分子诊断学重点1
分子诊断学 期末复习资料

1.(一)真核生物基因组:①有细胞核基因组和细胞器基因组之分;②核基因组以线状DNA分子的形式存在于染色质上;③基因多数为断裂基因,有内含子结构和大量重复序列;④单顺反子,基因家族;⑤核基因组由染色体DNA组成,分子量大,结构复杂,与蛋白质结合。
(二)原核生物基因组:①基因组相对较小,通常为一条双链DNA分子②功能相关的基因高度集中,构成操纵子③重复序列少,大多数的蛋白质基因保持单拷贝形式,而RNA基因常是多拷贝④没有内含子,基因是连续的⑤多顺反子,构成转录单位⑥绝大部分DNA都是用于编码蛋白质的,只有很少不编码序列⑦DNA分子中有各种功能区(三)病毒基因组:①只有一种核酸,4种类型,为双链DNA、单链DNA、双链RNA、单链RNA ②核酸大小差别较大③具有启动子和操纵子结构④具有重叠结构2.核酸提取总原则和过程:1)分离制备总原则:①保证核酸一级结构的完整性②尽量排除其他的污染,保证核酸的纯度2)核酸提纯步骤:①破碎细胞:超声破碎,匀浆法、低渗法②提取:采用酶学或化学试剂酚等去除蛋白质及其他大分子;③纯化:3.核酸分子杂交基本原理:利用核酸变性和复性的性质,使具有一定同源性的两条核酸单链在一定条件下按照碱基互补配对原则形成异质双链的过程。
(可发生在同源或异源的DNA链与DNA链之间,也可在RNA链与DNA链)4.理想探针的特点:①高度特异性②易于标记和检测③灵敏度高④稳定且制备方便5.核酸探针的种类和优缺点:1)基因组DNA探针:优点:制备方法简便、省时、易得,稳定不易降解,标记方法多样也较成熟缺点:杂交中可能会自我复性2)cDNA探针:优点:具有基因组DNA优点,且不存在内含子和其他高度重复序列,尤其适用于基因表达研究。
缺点:制备困难。
3)RNA探针:优点:不含高度重复序列,可降低非特异性杂交,复杂性低,杂交反应效率高;缺点:不稳定,标记方法复杂。
4)寡核苷酸探针:优点:①短的探针比长探针杂交速度快,特异性好;②可以在短时间内大量制备;③可以在合成过程中进行标记制成探针;④可合成单链探针,避免用双链DNA探针在杂交中的自我复性,提高了杂交效率;⑤可以检测小DNA片段缺点:长度有局限性,短链优于长链。
分子诊断复习

名词解释:Molecular diagnosis :分子诊断,是指通过检测基因的结构异常或其表达异常,对人体的健康状态和疾病做出诊断的方法,属于病因学诊断。
评估基因的存在和缺陷通常以DNA为材料,而评估基因的表达量则以基因转录或翻译的产物RNA或蛋白质为材料来分析评估个体的生理/病理状态。
目前PCR、寡核苷酸探针杂交、DNA测序仍然是分子诊断的主流技术。
Molecular cloning:分子克隆,是指按照人的意愿, 在体外将制备的DNA片段与载体重组, 然后导入受体细胞, 并在受体细胞中复制、扩增,以获得该DNA分子的大量拷贝。
此技术称为分子克隆技术或DNA重组技术。
Molecular hybridization:核酸分子杂交,是在核酸变性与复性的基础上建立起来的一种分子生物学技术。
具有一定同源性的两条DNA链或两条RNA链或一条DNA链和一条RNA链在一定条件下按照碱基互补配对原则形成异质双链的过程称为分子杂交。
PCR:聚合酶链反应,是已知特异性DNA序列的体外引物定向酶促扩增技术。
RT-PCR:逆转录PCR,先将RNA用逆转录酶逆转录成cDNA,然后再加入特异引物对目标片段进行扩增,扩增产物的分析与其他常见PCR方法类似。
常用的逆转录酶有AMV逆转录酶(最适温度42℃)和MoMLV逆转录酶(最适温度37℃),常用的逆转录引物有三种:随机引物、Oligo(dT)和特异性引物。
FQ-PCR:实时荧光定量PCR技术,是指在PCR反应体系中加入荧光基团,利用荧光信号累积实时监测整个PCR 进程,最后通过标准曲线对未知模板进行定量分析的方法。
Gene chip:基因芯片,又称DNA芯片或DNA微阵列,即将大量的基因片段有序地、高密度地固定排列在玻璃片、硅片或纤维膜等载体上制成点阵。
Genetic engineering:基因工程,是指将基因进行克隆, 并利用克隆的基因表达、制备特定的蛋白质或多肽产物, 或定向改造细胞和个体的特性所用的方法及相关工作。
01-绪论分子诊断学

第三节 分子诊断学在医学中的应用
感染性疾病的诊断
HBV病毒
HIV病毒
第三节 分子诊断学在医学中的应用
遗传性疾病的诊断
第三节 分子诊断学在医学中的应用
老年痴呆症可以通过基因诊断进行早期预测
第三节 分子诊断学在医学中的应用
白化病
PCR快速检测致食物中毒的细菌
选择性培养 基增菌
DNA提取
样品 增菌 PCR
2. 人体三大功能调控系统(神经、内分泌和免
疫)的分子生物学基础
3. 细胞增殖、分化和凋亡的分子基础
4. 基因的结构异常或表达异常与疾病的关系
5. 应用分子生物学技术进行基因诊断、基因治
疗、生物制药和卫生防疫。
第一节 分子诊断学的定义及其研究范畴
第一节 分子诊断学的定义及其研究范畴
分子生物学分支学科 • 医学分子生物学 • 药学分子生物学 • 神经分子生物学 • 肿瘤分子生物学 • 分子病理学 • 分子药理学 • 分子免疫学 • 分子遗传学 • 分子诊断学 • 分子病毒学 ……
二、分子诊断学发展简史
分子诊断学发展史就是
分子生物学技术发展史。
基因诊断概况
第二节 分子诊断学的发展简史
1978年Kan首次成功地将分子杂交技术用
于镰状细胞贫血的产前基因诊断;
基因诊断技术史:
第一阶段 第二阶段 第三阶段
代表技术-分子杂交技术 代表技术-常规PCR(1985年) 代表技术-生物芯片技术
• 黄培堂 等译2002分子克隆实验指南(第三版) 科学出版社
• Sambrook J.等, 1982 Molecular Cloning 1989(2nd edition 2000(3rd edition)
分子诊断1

名词解释分子诊断学(molecular diagnostics):分子诊断学是以分子生物学理论为基础,利用分子生物学的技术和方法来研究人体内源性或外源性生物大分子和大分子体系的存在、结构或表达调控的变化,为疾病的预防、诊断、治疗和转归提供信息和依据的一门学科。
基因组(Genome):指生物体全套的遗传信息。
基因组学是研究生物基因组的组成、组内各基因的结构、功能及表达调控的科学,包括基因组核苷酸序列测定、基因组作图、基因定位及基因功能分析等。
基因组学(genomics):是研究生物基因组的组成,组内各基因的结构、功能及表达调控的科学,包括基因组核苷酸序列测定、基因组作图、基因定位及基因功能分析等。
包括结构基因组学和功能基因组学。
人类基因组多样性(human genome diversity)同一种或不同种基因组均存在或多或少的差异,这种差异称人类基因组多样性。
微卫星DNA多态性:微卫星DNA的排列方式有的三种:完全重复(无间隔),不完全重复(有非重复序列的间隔)和混合重复(2个或多个重复序列彼此毗连连续出现)。
完全出现最多见的方式。
转座因子:能在基因组中从一个位点移至另一位点的DNA序列,称为转座因子或转座元件。
黏性末端:对于双链DNA病毒而言,基因组双链两端具有能够互补的单链DNA部分,称为黏性末端。
重叠基因(overlapping gene):是指两个或两个以上基因的ORF共有一段DNA序列,即某段DNA序列成两个或两个以上基因的共有的组成部位。
分段基因(segnented genome):是指病毒基因组中由几条不同的核酸分子组成,另见于tRNA病毒、RNA病毒及双链RNA病毒。
人类基因组计划(HGP):是旨在测出人类基因组30亿碱基对的核苷酸序列,发现所有人类基因并确定他们在染色体上的位置,破译人类全部遗传信息,发展基因组学新技术,探讨人类基因组研究的社会、法律和伦理等问题的一个国际性研究项目。
分子诊断知识科普

分子诊断知识科普分子诊断是一种基于分子生物学和遗传学原理的诊断方法,通过分析个体的基因、蛋白质或其他分子水平的信息,来判断其是否患有某种疾病或具有某种特定的遗传变异。
分子诊断可以通过检测基因突变、基因表达水平、蛋白质标记物等来识别疾病的存在或发展状态。
与传统的疾病诊断方法相比,分子诊断具有更高的准确性和灵敏度。
传统的诊断方法主要依靠临床症状、体征和影像学检查等,但这些方法往往无法提供足够的信息来进行准确的诊断。
而分子诊断则可以直接检测疾病相关的分子标记物,从而提供更为准确的诊断结果。
一、分子诊断的基本原理分子诊断的基本原理是通过检测和分析个体的基因组、转录组和蛋白质组等分子信息,来确定是否存在某种疾病或病理状态。
这种方法通常需要从患者的血液、体液或组织样本中提取并分析分子,并与正常个体或已知疾病个体的分子信息进行比对。
分子诊断的核心技术包括基因测序、PCR(聚合酶链式反应)、核酸杂交等。
其中,基因测序是一种通过测定DNA序列来获取个体基因信息的方法。
PCR是一种通过扩增DNA片段来增加检测灵敏度的方法。
核酸杂交则是一种通过将目标序列与一段互补的DNA或RNA序列结合来检测目标序列的方法。
通过这些技术,分子诊断可以检测到包括遗传疾病、感染病、肿瘤等在内的多种疾病。
例如,通过检测BRCA1和BRCA2基因的突变可以判断一个人是否患有乳腺癌或卵巢癌的遗传风险。
通过检测某种病原体的DNA或RNA可以确定感染者的感染状态。
通过检测肿瘤细胞中的特定基因突变可以确定肿瘤的类型和治疗策略。
二、分子诊断的应用领域分子诊断在医学领域有着广泛的应用。
下面将介绍一些常见的应用领域。
1. 遗传疾病诊断:分子诊断可以通过检测个体的基因突变来确定遗传疾病的存在和风险。
例如,通过检测孩子的基因突变可以确定其是否患有遗传性疾病,如先天性心脏病、遗传性失聪等。
2. 传染病诊断:分子诊断可以通过检测病原体的DNA或RNA来确定感染病的存在和类型。
分子诊断学知识点总结

分子诊断学知识点总结分子诊断学是指利用分子生物学的技术和方法,对生物体内的DNA、RNA、蛋白质等分子水平进行诊断和检测的一门学科。
随着分子生物学技术的不断发展和进步,分子诊断学在临床诊断、疾病预防和治疗等方面发挥着越来越重要的作用。
下面将对分子诊断学的基本原理、常见技术和应用进行概述。
一、基本概念1. DNA、RNA和蛋白质的基本结构和功能DNA是生物体内的遗传物质,包含了细胞的遗传信息,主要存在于细胞核中。
RNA是一种中间体分子,可以将DNA中的遗传信息转录成蛋白质。
蛋白质是生物体内的重要分子,是细胞结构和功能的基本单位。
2. 基因突变与疾病基因是决定生物性状的遗传信息的单位,基因突变是指基因序列发生了变化,可能导致蛋白质功能异常,甚至引发疾病。
3. 分子诊断学的基本原理分子诊断学利用分子生物学技术对生物体内的分子进行检测和分析,从而实现疾病的诊断、预防和治疗。
二、常见技术1. 聚合酶链式反应(PCR)PCR是一种在体外扩增DNA片段的技术,可以从少量的DNA样本中扩增出大量的DNA片段,是分子诊断学中常用的技术手段。
2. 核酸杂交技术核酸杂交技术是一种通过DNA或RNA的互补配对进行检测的方法,可以用于寻找特定基因或病毒的存在。
3. 蛋白质质谱分析蛋白质质谱分析是一种通过蛋白质的质量和结构来对蛋白质进行分析和检测的技术。
4. 基因测序技术基因测序技术是一种对DNA序列进行测定和分析的技术,可以帮助人们了解基因的结构和功能。
5. 基因芯片技术基因芯片技术是一种可以在一个芯片上同时检测多个基因的技术,可以用于疾病的诊断和预测。
三、应用领域1. 临床诊断分子诊断学在临床诊断中可以对各种疾病进行快速和精准的诊断,如肿瘤、遗传病、感染病等。
2. 疾病预防分子诊断学可以通过对病原体的检测和分析,帮助人们预防感染性疾病的发生和传播。
3. 个体化治疗分子诊断学可以根据个体的基因信息,为患者提供个性化的治疗方案,提高治疗的效果和减少副作用。
分子诊断学重点内容

分子诊断学重点内容(Navy)1、分子诊断学(molecular diagnostics)是利用分子生物学技术来研究机体外源性和内源性生物大分子和大分子体系的存在、结构或表达调控的改变,从而为疾病的预测、预防、诊治和转归提供分子水平信息。
2、基因:是一段携带功能产物(多肽,蛋白质,tRNA和rRNA和某些小分子RNA)信息的DNA片段,是控制某种性状的的遗传单位。
3、密码子偏爱(codon bias )指在不同物种的基因中经常为某种氨基酸编码的只是其中的一个密码子。
当鉴别到一个ORF时,密码子偏爱常常用来确定这个ORF是否是一个基因。
4、基因组(genome):指一个细胞或生物体的一套完整的单倍体遗传物质。
5、C值矛盾:生物体的进化程度与基因组大小之间不完全成比例的现象称为C值矛盾也称C值佯谬( C value paradox)6、N值矛盾:基因组中基因数目与生物进化程度或复杂程度的不对称性,称为N值矛盾或N值佯谬(N value paradox)。
7、基因组计划是指以获得某物种基因组全序列为主要目标的科学计划。
8、厘摩尔根(cM)即重组频率为1%的两个基因间的遗传距离9、基因组学(Genomics)指对所有基因进行基因组作图(包括遗传图谱、物理图谱、转录图谱)核苷酸序列分析,基因定位和基因功能分析的一门科学。
10、鉴别蛋白质编码基因的五项标准是什么?开放阅读框、密码子偏爱、序列保守性、转录产物、基因失活11、原核生物是细菌等原始生物的总称,是最简单的细胞生物体12、质粒:独立于细菌细胞染色体以外,能自主复制并稳定遗传的共价闭合环状DNA(cccDNA)分子,称为质粒(plasmid)13、琼脂糖凝胶电泳泳动速度:超螺旋DNA分子>线性DNA分子>半开环DNA分子14、接合作用(conjugation)当细胞与细胞、或细菌通过菌毛相互接触时,质粒DNA 从一个细胞(细菌)转移至另一细胞(细菌)的DNA转移称为接合作(conjugation)。
分子诊断学

研究对象
Artwork from /
Old Way
New Way
Patient’s tissue sample
Proteomics
Proteins
Pathology
Mass Spectrometry Proteomic image
Genomics Patient’s tissue sample or blood sample
DNA Gene chip Microarray image
Edited from the artwork by Jeanne kelly © 2002 Donna Kerrigan, M.S. Jeanne Kelly, Brian Hollen. Understanding Cancer and Related Topics Understanding Molecular
ISH(In-situ Hybridization,原位杂交) 3、测序技术 4、芯片技术
Microarrays(微阵列) Tissue Arrays(组织芯片) 5、质谱技术(Mass Spectroscopy)
核酸等温控制技术在呼吸道病 原菌检测中的应用
艾滋病的核酸检测与分析
动脉粥样硬化性重要疾病: 冠心病基因组研究进展
& The Association for Molecular Pathology
美国研究病理学会和分子病理学协会
中山大学
2002
Molecular diagnostics: a training and study guide Gregory J. Tsongalis William B. Coleman
第十五章 分子诊断的其它应用
第八章 蛋白质组学研究技术 第十六章 生物信息学在分子诊断中的应用
分子诊断重点

名词解释1、基因:基因是一段携带功能产物(多肽,蛋白质,tRNA和rRNA和某些小分子RNA)信息的DNA片段,是控制某种性状的的遗传单位。
2、类核:原核生物基因组DNA位于细胞的中央区,与支架蛋白和RNA结合在一起,以复合体的形式存在,经高度盘旋聚集形成一个较为致密的区域,称为类核。
质粒:是细菌细胞染色体以外的能独立复制并能稳定遗传的共价闭合环状DNA分子。
重叠基因(o v e r l a p p i n g g e n e):病毒基因组的一段D N A序列有两个或两个以上的开放读码框架,可以编码两种或两种以上的多肽链,称为重叠基因。
载体(vector):是指能携带目的基因进入宿主细胞进行扩增和表达的一类DNA分子。
指在P C R扩增过程中荧光信号开始由本底进入指数增长阶段的拐点所对应的循环次数。
荧光定量P C R:在P C R反应体系中加入荧光基团,利用荧光信号积累实时监测整个P C R进程,最后通过标准曲线对未知模板进行定量分析的方法。
淬火:将热变性DNA骤然冷却的处理过程。
核酸探针:是指能与待测的靶核酸序列互补杂交的已知序列的核酸片段。
一般带有某种适当的标记以便检测。
基因芯片:指在固相支持物上原位合成寡核苷酸或者直接将大量基因探针以显微打印的方式有序地固化于支持物表面,然后与标记的样品杂交,通过对杂交的检测分析,得出样品的遗传信(基因序列及表达的信息)。
S o u t h e r n印迹杂交:DNA/DNA杂交,即将DNA电泳、转印到固相支持物上,用探针进行检测的方法。
荧光原位杂交(F I S H):是一种利用非放射性的荧光信号对原位杂交样本进行检测的技术。
通过荧光标记的探针与待测标本的核酸进行原位杂交,在荧光显微镜下对荧光信号进行辨别和计数,从而对染色体或基因异常的细胞、组织标本进行检测和诊断。
运用分子生物学技术直接检出微生物的DNA/RNA,判断有无感染和何种病原体感染。
16、完整检出策略:运用分子生物学技术直接检出微生物的DNA/RNA,判断有无感染和何种病原体感染,并能对病原体进行分类、分型(亚型)和耐药性鉴定。
分子诊断学复习

分子诊断学复习名词解释1.DNA测序:基因是遗传信息的物理和功能单位,含有产生一条多肽链或功能RNA所必需的全部核苷酸序列,即一段DNA序列2.蛋白质组(proteome):源于蛋白质(protein)与基因组(genome)两个词的杂合,意指“一种基因组所表达的全套蛋白质”,即包括一种细胞或一个组织乃至一种生物所表达的全部蛋白质。
3.蛋白质组学(proteomics) :蛋白质组学就是对机体或组织或细胞的所有蛋白质进行鉴定和结构功能分析的一个研究领域。
基因:是一段携带功能产物信息的DNA片段,是控制某种性状的遗传单位。
4.基因组(genome):是指一个细胞或生物体的一套完整的单倍体遗传物质。
5.基因组学(Genomics):指对所有基因进行基因组作图(包括遗传图谱、物理图谱、转录图谱,核苷酸序列分析,基因定位和基因功能分析的一门科学。
6.基因扩增:定义:指基因拷贝数增加的现象,基因扩增是基因表达增加的一种重要机制。
7.聚合酶链反应(PCR):利用DNA聚合酶催化一对引物间的特定DNA片段在体外进行快速、大量扩增的方法,也称无细胞克隆技术。
8.增色效应:DNA变性后,双螺旋解体,碱基堆积不复存在,螺旋内部的碱基暴露,致变性后的DNA对260nm紫外光的吸光率明显增加,这种现象称为增色效应。
9.熔解温度:将DNA解链达到50%或OD260达到最大值的50%时的温度,称为解链温度或熔解温度。
10.溴化乙锭(ethidium bromide,EB)是一种荧光染料,可嵌入核酸双链的碱基对之间,在紫外线激发下发出红色荧光。
11.引物:与靶基因片段两侧互补的寡核苷酸,其本质是单链DNA,它决定扩增产物的特异性和长度。
12.短产物片段:的长度严格限定在2个引物链的5’端之间,是需要扩增的特定片段,以指数形式扩增(2n)。
13.长产物片段:比两引物限定区更长的延伸产物,是以原始模板DNA为模板的扩增反应产物,以倍数形式扩增(2n)。
分子诊断1,2

HER-2作为治疗的靶点 :
Herceptin 对Her-2基因扩增(HER-2蛋白高表达)的患者 有明确的疗效。
54
荧光原位杂交(FISH)检测HER-2
用已知的标记单链核酸 为探针,按照碱基互补 的原则,与待检材料中 未知的单链核酸进行异 性结合,形成可被检测 的杂交双链核酸。由于 DNA分子在染色体上是沿 着染色体纵轴呈线性排 列,因而可以探针直接 与染色体进行杂交从而 将特定的基因在染色体 上定位
体外诊断试剂盒
30
分子诊断的现状及在医学中的应用
感染性疾病的分子诊断
病原微生物感染导致的疾病,仍然是严重威胁人类健康的一个重要方 面。传统的病原体检测(微生物学检测、免疫学检测和血液学检测), 不易早期诊断,并受到灵敏度限制。 例如:肝炎系列、HIV,以及近年来一些严重的传染病疫情方面,分 子诊断都发挥了重要的作用。
15
蛋 白 质 合 成
ห้องสมุดไป่ตู้
核酸的分布
1.
2.
3.
DNA:细胞核(98%)、 线粒体、叶绿体 RNA:细胞核、细胞质 rRNA:核仁中合成,占 总RNA的80% tRNA:核内合成,占总 RNA的15% mRNA:核内合成,占 总RNA的5%
16
17
18
什么是分子诊断?
传统的诊断: 宏观→微观,大→小,外→里 分子诊断: 微观,生物大分子是构成机体和参与生命 活动的主要成份。
59
PCR的原理:
95℃
模板DNA
60
PCR的原理:
PCR的基本原理
DNA引物
PCR反应条件 50℃ 95℃ PCR过程 PCR的特点
分子诊断学重点

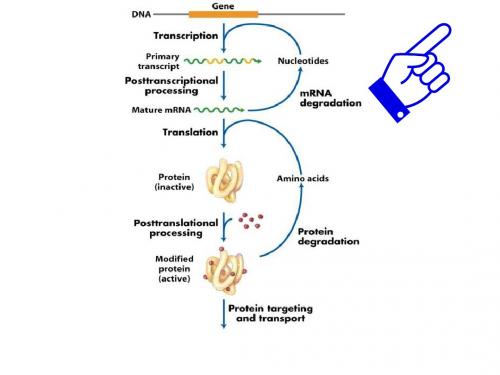
molecular diagnostics:It represents the application of the principles of basic molecular biology to the investigation of human disease processes.中心法则is the theory that DNA encodes RNA (the process of transcription) which encodes proteins (translation), but that information does not flow from proteins to DNA.DNA复制:the process by which the double helix is unwound and each strand acts as a template. Bases are matched to synthesize the new partner strands.转录:the process by which an RNA copy of a gene is made.逆转录:synthesis of a DNA using an RNA template.翻译:the process by which ribosomes use the information in mRNAs to synthesis proteins.基因: the basic unit of heredity. Usually, this is equated with the production of one RNA or one protein. A gene contains coding regions, introns, untranslated regions and control regions内含子:a region that interrupts the transcribed part of a gene. An intron is transcribed, but is removed by splicing during maturation of the transcript. The word refers to the intervening sequence in both the DNA and its RNA product.外显子: a region of a gene that is ultimately represented in that gene’s mature transcript.The word refers to both the DNA and its RNA product. RNA剪切:Remove the noncoding sequences from primary RNA transcript.选择性剪切:produce of many different proteins from one gene.从一个mRNA 前体中通过不同的剪切方式产生不同mRNA剪接异构体的过程,而最终的蛋白质也会不同。
分子诊断学复习资料

分子诊断学复习资料分子诊断学复习资料——参照中国医药科技出版社(第二版)编写1.分子诊断学:是以分子生物学理论为基础,利用分子生物学的技术和方法来研究人体内源性或外源性生物大分子和大分子体系的存在、结构或表达调控的变化,为疾病的预防、诊断、治疗和转归提供信息和依据。
①主要特点:直接以疾病基因为探索对象,属于病因学诊断,对基因的检测结果不仅具有描述性,更具有准确性;可准确诊断疾病的基因型变异,基因表型异常以及由外源性基因侵入引起的疾病。
灵敏度高,便于早期诊断及疾病预防;以基因分析为基础,特异性高。
②发展简史(三个阶段)第一个阶段:准备和酝酿阶段1953年前,蛋白质是生命的物质基础,DNA是遗传物质基础。
(三个实验:肺炎双球菌转化实验、体外转化实验、噬菌体转导实验)、第二个阶段:DNA双螺旋结构、中心法则、PCR技术第三个阶段:2001年,首张人类基因组序列图谱及其他物种基因组序列的公布。
基因组分、蛋白质组学等方面的巨大技术进步,促进进入第三阶段,即以生物芯片技术为代表的高通量密集型技术。
③主要应用:感染性疾病、遗传性疾病、肿瘤的分子诊断,个体化医疗,其他如耐药性、疗效监测,多基因疾病2.基因:是有功能的DNA片段,含有合成有功能蛋白质多肽链或RNA所必学的全部核苷酸序列,是遗传的结构和功能单位。
分为结构基因和调控基因。
①结构基因:编码蛋白质或RNA的编码序列调控基因:保证转录功能起调控作用的非编码序列②操纵子(operon)操纵基因与其控制下的结构基因共同组成的功能单位③断裂基因:指基因的内部存在间隔区,间隔区的DNA序列与该基因所决定的蛋白质没有关系。
间隔区又称为内含子。
出现在成熟RNA中的有效区段为外显子。
重叠基因:指基因的开放阅读框(ORF)存在一个或多个核苷酸重叠的基因跳跃基因:又称转座子,基因在染色体上的位置不固定,能由一条染色体跳到另一条染色体上。
必须基因:生物体中存在的一些维持生物细胞生长所必需的基因,缺少或突变这些基因均能导致生物体死亡3.基因组(gnome):细胞中一套完整单倍体的遗传物质的总和基因组结构主要指不同的DNA功能区在DNA分子中的分布和排列4.C值:基因组的大小又称C值,常用碱基数目或碱基对数目来描述①C值是特异的,物种不同,差异极大,真核生物中,一般随着进化,C值增大②C值矛盾:又称C值悖论,生物的进化程度与基因组大小不完全成比例的现象5.真核生物基因组1)基本特征:①有细胞核基因组和细胞器基因组之分;②核基因组以线状DNA分子的形式存在于染色质上;③基因多数为断裂基因,有内含子结构和大量重复序列;④单顺反子,基因家族;⑤核基因组由染色体DNA组成,分子量较大,结构复杂,与蛋白质结合。
分子诊断重点

1、基因——DNA分子中含有特定遗传信息的核苷酸序列,是遗传物质的最小功能单位。
合成有功能的多肽链或RNA所必需的全部核酸序列(通常是DNA序列)。
2、基因组:广义:一物种的全部遗传物质及其携带的遗传信息。
狭义:单倍体细胞中的全套染色体(人:22条常染色体+ X,Y + 线粒体DNA)3、断裂基因:基因的编码序列在DNA上不是连续的,而是被不编码的序列隔开。
4、重叠基因:基因组DNA中某些序列被两个或两个以上的基因所共用。
基因重叠现象在病毒基因组中普遍存在。
5、跳跃基因:即转座子,在哺乳动物体内一般含有几十万个跳跃基因DNA;6、基因家族指核苷酸序列或编码产物的结构具有一定程度同源性的一组基因7、假基因(pseudogene): 在多基因家族中有的成员并不能表达出有功能的产物,用ψ表示。
8、SNP:单核苷酸多态性指单个核苷酸变异而形成的DNA分子多态9、质粒:细菌细胞染色体以外,能独立复制并稳定遗传的共价闭合环状分子10、转座因子又称转座元件(transposable element),是一类在细菌染色体、质粒或噬菌体之间自行移动并具有转位特性的独立的DNA序列11、微卫星:广泛分布在原核和真核生物基因组中,常出现在基因的非编码区和染色体末端,重复序列长度仅为1—6bp,呈串联重复排列。
12、黏性末端:对于双链DNA病毒而言,基因组双链两端具有能够互补的单链DNA部分,称为粘性末端13、原核生物基因组的特点1.原核生物基因组:DNA原核生物基因组DNA较小,基因数目较少原核生物基因组中只有一个DNA复制起点;2原核生物的操纵子结构;操纵子结构是原核生物基因组的功能单位, 原核生物的结构基因大多数按功能相关性成簇地串联排列于染色体上;3.原核生物的结构基因多数是单拷贝基因,只有编码rRNA和tRNA的基因有多个拷贝;4原核生物结构基因的编码顺序一般不重叠;5.具有编码同工酶的不同基因6.GC含量差异大7.非编码区内主要是一些调控序列;8.具有可移动的DNA序列第三章分子克隆1、分子克隆:按照人的意愿,在体外将制备的DNA片段与载体重组,然后导入受体细胞,并在受体细胞中复制、扩增,以获得该DNA分子的大量拷贝,此过程称为分子克隆,也称基因克隆或DNA克隆或重组DNA (recombinant DNA) 。
分子诊断学_绪论感染性疾病的分子诊断
2. 完整检出策略:不仅对病原体做出判断,还要 进行分型(包括亚型)和耐药性方面的检测。 思路:诊断—分型—亚型—耐药性检测
二、感染性疾病分子诊断的常用方法
1. 信号放大检测技术方法 不涉及靶分子扩增,采用多酶、多探针或二者结 合等方式增加探针标记物的浓度从而使信号得 到放大,提高检测的灵敏度。如:液相杂交、 杂交捕获及支链DNA(bDNA)等
HIV基因的检测 人类免疫缺陷病毒(human immunodeficiency virus, HIV)
参考NSFC申请材料
单纯疱疹病毒(herpes simplex virus,HSV)
巨细胞病毒(cytomegalovirus virus, CMV)
Section 3 病原菌的基因检测
必须在规范的基因扩增实验室中进行。
应采取必要的质控规程,包括阳性对照和阴性 对照。
阳性结果时必须对原始样本重复检验: 或者扩增另一个基因片段 或在另一个实验室对同一样本进行检测。
肝炎病毒基因的检测(hepatitis B virus, HBV)
乙肝病毒基因组中有4个ORF,分别称为S、C、 P和X。S区编码HBV的包膜蛋白,C区编码含 HBeAg和HBcAg的多肽,P区编码依赖RNA的 DNA聚合酶。
阳性结果解释
检测质控是否正常。 注意检测的特异性和可能受到的污染等情况。 产生假阳性的因素:引物探针的特异性、样本 在采集、运送和处理等不同环节中的交叉污染问 题。
定性检测结果解释 考虑定性检测的局限性
考虑特定情况下,核酸样本的稳定性和不易降 解性。
疾病状况的判断
病原体存在与患病之间的相关关系 为正确判断疾病应:选择具有代表性的标本, 采用具有说服力的检测方法,建立适量的临界值。
分子诊断学`1

分子诊断学:以分子生物学理论为基础,利用分子生物学技术和方法研究人体内/外源性生物大分子体系的存在、结构或表达调控变化,为疾病的预防诊断治疗和转归提供信息和依据。
断裂基因:指基因的内部存在间隔区,间隔区的DNA序列与该基因所决定的蛋白质没有关系。
间隔区又称为内含子。
出现在成熟RNA中的有效区段称为外显子。
SNP:是指单个核苷酸变异而形成的DNA 分子多态性。
根据SNP在基因组中的位置,可分为编码区SNP(cSNP)、基因周边区SNP(pSNP)、基因间SNP(iSNP)。
转座因子/可转座元件:能在基因组中从一个位点移至另一个位点的DNA序列。
克隆:细胞通过无性繁殖所产生的与亲代细胞完全相同的子代群体称克隆。
分子克隆:将某一特定的DNA片段通过重组DNA技术插入到一个载体中,然后在宿主细胞中进行自我复制所得到的大量完全相同的该DNA片段的“群体”。
基因工程:将基因进行克隆,并利用克隆的基因在受体细胞内复制、转录和翻译表达,制备特定的蛋白质或多肽产物,或定向改造细胞和个体的特性所用的方法及相关工作。
载体:是携带靶DNA片段进入宿主细胞进行扩增和表达的运载工具,本质为DNA。
克隆载体:能将目的基因在受体细胞中复制扩增并产生大量目的基因的载体。
表达载体:能将外源基因在受体细胞中有效转录和正确翻译的载体。
穿梭载体:能够在两类不同宿主中复制、扩增和选择的载体。
转染:将噬菌体、病毒或以他们为载体的DNA重组体导入真核细胞的过程转化:将重组的DNA 分子引入细菌(原核细胞),使其在细菌体内扩增及表达的过程。
DNA复性:当变性因素解除后,两条DNA链又可通过碱基互补配对形成DNA双螺旋结构。
核酸探针:能与特定靶基因序列发生特异性互补结合,并可用特殊方法检测的被标记的已知核苷酸序列。
核酸变性:指维系核酸双链互补碱基对之间的氢键断裂变成单链的过程荧光阈值:指设定在荧光扩增曲线上处于荧光信号指数扩增阶段时的任意一个值。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
分子诊断:应用分子生物学方法检测患者体内遗传物质的结构或表达水平的变化而做出诊断的技术,称为分子诊断。
分子诊断学:是临床检验诊断学的一个重要分支,它利用分子生物学技术来研究机体外源性和内源性生物大分子和大分子体系的存在、结构或表达调控的改变,从而为疾病的预测、预防、诊治和转归提供分子水平信息。
基因:基因是一段携带功能产物(多肽、蛋白质、tRNA、rRNA和某些小分子RNA)信息的片段基因组:是指生物体的一套完整的单倍体遗传信息,包括所有基因和基因间的区域。
基因组学:指对所有基因进行基因组作图(包括遗传图谱、物理图谱、转录图谱)、核苷酸序列分析、基因定位和基因功能分析的一门科学。
人类基因组多样性:即人类基因组的个体差异。
生物大分子:指分子体内由分子量较低的基本结构单位首位相连形成的多聚化合物。
CsCl-EB密度梯度超速离心法:CsCl-EB法是一种沉降平衡离心法,经超速离心,离心介质CsCl形成一连续的密度梯度,在过量EB存在的条件下,各种不同密度的物质经离心平衡后得以分开。
(CsCl-EB法分离质粒和染色体DNA就取决于溴化乙锭与线状及闭环DNA分子的结合量有所不同)。
Western印迹:蛋白质印迹,又称免疫印迹,是一种通过标记的抗体检测经SDS-PAGE分离的特定蛋白质的技术。
克隆载体:能携带目的基因进入宿主细胞进行扩增和表达的一类DNA分子,用于在宿主细胞中克隆和扩增外援DNA片段表达载体:能携带目的基因进入宿主细胞进行扩增和表达的一类DNA分子,用于在宿主细胞中获得外援基因的表达产物。
重组DNA技术:是指在基因水平上,将目的DNA在体外重组于能自我复制的载体DNA分子上,然后将重组DNA导入宿主细胞中进行增生,以获得大量的目的DNA片段,或以此为基础,通过基因的诱导表达,得到大量相应蛋白质产物的过程。
基因组文库:将某种生物的基因组DNA切割成一定大小的片段,并与合适的载体重组后导入宿主细胞进行克隆。
这些存在于所有重组体内的基因组DNA片段的集合,即基因组文库,它包含了该生物的所有基因。
cDNA文库:提取组织细胞的mRNA,逆转录成Cdna, 与适当载体连接后转化受体细胞,得到含重组DNA的噬菌体或细菌克隆,从而构建成某种生物的cDNA 文库。
基因工程:又称基因拼接技术和DNA重组技术,是以分子遗传学为理论基础,以分子生物学和微生物学的现代方法为手段,将不同来源的基因(DNA分子),按预先设计的蓝图,在体外构建杂种DNA分子,然后导入活细胞,以改变生物原有的遗传特性、获得新品种、生产新产品PCR即聚合酶链反应,是利用DNA聚合酶(如TaqDNA聚合酶)等在体外条件下,催化一对引物间的特异DNA片断合成的基因体外扩增技术。
核酸分子杂交:不同来源的单链核酸分子在适合的温度和离子强度下,通过碱基互补形成双链杂交体的过程生物芯片:是采用类似集成电路制作中的微加工技术,将生命科学研究中的若干不连续过程集成到一块几平方厘米大小的芯片上,并使这些分散的过程连续化与微型化,以此来实现对大量生物样品中各数据的快速自动和并行处理。
基因芯片:将大量的基因片段有序地,高密度的排列在玻璃片或纤维膜等载体上,称之为基因芯片。
第二章1分子诊断的主要特点是什么?主要特点:直接以疾病基因及其表型为检测对象,特异性高,属于病因学诊断,其检测结果不仅具有描述性,且具有准确预测性;分子诊断不仅可准确诊断有基因表型异常的疾病,更重要的是其可对疾病基因型的变异作出判断。
2、分子诊断学经历了哪几个发展阶段?每个阶段各有什么特征性技术得到建立及应用?第一阶段:主要利用DNA分子杂交方法进行遗传病的基因诊断。
第二阶段:创建并利用PCR技术。
第三阶段:以生物芯片为代表的高通量密集型技术。
4、真核生物基因组结构特点有哪些?与原核比较,主要区别是什么?真核生物基因结构特点:1)基因组庞大2)线状双链DNA和二倍体3)非编码区远多于编码区4)功能基因大多不连续5)重复序列5、真核生物线粒体基因组有何特点?①非孟德尔的母性遗传②高突变率③异质性和复制分离④阈值效应⑤半保留复制与协调作用6、什么是人类基因组多样性?产生多态性的方式主要是什么?人类基因组多样性:即人类基因组的个体差异。
产生的主要方式:(1)重复序列单元的拷贝数变异(2)可转座元件导致的分子多态(3)单个核苷酸的变异第三章2、酚抽提法制备真核基因组DNA的原理及相关试剂的作用。
酚抽提法:主要利用基因组DNA较长的特性,将其与细胞器或质粒等小分子DNA分离。
首先在含EDTA、SDS及无DNA酶的RNA酶的条件下裂解细胞,经蛋白酶K处理后,用pH8.0的Tris饱和酚抽提DNA,加入一定量的异丙醇或乙醇,基因组的大分子DNA即沉淀形成纤维状絮团漂浮其中,可用玻棒将其取出,而小分子DNA则只形成颗粒状沉淀附于壁上及底部,从而达到提取的目的。
相关试剂作用:EDTA为二价金属离子螯合剂,可用螯合DNA酶激活所需要的Mg2+ Ca2+等二价金属离子,从而抑制DNA酶的活性,并具有降低细胞膜稳定性的作用;SDS 为阴离子去垢剂,其作用是降解细胞膜及乳化脂质和蛋白质,使与其结合的物质沉淀从而达到分离作用,另外SDS具有使蛋白质变形、解聚和降解DNA酶的作用;RNA酶主要有效水解RNA;蛋白酶K则可水解各种蛋白质及裂解细胞酚使一种强蛋白变性剂,可使蛋白质变性沉淀,也可抑制DNA酶的活性;pH8.0的Tris溶液主要是使DNA能顺利进入水相,而避免其滞留于蛋白质层。
3、碱裂解法制备质粒DNA的原理。
碱裂解法主要利用在NaOH提供的高碱性(12.0—12.6)条件下,强阴离子去垢剂SDS 破坏细胞膜,使细胞裂解,经处理后使宿主细胞染色体DNA缠绕在细胞膜碎片上形成的大分子复合物,在高钾盐条件下形成沉淀,而质粒DNA保留于上清中容易通过离心将其分离,当PH调至中性时,宿主染色体DNA片段也不能再复性。
而质粒DNA链重新恢复其天然状态。
通过无水乙醇沉淀质粒DNA,并用70%的乙醇洗涤,得到的质粒DNA可用于限制酶图的绘制、细菌转化、特定DNA片段的分离、DNA测序和PCR等试验。
第三章生物大分子的分离纯化4在对真和细胞RNA进行分离纯化时,如何避免和取出Rnase的污染?酸性异硫氰酸胍-酚-氯仿一步法分离总RNA的原理。
一要全力避免细胞外RNase的污染并抑制其活性,二要尽快地抑制细胞内RNase的活性并极力地去除RNase。
对广泛存在的细胞外RNase,应在RNA制备的全过程中保持高度的警惕,并采取严格的措施以避免其污染和抑制其活性。
空气中弥漫的烟雾与飞扬的灰尘都可能因携带细菌、真菌等微生物而带来RNA酶的污染,应选择一个洁净的实验室进行操作。
操作者是RNA酶污染的一个重要来源,全部试验过程中均需戴手套与口罩操作并经常更换,所用的玻璃器皿需置于干燥烘箱中200°C烘烤2h 以上。
凡是不能用高温烘烤的材料如塑料容器等皆可用0.1%的焦炭酸二乙酯(DEPC)水溶液处理。
除DEPC外,其他RNA酶如钒氧核苷酸复合物,这种由氧钒离子和4种核糖核苷之中的任意一种所形成的复合物,能与多种RNA酶结合并几乎能完全抑制RNA酶的活性。
原理:以4mm/L的异硫氰酸胍为蛋白变形剂,0.14mol/L的β-巯基乙醇可切断蛋白分子内二硫键来提高蛋白变形,然后在pH4.0的酸性条件下,经酚/氯仿抽提裂解溶液,通过异丙醇沉淀RNA与75%的乙醇洗涤来制备RNA。
、5.蛋白质分离纯化的方法、含量的测定及纯度的鉴定蛋白质分离:二维凝胶电泳(1等电聚焦凝胶电泳2SDS-聚丙烯酰胺凝胶电泳3图像分析技术)第四章分子克隆克隆载体必须具有四个条件:①具备复制能力,保证重组DNA在宿主细胞内独立复制②有一个或多个限制性内切酶的单一识别点,便于目的基因的插入③具备多个拷贝数,易与宿主细胞的染色体分开,便于分离提纯④有一定的标记基因,便于进行筛选⑤有较高的遗传稳定性基因克隆的载体类型:质粒载体,噬菌体载体,黏粒载体,人工染色体载体,噬菌粒载体质粒特点如下:1、分子相对较小(3∽10kb);2、松驰型复制;3、具有适当的多克隆位点以便外源DNA 插入;4、具有插入失活筛选标志,便于从平板上直接筛选阳性重组子;5、质粒能携带的外源DNA 片段一般较小(<15kb基因克隆过程包括以下步骤:1、获得待克隆的DNA 片段(基因);2、载体的选择3、目的基因与载体连接成重组DNA4、重组DNA导入受体细胞5、重组体的筛选酶主要用途限制性核酸内切酶识别DNA特定序列,DNA聚合酶I或其大片段(1)缺口平移制作标记DNA探针(2)合成cDNA第二链(3)填补双链DNA3′凹端(4)DNA序列分析逆转录酶将mRNA转录成cDNA以制备基因片段DNA连接酶连接两个DNA分子或片段2. ColE1:天然质粒,属高拷贝型。
特点是在培养基中加入氯霉素抑制细菌蛋白质合成后,质粒仍然能复制达到每个细胞1000-3000拷贝之多。
选择标记:大肠杆菌素(colicin)E1和对E1免疫的基因(immE1)3. pBR322该质粒具有以下优点:1)分子大小4363bp,容易纯化。
2)含有2个抗生素抗性基因,可以作为选择标记。
而且每一个标记基因都含有单一的酶切位点,可以插入DNA,amp基因内可被Pst I, Pvu I, Sac I切开,而四环素抗性基因可被BamH I, Hind III切开,通过插入失活筛选重组子。
3)受体细胞内,pBR322以多拷贝存在,一般一个细胞内可达到15个,而在蛋白质合成抑制剂存在条件下,可达到1000-3000拷贝,如氯霉素。
可以产生大量的重组pBR322分子4. pUC系列pUC8的优点:1)突变位点位于复制起点,复制前可以使细胞中质粒的拷贝数达到500-700个,可以提高载体的产量。
2)重组子的鉴定可一步完成,固体培养基中添加amp和X-gal, IPTG,节约了一半的时间。
3)pUC8的多克隆位点,可以使具有不同粘端的DNA片段插入载体,而不必连接linker。
4)载体中的多克隆位点与M13mp系列的载体是相同的,因此,插入pUC系列的克隆DNA 可以直接转入M13mp载体,可以进行DNA测序和体外定点突变。
DNA链末端合成终止法也称为Sanger法,它的基本原理是将2ˊ,3ˊ﹣双脱氧核苷酸(ddNTP)参入到合成的DNA链中,由于脱氧核糖的3ˊ位碳原子上没有羟基,因此不能与下一位核苷酸反应形成磷酸二酯键,DNA合成反应即终止。
在测定时,首先将模板分为四个反应管,分别加入引物和DNA聚合酶,将32P或35S标记的dNTP(仅标记一种即可)作为底物参入到新合成的DNA链中。
反应一定时间后,每一管内加入四种ddNTP 中的一种,就可获得一系列在不同部位终止的、大小不同的DNA片段。