器械材料和目标可沥滤物发生化学变化。
3.2 样品选择科学的采样是获得代表性样品的关键,因产品的加工制造过程会对可沥滤物残留量产生影响,所以试验样品优先选择最终产品或取自最终产品中有代表性的样品或经论述的与最终产品相同的工艺过程制得的材料制备的适合浸提液。
但某些情况下,因产品大型和/或复杂的器械使得无法在终产品上进行浸提时,实验室应建立完整的采样操作规程,特别注意样品的代表性。
一般可选取有代表性的部分进行浸提,然后推导出整个器械的结果,代表性的部分可采用如下方法:如果含有几种不同的材料,选取的样品中每一组分占样品的比例宜与该组分占被测器械的比例一致,或选择经评价证明是器械上残留含量最高的一个组成部分进行试验,有时还可以通过相同原材料在相同工艺条件下加工成的最终品检测等方式进行,采样方法应经过确认并保留相关理由、记录,必要时,提供相关支持性资料。
值得注意的是,某些产品需要在使用前现场制备,比如某些需要通过光固化、化学固化的口腔修复类产品等,该类产品的样品应严格按照产品使用说明书中规定的时间、浓度、剂量等要求制备后获得试验样本。
3.3 浸提介质浸提介质的选择需要考虑以下因素:(1)临床接触介质性质(酸性、碱性、极性、非极性等)。
例如对于一次性使用输注器具拟输注的药物是最佳的提取溶剂,但是输注器具和拟输注的药品种类繁多,且不同输注器械在临床的应用情况有很大的差异。
因此对于无法按照临床使用情况制备检验液的样品,或者药品中某些成分对已知可沥滤物分析存在干扰时,可以使用替代溶剂开展研究。
如对于聚氯乙烯输注器具中增塑剂DEHP 的研究,可通过充分的论证或实验研究分析后一般情况下可采用乙醇/水(ρ=0.9373g/ml ~0.9378g/ml)进行浸提研究。
(2)已知物的溶解特性,结合临床接触介质性质,初步确定拟采用的替代溶剂。
(3)根据已知物的毒理学推导TE值及浸提液体积,计算得到的浸提液中的允许浓度,确认该已知物在初步确定的替代溶剂中的溶解度能够满足该允许浓度的要求。
(4)替代溶剂的选择,适用时可采用试验证明替代溶剂的提取能力应高于实际临床接触介质。
如采用替代溶剂获得的结果不能满足毒理学风险评估要求时,可采用经论述的更加接近临床实际的方式进行评估。
替代溶剂的选择应综合考虑上述4个因素。
对于无机物(金属元素)可选择水或弱酸性介质作为浸提液,但还要考虑实际使用环境,比如因酸性饮料等的使用,会使得口腔内医疗器械常接触较酸的液体环境,因此在进行口腔产品的金属离子释放研究时,可采用较酸的浸提液,具体可参考相关标准及指南规定。
除此之外还应注意,在进行溶剂选择时,还应避免所用溶剂是否会与要研究的物质发生反应使得检测不出或低于其实际的量。
比如,异氰酸酯是合成一系列性能优良的聚氨酯材料的单体,但由于异氰酸酯类单体易与水发生水解反应生成胺类物质,因此当对含聚氨酯材料产品中进行异氰酸酯单体残留研究时采用含水溶剂,如某些药物的生理盐水溶液、乙醇/水混合溶剂等,这种情况下获得的结果一般是不被认可的。
3.4 浸提体积浸提体积的选择首先应保证浸提液能够完全浸没浸提样品,其次浸提液中可沥滤物浓度能够满足检测灵敏度要求,同时还应避免因其浓度过大影响被浸提物质的进一步析出。
如果供试品溶液中待测成分浓度超过线性范围,一般需通过稀释的方法降低待测成分浓度后再行测定。
3.5 浸提温度对于浸提温度,首先应考虑器械临床实际的使用温度,如选择提高温度进行加速浸提、加严浸提或极限浸提时应注意,浸提温度不应引起器械材料和目标可沥滤物发生化学变化。
3.6 浸提时间对于浸提时间,应按照器械作用于患者的性质和接触时间来确定并尽可能模拟产品临床实际,特别是临床最大可能接触时间。
3.7 其他因素除上述条件外,还应考虑是否需要采用动态模拟、浸提液的循环速度等因素,无论采取何种浸提条件,均需证明器械所选用的浸提条件代表产品在预期使用中带给患者的最大风险。
4.分析测试方法对于某些已经建立起相关标准检测方法的研究物质,优先选用标准方法,如国际标准、区域标准或国家标准发布的方法,或由知名技术组织或有关科技文献或期刊中公布的方法,在使用有关科技文献或期刊中公布的方法之前,应参照本指南中方法学验证参数进行验证,以确保方法的适用性。
如果不能确定方法的适用性,应开发合适的新方法。
对于无标准检测方法的可沥滤物,企业需开发新的检测方法并进行方法学验证及确认工作。
开发新的可沥滤物检测方法应根据拟研究物质的理化性质(包括极性、稳定性、溶解特性、环境敏感性等)、浸提选用溶剂、法规或毒理学评估所需的精度等合理选择检测仪器及检测方法,同时,应进行必要的方法学验证以保证方法的可靠性满足安全性评价要求。
对于无机物(元素)检测方法有:电感耦合等离子体原子发射光谱法(ICP-OES),电感耦合等离子体-质谱法(ICP-MS),原子吸收分光光度法(AAS)、离子色谱(IC)等;对于有机物的主要检测方法有:高效液相色谱/质谱(HPLC/MS)、离子色谱法/质谱(IC/MS)、气相色谱/质谱(GC/MS)、傅里叶变换红外光谱法(FTIR)等。