CLNK工艺装备验证记录
清洗验证方案模板
8.5 Recovery rate回收率 17 9. Equipment description 设备描述 17
9.1 List of equipment areas in direct contact with the active ingredients与活性成分直接接触设备面积清单 18 9.2 Sampling location selection rationale取样位置选择依据 19 9.3 Sampling Plan取样计划 19 10. Acceptance criteria 可接受标准 21 10.1 Visual checks目视检查 21 10.2 Active pharmaceutical ingredient residues活性成分残 留 21 10.2.1 Principles原理 21 10.2.2 Target Product Selection目标产品选择 22 10.2.3 Minimum daily therapeutic dosage criteria最低日治疗剂 量标准 22 10.2.4 10ppm criteria 10ppm标准 23 10.2.5 Comparison of calculation results计算结果对比 24 10.2.6 Calculation of residue of active ingredients per unit area 单位面积活性成分残留的计算 24 10.2.7 Others其它 25 10.3 Microbiological limit 微生物限度 25 10.3.1 Microbial limit微生物限度 25 10.3.2 Treatment and testing of microbial samples微生物样品处
工艺验证及清洁验证检验方法(PPT76张)

备注:通常对于盲管或垂直管路,为了清洁干净要求管道中水的流速要大于湍 流所需要的流速1.52米每秒 防止容器底部积水 底部出口的大小(英寸) 0.5 1.0 1.5 2.0 5.0 45.5 140.9 268.2 容器排水速率估算(LM)
2.5
454.6
喷淋球覆盖率的检查
• • • • • • • 核黄素覆盖测试检查法: 1.把核黄素均匀涂布在设备内表面 2.干燥 3.按照预定的清洗规程启动CIP 4.干燥 5.采用黑光灯检查 6.合格标准:无核黄素残留,在黑光灯照 射下无银光
管道盲管
为有效和可重复的清洗通常要求管路系统 中的分叉口和交接口的L/D小于2.0
仪表 阀门 D L
制定清洁规程
• 制定SOP的清洁验证的先决条件 • 通常参照设备的说明书制定详细的规程,规定每 台设备的清洗方法,并保证具有良好的重现性。 • 清洁规程的要点:
• • • • • • • • 系统的拆卸 预清洁 清洁剂、浓度、溶液量、水质量; 时间、温度、流速、频次、压力 消毒(浓度、方法、用量) 装配(按说明书要求) 干燥(明确干燥方式和参数) 检查(符合预定的标准)
DQ.IQ.OQ.PQ的联系与生产工艺
PQ包括以下内容:
实际的产品和在OQ中确立的运行参数和程序 确认在OQ中确立的工艺过程与实际能力 产品的可接受性 工艺过程的可重现性与可靠性
欧盟和中国 对工艺验证要求的差异
1、从验证的分类来看:
欧盟对工艺验证执行回顾性验证(收集批号至少 20批)或是同步验证(收集批号至少3批)没有 特别的要求。 但部分的欧盟检查官认为,API最先是在实验室 合成成功以后,转入生产的,因此应有前验证, 并在试验开发阶段确立关键工艺参数,但当解释 到我们多为老产品,检查官对前验证没有更多要 求。但要求在实验室条件下,评估关键工艺参数 。
工艺验证清洁验证检验方法验证

3、欧盟非常关注对变更的验证:
欧盟非常关注对变更的验证,特别是对起始物料 和关键工艺参数的变更。
这一类变更,一定要反映出变更前后杂质档案的 变化。
变更起始物料和关键工艺参数,一条重要的标准 是:物料的杂质档案(原料的杂质档案,成品的杂 质档案)没有发生变化,没有新杂质产生。
工艺性能和产品质量监控在整个 产品生命周期内的应用
技术转移
生产
生产终止
工艺放大活动 应运用良好 一旦生产终止
的监控能为工 的工艺性能 ,如稳定性研
艺性能和为成 和产品质量 究等监控应继
功整合到生产 监控体系来 续以完成该研
中去提供初步 确保性能受 究。应根据区
的提示。转移 控并确定改 域法规要求继
和工艺放大活 进领域
• 清洁验证
清洁验证主要的5个内容
• 1.法规对清洁验证的要求及背景 • 2.清洁方法的开发设计 • 3.清洁分析方法的验证 • 4.清洁验证示例及分析 • 5.清洁验证审计中经常出现的问题
法规对清洁验证的要求
• 新版GMP的要求 • 第一百五十五条
• 清洁方法应经过验证,证实其清洁的效果,以有 效防止污染和交叉污染,清洁验证应综合考虑设 备的使用情况、所使用的清洁剂和消毒剂、取样 方法和位置以及相应的取样回收率、残留物的性 质和限度,残留物检验方法的灵敏度因素。
验证方法和持续改进的基础。
生产活动的目标包括达到产品实现,建立和维持控制状态及促进持续改 进。制药质量体系应确保能经常达到理想的产品质量,能获得适宜的 工艺性能,控制系列是合适的,确定改定机会,且知识能持续拓展。
产品终止
产品终止活动的目标是有效地管理产品生产周期的最后阶段
金属素材端子IQC检验记录表样板
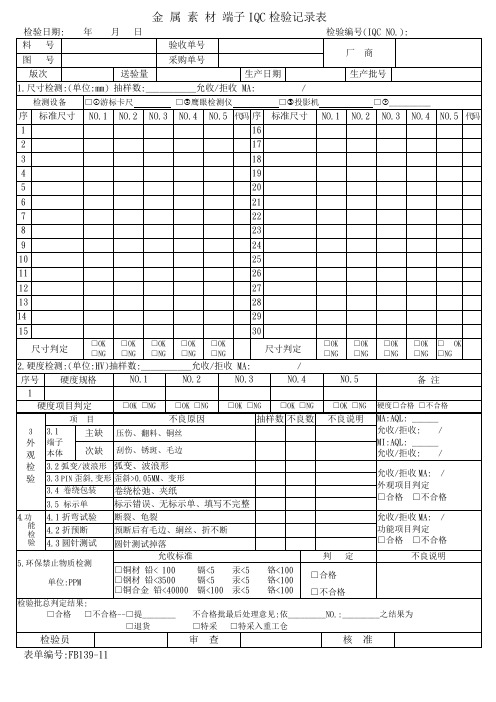
代码
序
标准尺寸
NO.1
NO.2
NO.3
NO.4
NO.5
代码
1
16
2
17
3
18
4
19
5
20
6
21
7
22
8
23
9
24
10
25
11
26
12
27
13
28
14
29
15
30
尺寸判定
□OK□NG
□OK□NG
□OK□NG
□OK□NG
□OK□NG
尺寸判定
□OK□NG
□OK□NG
□OK□NG
□OK□NG
□OK□NG
验
4.1折弯试验
断裂、龟裂
允收/拒收MA: /
功能项目判定
□合格□不合格
4.2折预断
预断后有毛边、絧丝、折不断
4.3圆针测试
圆针测试掉落
5.环保禁止物质检测
单位:PPM
允收标准
判定
不良说明
□铜材铅< 100镉<5汞<5铬<100
□钢材铅<3500镉<5汞<5铬<100
□铜合金铅<40000镉<100汞<5铬<100
MI:AQL: ______
允收/拒收: /
3.1
端子
本体
主缺
压伤、翻料、铜丝
次缺
刮伤、锈斑、毛边
3.2弧变/波浪形
弧变、波浪形
允收/拒收MA: /
外观项目判定
□合格□不合格
3.3PIN歪斜,变形
CL检查表模板

试剂与标准物质的验收
采购的试剂与标准物质是否检查标签、证书或其他证明文件的信息?
必要与可行时,是否通过适当的检测手段,以确保满足检测方法的要求?
对于痕量分析,是否关注试剂空白对检测结果的影响?
b) 如果检测方法中规定了内部质量控制方案与程序,包括规定限值,实验室是否严格执行?
如果检测方法中无此类方案,适用时,实验室是否采取以下方法:
(1)空白
(2)实验室控制样品
(3)加标
(4)重复检测
注:参考CNAS-CL10:2021 5.9 b〕注
c〕适用时,实验室是否使用控制图监控实验室能力?
质量控制图与戒备限是否基于统计原理?
是否选择适当的设备用于二级抽样、包装、提取等,以防止影响检测结果?
注:参考CNAS-CL10:2021 5.8 c〕注
d)是否对进入样品储存区的人员进展控制?
样品的保管人是否被授权并能履行其工作职责?
注:参考CNAS-CL10:2021 5.8 d〕注
e)实验室是否保存过期样品的处理与处置记录?
5.9检测与校准结果的质量保证
a〕实验室是否建立与实施充分的内部质量控制方案,以确保并证明检测过程受控以及检测结果的准确性与可靠性?
质量控制方案是否包括空白分析、重复检测、比对、加标与控制样品的分析?
方案中是否还包括内部质量控制频率、规定限值与超出规定限值时采取的措施?
质量控制方案是否覆盖申请认可或已获认可的所有检测技术与方法?
e) 实验室是否建立方案,尽可能参加能力验证或实验室间比对以验证其能力?
其频次是否与所承当的工作量相匹配?
工艺验证标准操作规程

XXXX有限公司现行文件1.目的:建立工艺验证的标准操作规程,并规范其操作。
2.范围:适用于公司所有生产产品工艺的验证。
3.责任:3.1生产部门负责验证方案与报告的编写;3.2化验室负责验证过程中的检测工作;3.3质量部负责验证方案与报告的批准与其中的偏差调查。
4.规程:4.1验证的类型:工艺验证通常可以按照以下三种方式进行:前验证、同步验证、回顾性验证。
4.1.1前验证:针对新的生产工艺或当工艺发生重大变化时所进行的工艺验证应采用前验证的方式,在验证成功结束之后才可以以放行产品。
工艺验证中所生产的产品批量应与最终上市的产品批量相同。
通常,工艺验证要求进行连续三个成功批次的生产。
4.1.2同步验证:在某些非常特殊的情况下也可以接受通过同步验证的方式进行工艺验证,即在常规生产过程中进行验证。
同步验证中生产的产品如果符合所有验证方案中规定的要求,可以在最终验证报告完成之前放行。
进行同步验证的决定必须合理、有文件记录并且经过质量部门批准。
同步性验证方法适用于以下情况:4.1.2.1由于需求很小而不常生产的产品;4.1.2.2生产量很小的产品,如放射性药品;4.1.2.3从前未经验证的遗留工艺过程,没有重大改变的情况下;4.1.2.4已有的、已经验证的工艺过程发生较小的改变时;4.1.2.5已验证的工艺进行周期性再验证时。
4.1.3回顾性验证:有些历史遗留的产品未进行工艺验证。
4.1.3.1这些工艺过程在满足以下条件可以通过对历史数据回顾的方式进行回顾性验证:4.1.3.1.1一直按照市售产品批量规模进行生产,能够很好的理解生产中的工艺过程并都记录下来;4.1.3.1.2有通过药典规定或经过验证实验方法进行检测所得到的充足可靠的验证数据;4.1.3.1.3对关键程序参数和关键质量特性做了规定并进行了控制;4.1.3.1.4建立了工艺过程的中间控制和可接受标准;4.1.3.1.5没有由于操作失误和设备故障之外而引起的任何工艺过程或产品失败;4.1.3.1.6在产品生产中应用的药物活性成分的杂质谱已经建立;4.1.3.1.7同时还应具备:工艺过程没有重大的历史改变:所有关键工艺参数和关键质量特征都可以作为有代表性的历史数据;执行回顾性验证的决定应得到质量部门批准。
固体生产线设备、容器具清洁残留验证及时限确认方案
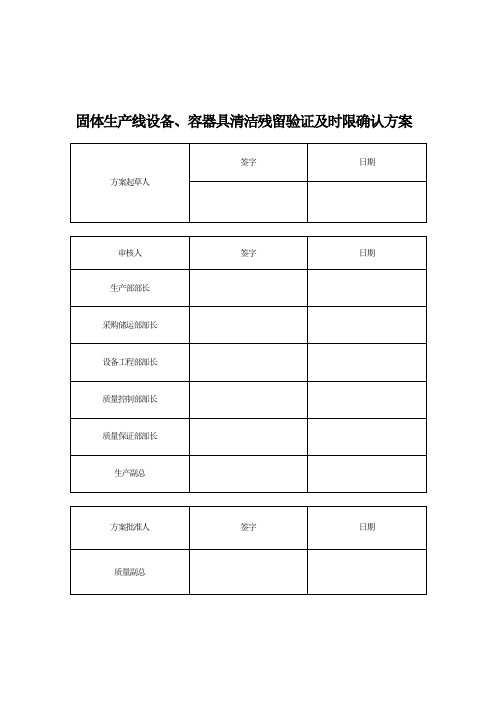
固体生产线设备、容器具清洁残留验证及时限确认方案目录1、概述2、目的3、范围4、职责5、时间进度6、人员培训7、文件/资料确认8、验证依据9、验证用仪器的确认10、清洁合格标准的制定方法通则11、取样方法12、最难清洁物(参照物)的确定13、清洁接受标准14、本次清洁验证标准汇总15、验证过程16、偏差与变更17、结果分析及评价、建议18、再验证周期19、附件清单附表一附表二附表三附表四附表五附表六附表七1、概述1.1为保证产品质量、避免污染及交叉污染,建立了D级洁净区容器具清洁消毒操作规程及生产线设备清洁操作程序,清洁目的是减少微粒、微生物、药物残留对后续药品质量的影响,保证产品的疗效、质量和安全性。
为对清洁程序实施的效果作一个正确评价,确保按规定的清洁程序操作后的设备、容器具,能真正达到防止污染及交叉污染的效果,我们对D级洁净区容器具清洁消毒操作规程、设备清洁操作规程进行验证。
2、目的设备、容器具按制定的清洁程序清洁后,通过目检、化学检测、微生物检测,确认固体制剂生产线设备、容器具经清洁后,不存在来自上批产品或清洁过程中带来污染的风险,以证实设备、容器具清洁程序的有效性。
确认当设备、容器具按已制订的清洁规程进行清洁后,能始终如一地达到预定的清洁标准要求。
3、范围本方案适用于按固体车间生产线设备清洁操作规程及《D级洁净区容器具清洁消毒操作规程》(SOP-PM-0000700)对设备、容器具进行清洁残留的验证及清洁时限的确认。
5、时间进度计划验证时间:2013年8月~2013年9月6、人员培训在本方案实施前,应对方案实施过程中涉及人员进行培训,以保证方案顺利实施,并做好培训记录,见附表一7、文件/资料确认检查操作文件及培训记录是否齐全,所用规程编写是否符合要求;文件资料是否符合GMP管理的要求,见附表二-1、二-28、验证依据《药品生产质量管理规范》(2010年修订)《药品GMP指南》口服固体制剂7.3 清洁验证《验证总计划》(SMP-QA-0200600)9、验证用仪器的确认检查验证用仪器是否完好,能否处于正常状态,见附表三10、清洁合格标准的制定方法通则❖(1)下一品种的批量(产品分组中的最小批量)--------A❖(2)取样面积(25cm2/棉签)---------C❖(3)与产品接触的总表面积(cm2)----------D❖(4)取样回收率(按>70%计,具体数值根据取样回收率试验计算)----------E ❖(5)上一品种生物活性日最低治疗剂量---------F❖(6)下一品种每日最高剂量----------G❖(7)安全因子(取10)----------H10.1化学残留量限度标准计算方法10.1.1分析检测浓度:10ppm擦拭法:表面残留物限度≤10ppm×A×E÷D×C÷H10.1.2生物活性的限度:最低日治疗剂量的1/1000擦拭法:表面残留物限度≤0.001×F÷G×A÷D×C×E12、最难清洁物(参照物)的确定12.2参照药典标准列出所有产品成份的溶解情况(表一),清洁过程为溶解过程,所以选择溶解度最差的成份作为清洁参照物,故初步选定联磺甲氧苄啶片中的磺胺嘧啶、磺胺甲噁唑及甲氧苄啶为清洁参照物。
TS复方板蓝根颗粒工艺验证方案审

建议与改进措施
优化工艺参数
根据实际生产情况,对工艺参数进行进一步优化,提高产品质量和 产量。
加强设备维护
定期对生产设备进行维护和检修,确保设备的稳定性和可靠性。
完善质量管理体系
加强原材料的质量控制,完善产品质量检测体系,确保产品的安全 性和有效性。
07参考文献与附件来自参考文献• [请在此处插入参考文献]
数据记录与整理
数据记录
01
对验证过程中的数据进行详细记录,包括工艺流程、
参数、质量检测等指标的数据。
数据整理
02 对记录的数据进行整理和分析,以评估TS复方板蓝根
颗粒工艺的稳定性和可靠性。
数据报告
03
根据数据分析结果编写验证报告,报告内容包括验证
结果、结论和建议等。
05
验证结果分析与评估
数据统计与分析
颗粒制备过程的质量控制
工艺流程
明确颗粒制备的工艺流程,包括 投料、混合、制粒、干燥、整粒
等步骤。
关键控制点
确定每个步骤的关键控制点,并明 确每个控制点的操作方法和判定标 准。
过程记录
建立每个步骤的过程记录,包括投 料记录、混合记录、制粒记录、干 燥记录、整粒记录等,以确保可追 溯性。
颗粒成品的质量标准及检验方法
产品质量可靠性
评估产品的质量稳定性和一致性,以及产品 在储存和使用过程中的变化情况。
06
验证结论与建议
验证结论
工艺流程合理
TS复方板蓝根颗粒的工艺流程设计合理,各工序环节紧密衔接 ,产品质量稳定。
工艺参数有效
工艺参数如温度、压力、时间等控制得当,能有效地保证产品质 量和产量。
设备运行良好
生产设备运行稳定,维护简单,能够满足生产需求。
板蓝根颗粒工艺验证

SOP-YZ-843-00 板蓝根颗粒工艺验证2 0 0 6年哈尔滨泰华药业股份有限公司板蓝根颗粒工艺验证验证目录1、验证立项申请表2、验证方案审批表3、验证方案4、验证记录5、验证报告6、验证证书验证立项申请表验证方案审批表板蓝根颗粒工艺验证方案哈尔滨泰华药业股份有限公司年月日验证方案目录1、概述1.1背景1.2目标1.3验证小组组成及职责1.3.1组成1.3.2职责1.4对象:生产工艺流程图2.验证方案内容2.1板蓝根颗粒各工序的工艺验证2.1.1工艺验证目标2.1.2设备2.1.3相关文件2.1.4生产系统要素和工艺变量的评价3.记录4.附表1.概述1.1背景板蓝根颗粒为治疗清热解毒类疾病的口服药。
是我厂的老产品。
我厂按GMP要求改造和扩建厂房、并更新设备,增加了现在国内先进的生产颗粒剂的关键设备,现以板蓝根颗粒拟生产进行工艺验证。
在验证之前,对纯化水系统、洁净空调系统(三十万级)、设备的安装,运行和性能验证,设备的清洗验证,检验仪器和仪表等均经过校正。
本次工艺验证以板蓝根颗粒工艺规程为依据。
在设备运行正常,以生产三批板蓝根颗粒的生产过程中实施(1万袋/批),对板蓝根颗粒的生产工艺条件,工艺参数及相关操作SOP进行同步验证。
1.2目标本产品工艺验证方案的目的在于为评价板蓝根颗粒生产系统要素和生产过程中可能影响产品质量的各种生产工艺变化因素,提供系统的技术参数,确认生产过程中的稳定性及生产系统的可靠性。
保证实现在正常的生产条件下,能始终如一地生产出符合预定规格质量标准的产品。
1.3验证小组组成及职责1.3.1组成组长:关欣组员:(生产技术部)闫文泉(质保部)张丽华、张弘(固体制剂车间)关欣、刘彦凯(物控部)何福春1.3.2职责1.3.2.1质保部、生产技术部负责验证小组的管理,验证报告的批准及合格证书的发放。
1.3.2.2验证小组部门的负责人对验证方案,验证报告进行审核会签。
1.3.2.3生产车间负责协助验证小组按验证方案组织验证的实施。
电机马达检验记录表

Db
三
功能
9 转向
从轴端视 顺时针转
噪音仪 目视
10 堵转2PCS
1次/
2次/
3次/
4次/
5次/
不可冒烟、起火、扎带断裂
1次/
2次/
3次/
4次/
5次/
目视
11 空载火花( ≤1.5级)
目视
12 震动 ( mm/s )
震动仪
13 引线(参照安规)
目视
14 Y电容(参照安规)
目视
15 温控器(参照安规)
I: 抽样计划:
检验项目
产品名称:
电机(马达)
批量:
产品型号:
抽样数:
产品规格:
箱号:
GB2828.1-2012/MIL-STD-105E表/Ⅱ级进行抽样(外观:一般检验水准Ⅱ
编号
项目
检验标准
规格
检验数据
1#
2#
1 表面不可生锈及残留过多防锈油;
AQL:致命=0 ;重要 = 1.0 ; 次要 = 2.5;尺寸:5PCS)
目视
序号:
16 其它
Defective Classification:(缺陷分类) Detail of Defectives(不良描述)
CR 致命
Ma(严重)
Mi(轻微)
1
2
3
4
5
判定结果:
□ 接受 □ 退货
Total Found:(不良总数)
检验员
确认
审核
目视
8 其它
1 螺孔(如: 4-M4右旋 )
螺纹栓规
2 引线长度
卡尺
3 引线长度
卡尺
4 引线长度
制药人必读--一文读懂工艺验证(上)

制药人必读--一文读懂工艺验证(上)第一节--工艺验证概述及传统工艺验证1工艺验证的定义工艺验证应当证明一个生产工艺按照规定的工艺参数能够持续生产出符合预定用途和注册要求的产品。
工艺验证可以有不同的验证方法,一般包括:传统工艺验证(前验证、同步验证)以及基于生命周期的工艺验证(工艺设计、工艺确认、持续工艺确认)。
工艺验证不应该是一次性的事情。
鼓励药品生产企业采用新的工艺验证方法,即基于生命周期的方法,将工艺研发、商业生产工艺验证、常规商业化生产中持续工艺确认相结合,来确定工艺始终如一的处于受控状态。
李永康老师手把手教您把控技术转移、工艺验证和清洁验证的相互关系,欢迎现场与讲师互动交流。
2工艺验证的一般原则工艺验证的方法和方针应该有文件记录,例如,在验证总计划中规定。
采用新的生产处方或生产工艺进行的首次工艺验证应当涵盖该产品的所有规格。
企业可根据风险评估的结果采用简略的方式进行后续的工艺验证,如选取有代表性的产品规格或包装规格、最差工艺条件进行验证,或适当减少验证批次。
工艺验证批的批量应当与预定的商业批的批量一致。
企业应当根据质量风险管理原则确定工艺验证批次数和取样计划,以获得充分的数据来评价工艺和产品质量。
企业通常应当至少进行连续三批成功的工艺验证。
对产品生命周期中后续商业生产批次获得的信息和数据,进行持续的工艺确认。
企业应当有书面文件确定产品的关键质量属性、关键工艺参数、常规生产和工艺控制中的关键工艺参数范围,并根据对产品和工艺知识的理解进行更新。
工艺验证一般在支持性系统和设备确认完成后才可以开始。
在某些情况下,工艺验证可能与性能确认同步开展。
用于工艺验证的分析方法已经过验证。
用于工艺验证批次生产的关键物料应当由批准的供应商提供,否则需评估可能存在的风险。
日常生产操作人员及工艺验证人员应当经过适当的培训。
工艺验证在执行前应进行适当的风险评估,以确定存在的风险点。
如企业从生产经验和历史数据中已获得充分的产品和工艺知识并有深刻理解,工艺变更后或持续工艺确认等验证方式,经风险评估后可进行适当的调整。
工艺验证标准操作规程

XXXX有限公司现行文件1.目的:建立工艺验证的标准操作规程,并规范其操作。
2.范围:适用于公司所有生产产品工艺的验证。
3.责任:3.1生产部门负责验证方案与报告的编写;3.2化验室负责验证过程中的检测工作;3.3质量部负责验证方案与报告的批准与其中的偏差调查。
4.规程:4.1验证的类型:工艺验证通常可以按照以下三种方式进行:前验证、同步验证、回顾性验证。
4.1.1前验证:针对新的生产工艺或当工艺发生重大变化时所进行的工艺验证应采用前验证的方式,在验证成功结束之后才可以以放行产品。
工艺验证中所生产的产品批量应与最终上市的产品批量相同。
通常,工艺验证要求进行连续三个成功批次的生产。
4.1.2同步验证:在某些非常特殊的情况下也可以接受通过同步验证的方式进行工艺验证,即在常规生产过程中进行验证。
同步验证中生产的产品如果符合所有验证方案中规定的要求,可以在最终验证报告完成之前放行。
进行同步验证的决定必须合理、有文件记录并且经过质量部门批准。
同步性验证方法适用于以下情况:4.1.2.1由于需求很小而不常生产的产品;4.1.2.2生产量很小的产品,如放射性药品;4.1.2.3从前未经验证的遗留工艺过程,没有重大改变的情况下;4.1.2.4已有的、已经验证的工艺过程发生较小的改变时;4.1.2.5已验证的工艺进行周期性再验证时。
4.1.3回顾性验证:有些历史遗留的产品未进行工艺验证。
4.1.3.1这些工艺过程在满足以下条件可以通过对历史数据回顾的方式进行回顾性验证:4.1.3.1.1一直按照市售产品批量规模进行生产,能够很好的理解生产中的工艺过程并都记录下来;4.1.3.1.2有通过药典规定或经过验证实验方法进行检测所得到的充足可靠的验证数据;4.1.3.1.3对关键程序参数和关键质量特性做了规定并进行了控制;4.1.3.1.4建立了工艺过程的中间控制和可接受标准;4.1.3.1.5没有由于操作失误和设备故障之外而引起的任何工艺过程或产品失败;4.1.3.1.6在产品生产中应用的药物活性成分的杂质谱已经建立;4.1.3.1.7同时还应具备:工艺过程没有重大的历史改变:所有关键工艺参数和关键质量特征都可以作为有代表性的历史数据;执行回顾性验证的决定应得到质量部门批准。
针管装配,点胶,硅化,固化工艺验证方案

针管装配,点胶,硅化,固化工艺验证方案江西***医用器械有限公司1 验证目的对留置针针管装配,点胶,硅化,固化工艺验证,验证采用的针管装配机,立式烘干机和UV固化机和点胶工艺,硅化工艺和固化工艺符合产品的使用要求;确定工艺文件。
2 适用范围本验证方案适用于江西丰临医用器械有限公司留置针车间留置针系列产品针管装配,点胶,硅化,固化工艺验证。
3 验证设备:针管装配机,立式烘干机,UV固化机。
4 本次验证使用点胶和硅化材料:(2)立式烘干机,型号规格:DHG-9432A.(3)360(1)低温固化潜伏环氧胶,产品型号:QJ-02型。
和4159硅油加稀释剂5 验证时间2012年10月21日起6 验证依据、引用标准和参考文献:验证依据:一次性使用静脉留置针、一次性使用无针连接式正压留置针注册标准引用标准和参考文献:GB/ T 2828.1 计数抽样检验程序第一部分:用于逐批检验按可接受质量界限(AQL)检索的抽样计划7 验证7.1运行确认(OQ):A 验证设备的功能操作;B 确认当设备运行于设定条件、上限设定条件、下限设定条件时的组装过程,并对产品的实际组装,点胶,硅化和固化过程进行评估。
7.2. 环氧胶,,360和4159加稀释剂配比硅油的选择评价确定每支针管环氧胶的标准量,360和4519加稀释剂的最佳配比浓度和硅化时间。
7.3热烘箱烘干温度设定和 UV固化条件设定(见吴刚UV紫外线固化验证)。
7.4性能确认1.针管加针座组装到位的长度标准确认2.各配比硅油浓度和静置时间的穿刺力测试对比。
3.环氧胶的连接牢固度测试4.立式烘干箱和UV固化机的烘干效果对比。
8.总结验证结果并写出可行性报告。
验证人: **** 日期:2012.10.20。