引物测序直播问题及回答
一代测序常见问题及解决策略知识分享

一代测序常见问题及解决策略知识分享测序常见问题及解决策略一、PCR常见问题1.假阴性,不出现扩增条带PCR出现假阴性结果,可从以下几个方面来寻找原因:1)模板:①模板中有杂蛋白;②模板中有Taq酶抑制剂;③在提取制备模板时丢失过多;④模板核酸变性不彻底。
2)酶:酶失活或反应时忘了加酶。
3)Mg2+浓度:Mg2+浓度过高可降低PCR扩增的特异性,浓度过低则影响PCR扩增产量甚至使PCR扩增失败而不出扩增条带。
4)反应条件:变性对PCR扩增来说相当重要,如变性温度低,变性时间短,极有可能出现假阴性;退火温度过低,可致非特异性扩增而降低特异性扩增效率退火温度过高影响引物与模板的结合而降低PCR扩增效率。
5)靶序列变异:靶序列发生突变或缺失,影响引物与模板特异性结合,或因靶序列某段缺失使引物与模板失去互补序列,其PCR扩增是不会成功的。
2.假阳性假阳性:出现的PCR扩增条带与目的靶序列条带一致,有时其条带更整齐,亮度更高。
常见原因有:1)引物设计不合适:选择的扩增序列与非目的扩增序列有同源性,因而在进行PCR扩增时,扩增出的PCR产物为非目的性的序列。
靶序列太短或引物太短,容易出现假阳性。
需重新设计引物。
2)靶序列或扩增产物的交叉污染:这种污染有两种原因:一是整个基因组或大片段的交叉污染,导致假阳性。
这种假阳性可用以下方法解决:操作时应小心轻柔,防止将靶序列吸入加样枪内或溅出离心管外。
二是空气中的小片段核酸污染,这些小片段比靶序列短,但有一定的同源性。
可互相拼接,与引物互补后,可扩增出PCR产物,而导致假阳性的产生,可用巢式PCR方法来减轻或消除。
3.出现非特异性扩增带PCR扩增后出现的条带与预计的大小不一致,或大或小,或者同时出现特异性扩增带与非特异性扩增带。
非特异性条带的出现,其原因:一是引物与靶序列不完全互补、或引物聚合形成二聚体。
二是Mg2+离子浓度过高、退火温度过低,及PCR循环次数过多有关。
DNA测序常见问题分析及解决办法

客户自己提供2ug纯化好的质粒
PCR产物定量
极低
重新电泳检测已纯化的PCR产物,确认有足够的量,或提供PCR原液由公司进行纯化
测序结果正常,与预期不符
找不到引物
质粒模板
检测是否为空载体,从其互补链上寻找,克隆位点离测序引物太近,长插入片段未测通。
PCR模板
不可能找到所用的测序引物,短片段可以从互补链上找到另一段的引物,长片段由于测不通,无法找到相应序列想得到全序列,短片段可以从两端进行测序,长片段需要经克隆后进行测序。
现象
可能原因
是否收费
收
不收
反应无信号或全部杂峰
引物与模板结合不好(引物或模板原因)
不收
质粒抽提未成功
质粒太大或活化方式不好、转化不好
不收
鉴定质粒或PCR浓度
建议50ul(pcr)/20ul(质粒)去离子水溶解
不收
帮助客户设计引物和序列拼接
不收
300bp以前中断或衰减
高级结构GC结构重复序列
PCR除外
收
客户提供引物导致测序结果不理想
引物不纯
收
质粒抽提成功率60%以下(大单)
质粒太大或活化方式不好、转化不好
质粒抽提不成功的,加收3元/质粒
验证实验结果,两次结果
一致
收2次费
不一致
收1收费
PCR原液纯化后
无反应结果
引物与模板结合不好(引物或模板原因)
收纯化费
不收测序费
测反向互补序列,AC结构不影响测序
严重的重复结构
测反向互补序列
模板特性决定的
根据具体情况决定
信号极弱或无信号
信号极弱或无信号
模板质量差
文库测序十问十答

文库测序十问十答从核酸提取、文库构建,到上机测序、数据分析,越来越多的实验室选择将这些决定实验命运的关键步骤掌控在自己手中。
下面就由小编带大家看一看自建库和测序过程中有哪些需要特别注意的环节,干货满满,不要错过哦~问:常规Illumina NGS文库长什么样图1 单端index文库测序模式图图2 双端index文库测序模式图(NovaSeq 6000平台)图3 双端index文库测序模式图(HiSeq X平台)答:如图,文库结构可分为以下几个部分:插入片段,P5、P7接头,测序引物结合位点及index。
中P5、P7接头位于文库两端,可以与flowcell上的寡核苷酸结合,在簇生成和测序过程中可作为引物或起到固定模板链的作用。
Index是不同样本的区分依据,当同一条lane中混入多个样本测序时,即可根据index区分来自不同样本的reads。
根据建库时使用接头结构不同,又分为单index文库和双index文库。
随着测序通量的不断增加,每条lane可以容纳的样本量也越来越多,双index可以变化出更多种组合,且能够降低标签串扰的比例,因此一些对灵敏度要求较高的检测通常会构建双index文库[1]。
图中黄色和蓝色的部分是测序引物结合位点,相信机智的你一定发现了:index5在NovaSeq 6000和HiSeq X平台的测序方向是不同的。
完成Read1、index7测序之后,NovaSeq 6000平台会继续以这条链为模板进行index5的测序,测序引物是flowcell上的P5接头,因此index5的测序方向和Read1、index7是一致的。
而HiSeq X平台的index5、Read2测序则是在末端翻转后进行的,因此index5的测序方向与Read2一致,而与Read1、index7相反,同样的index5在HiSeq X和NovaSeq 6000平台测得的序列是反向互补的,因此在填写文库信息的时候一定要注意测序平台和序列的对应关系。
一代测序常见问题及解决策略

测序常见问题及解决策略一、PCR常见问题1.假阴性,不出现扩增条带PCR出现假阴性结果,可从以下几个方面来寻找原因:1)模板:①模板中有杂蛋白;②模板中有Taq酶抑制剂;③在提取制备模板时丢失过多;④模板核酸变性不彻底。
2)酶:酶失活或反应时忘了加酶。
3)Mg2+浓度:Mg2+浓度过高可降低PCR扩增的特异性,浓度过低则影响PCR扩增产量甚至使PCR扩增失败而不出扩增条带。
4)反应条件:变性对PCR扩增来说相当重要,如变性温度低,变性时间短,极有可能出现假阴性;退火温度过低,可致非特异性扩增而降低特异性扩增效率退火温度过高影响引物与模板的结合而降低PCR扩增效率。
5)靶序列变异:靶序列发生突变或缺失,影响引物与模板特异性结合,或因靶序列某段缺失使引物与模板失去互补序列,其PCR扩增是不会成功的。
2.假阳性假阳性:出现的PCR扩增条带与目的靶序列条带一致,有时其条带更整齐,亮度更高。
常见原因有:1)引物设计不合适:选择的扩增序列与非目的扩增序列有同源性,因而在进行PCR扩增时,扩增出的PCR产物为非目的性的序列。
靶序列太短或引物太短,容易出现假阳性。
需重新设计引物。
2)靶序列或扩增产物的交叉污染:这种污染有两种原因:一是整个基因组或大片段的交叉污染,导致假阳性。
这种假阳性可用以下方法解决:操作时应小心轻柔,防止将靶序列吸入加样枪内或溅出离心管外。
二是空气中的小片段核酸污染,这些小片段比靶序列短,但有一定的同源性。
可互相拼接,与引物互补后,可扩增出PCR产物,而导致假阳性的产生,可用巢式PCR方法来减轻或消除。
3.出现非特异性扩增带PCR扩增后出现的条带与预计的大小不一致,或大或小,或者同时出现特异性扩增带与非特异性扩增带。
非特异性条带的出现,其原因:一是引物与靶序列不完全互补、或引物聚合形成二聚体。
二是Mg2+离子浓度过高、退火温度过低,及PCR循环次数过多有关。
三是酶的质和量,往往一些来源的酶易出现非特异条带而另一来源的酶则不出现,酶量过多有时也会出现非特异性扩增。
测序常见问题分析解答

测序常见问题分析主讲人: 张隽辉LOGO1. 序列结构对测序的影响图例:poly结构原因: 主要原因是在polyT结构后,测序酶容易在模板上 滑动,导致随后的峰型变乱 或者移码解决办法: 此类样品通过对其反向互补序列进行测序,通过 拼接可以得到模板全长序列.图例:重复结构原因: 重复结构会导致测序复制框的滑移,另外测序试剂采用 了I, U 代替G, T, 与模板结合能力较弱, 测序酶很难延伸, 信号会迅速衰减或峰谱很乱解决办法: 使用反向引物对模板进行测序,测到重复序列反向互 补位置时,即可完成模板全长的拼接,一般测A\C重复 序列比G\T要容易一些图例:回文结构位点94至137碱基前后互补, 形成回文结构,该结构导致 后面的信号衰减,出现错误的判读。
图例: 特殊结构现象: 信号在73bp(信号峰图2700处)突然中断, 测序PCR反应终止原因: 变性后的单链在该处很可能互补或在全序列中有大的 特殊结构,造成严重的二级结构,使dNTP 和 ddNTP不 能和模板结合, 测序酶无法正常延伸解决方法: 酶切,做亚克隆测序2.测序常见图谱分析图例:双克隆1现象: T载体序列峰图正常, 连接位置处出现套峰原因: 1.有插入片段的载体和空载体同时存在 2.PCR产物用T载体进行连接时,其片段能以正反两个方向 插入T载体解决办法: 重新挑取单克隆备注: 重新进行PCR反应或者酶切鉴定仅能证明该克隆含有插入片段, 并不足以证明模板的单一图例:双克隆2现象: 载体序列及插入片段前半部分峰图单一,套峰出现在插入片段中间原因: 1. pcr产物不纯,且产物的引物结合区以及峰图单一的序列 都一样 2. 部分模板碱基发生突变,双峰出现的位点是由碱基突变 的位置决定的,有时会一端正常一端双峰(插入片断超过一 个反应测序长度(800~900bp)), 或者两个反应都双峰解决方法: 重新挑单克隆图例:非特异扩增现象: 峰图起始即双峰 原因及解决方法: 1. 对于质粒样品, 引物在序列上有两个结合位点(载体上和插入片段上),换一个引物测序 2.对于PCR产物,有两个模板,且两个模板都能和引物结合扩增, 换引物或克隆测序图例:等位基因双模板现象:序列的80bp到120bp之间有两套峰存在,没有发生移码突变解决方法: 采取克隆的方法将两套模板分开,分别进行测序图例:碱基缺失碱基缺失常见在PCR产物中,特别是从基因组中扩增得到的PCR片段, 上图的186位缺失两个连续的T。
测序常见问题解答

测序常见问题解答1.为什么需要新鲜的菌液?首先,新鲜的菌液易于培养,可以获得更多的DNA,同时最大限度地保证菌种的纯度.2.如何提供菌液?如果您提供新鲜菌液,用封口膜封口以免泄漏;也可以将培养好的4—5ml菌液沉淀下来,倒去上清以方便邮寄。
同时邮寄时最好用盒子以免邮寄过程中压破.3.如何制作穿刺菌?用灭菌过1.5ml或2ml离心管加入LB琼脂(7g/L)斜面凝固,用接种针挑取分散良好的单菌落穿过琼脂直达管底,不完全盖紧管盖适当温度培养过夜,然后盖紧盖子加封口膜,室温或4度保存。
4.PCR产物直接测序有什么要求?1).扩增产物必须特异性扩增,条带单一.如果扩增产物中存在非特异性扩增产物,一般难以得到好的测序结果;.2)必须进行胶回收纯化;3)DNA纯度在16—2.0之间.浓度50ng/ul以上.5.为什么PCR产物直接测序必须进行Agarose胶纯化?如果不进行胶纯化而直接用试剂盒回收,经常会导致测序出现双峰甚至乱峰。
这主要是非特异性扩增产物或者原来的PCR引物去除不干净所导致。
大多所谓的PCR"纯化试剂盒"实际上只是回收产物而不能起到纯化的作用的。
对于非特异性扩增产物肯定无法去除,而且通常他们不能够完全去除所有的PCR引物,这会造成残留的引物在测序反应过程中参与反应而导致乱峰。
6.如何进行PCR产物纯化?PCR产物首先必须用Agarose胶电泳,将特异扩增的条带切割下,然后纯化。
使用凝胶回收试剂盒回收.产物用ddH2O溶解。
7.PCR产物直接测序的好处?A) PCR产物直接测序可以反映模板的真实情况.B) 省去克隆的实验费用和时间.C) PCR产物测序正确的片段进行下一步克隆实验使结果更有保障.D) 混合模板进行PCR的产物直接测序可以发现其中的点突变.8.对用于测序的质粒DNA的要求有哪些?对测序模板DNA的一般要求:1).DNA纯度要求高,1.6—2.0之间,不能有混合模板,也不能含有RNA,染色体DNA,蛋白质等;2).溶于ddH2O中,溶液不能含杂质,如盐类,或EDTA等螯合剂,将干扰测序反应正常进行。
引物设计常见问题与解答(一)

2. 引物纯化方式有哪些,如何选择?
◆ C18柱脱盐:有人称其为简易反相柱,它对DNA有特异性的吸附,可以被有机溶解洗脱,但不会被水洗脱,所以能有效地去除盐分。它不能有效去除比目的片段短的小片段。实际上,它是一种脱盐的作用。这种方法一般不会对普通PCR反应产生影响。对于需要用于测序、克隆的引物不能使用这个级别。
4. 投诉定量不准是怎么回事儿?
答:我们偶尔收到用户投诉定量不准的报告,出现这种情况的可能性有(1)生产人员定量错误。这种可能性有,但是不大,因为我们的生产人员都是经过严格的培训,程序化规范操作和换算。公司内部的考核机制也使得分装人员没有必要故意少给用户OD数,因为无论OD数是否达到定单要求,我们都统计为工作量,没有达到OD数的,分装人员将清单及时报到序列录入部门安排重合就可以了。合成产量的高低是序列录入部门人员的考核指标之一。一般情况下,引物都有留样。接到用户投诉后,我们都会找出留样重新定量,一般都没有问题。(2)分装没有问题,但引物抽干或收样过程中,引物干粉可能意外丢失。这种情况很少见。(3)系统误差,我们认为10%左右为允许误差。使用过程中,引物工作浓度范围很宽,定量上的少许偏差不影响实验。(4)用户没有能够正确理解引物OD数的含义,没有能够正确使用分光光度计,特别是使用微量测定;用户没有将OD读数,正确地转换成母液中OD数。这种情况比较常见。(5)用户收到引物干粉时,打开引物管盖前没有离心或其他误操作导致引物干粉部分丢失。
1. 引物是如何合成的?
目前引物合成基本采用固相亚磷酰胺三酯法。DNA合成仪有很多种, 主要都是由ABI/PE 公司生产,无论采用什么机器合成,合成的原理都相同,主要差别在于合成产率的高低,试剂消耗量的不同和单个循环用时的多少。申能博彩公司采用的合成仪主要机型为ABI3900高通量合成仪,合成长链主要采用Beckman 1000M和PE8909 DNA合成仪,引物修饰和高OD数合成采用ABI394等。
测序常见问题分析

测序常见问题分析测序常见问题分析1.图谱无信号1.1、质粒培养,转化或其他随机原因使质粒上引物序列发生变化,导致无结合位点1.1.1同一模板两端引物测序均无信号,浓度不好安排重抽,反之停止实验,建议客户确认后重新送样1.1.2同一模只一端无信号,确认不是空载体,安排重新实验或换同序列其他引物,两次测序无信号则停止实验,客户要求测通的安排单向测通。
反之建议客户单向测通1.1.3客户提供序列我司合成引物,二次实验仍无信号则停止实验,建议客户提供载体图谱或目的基因我处给他设计引物测序1.1.4客户提供序列我司设计并合成引物核实设计无误可安排重新实验,仍不成功则停止实验,请客户确认提供的序列是否和模板相匹配1.1.5我司设计并合成的步行引物核实设计无误可安排重新实验,仍不成功可重设引物或换另一端测通。
1.1.6某一通用引物测序无信号先查看当天该引物的其他实验或近期该引物的实验情况,均无信号则换管引物重新实验,当天该引物的其他实验有成功的,该反应重做1.1.7一订单多样品用同一对引物测序,某条引物测序均无信号,若为客户提供引物则停止实验。
若为通用引物有同序列其他引物,在确认不是空载载体的情况下,安排换引物试作三至四个反应,效果好全部安排用该引物测序;效果不好都停止实验。
客户要求测通的安排单向测通,反之建议单向测通。
1.1.8重摇重抽测序无信号则取消,建议重新提供样品1.1.9客户提供质粒直接测序无信号则停止实验,建议客户提供新鲜菌液1.1.10用PCR引物测序无信号可换备用引物测序,无备用引物停止实验,建议用载体上引物测序。
1.2、PCR 产物1.2.1同一模板两端引物测序均无信号则停止实验,建议客户克隆后测序1.2.2 同一模只一端无信号,安排重新实验, 仍无信号则停止实验,客户要求测通的安排单向测通,反之建议步行测通2. 图谱信号弱可参考原始峰图,对不能出报告的峰图的处理如下:2.1 同一模板某一端,可安排提高浓度梯度重新实验2.2 某一模板或一订单多个模板中的一个模板2.2.1 鉴定浓度正常可安排提高浓度梯度重新实验,效果仍不好可作弱stop处理2.2.2 一次抽提出的质粒亮度弱,可安排重新抽提质粒后测序2.2.3 二次抽提出的质粒亮度弱,可停止实验,建议重新提供样品3 双峰3.1 PCR 产物测序3.1.1 引物不纯,引物客户提供该结果可出,引物我司合成安排重合引物3.1.2 引物特异性差,有双或多结合位点,可换备用引物测序,无备用引物即出结果3.1.3 polyT/A/G/C 出结果,单向建议客户换另一端测序。
测序常见问题及其分析

图2
解决方法:主要原因是PCR产物没有纯化,含有部分序列一致的两种以上长度不一的片段,将PCR产物进行PAGE纯化(至少琼脂糖充分电泳后切胶纯化)后再进行测序,便可解决。
问题图3(测序引物有碱基缺失)
测序引物有碱基缺失(一般是引物的5'端缺失),和模板的碱基缺失即图1有些类似,所不同的是模板碱基缺失一般是在一段正常测序序列后才出现移码,而引物碱基缺失的话,则从测序一开始就出现移码,表面在图形上便是一开始就是严重的峰形重叠。
解决方法:重新合成引物,或将引物进行PAGE纯化
2、克隆测序时出现峰形重叠
原因:所挑选的重组子不是单克隆,所提供的测序用质粒中含有两种以上插入片段不同的质粒;或是是送测序的菌液污染
解决方法:重新挑单克隆的菌落(划线分离单菌落),提质粒或送菌液。
业务员测序常见问题解答

测序常见问题解答1.为什么提供新鲜的菌液?如何提供新鲜的菌液?2. DNA测序样品用什么溶液溶解比较好?3. 提供DNA测序样品时,提供何种形态的比较好?4. 提供的测序样品为菌体时,以什么形态提供为好?5. PCR产物直接测序有什么要求?6. 为什么PCR产物直接测序必须进行Agarose胶纯化?7. 如何进行PCR产物纯化?8. 对于测序用的质粒DNA的要求有哪些?9. 如何鉴定质粒DNA浓度和纯度?10. 对测序引物的要求有哪些?11. 为什么测序引物必须特异地与DNA模板结合?12. 测序结果有很多套峰(出现很多N),还照常收费,为什么?13. 为什么用PCR产物测序时,经常会出现套峰现象?14. 出现套峰的原因是什么?15. 测序结果不到800 Bases,还照常收费了,为什么?16. 为什么在测序报告上找不到引物序列?17. 在测序结果上,找不到测序用引物后面的序列,为什么?18. 怎样使用擎科提供的测序报告?19. PCR片段直接测序和PCR片段经克隆后测序的结果有何区别?20. 测序结果和文献资料不一样,为什么?21. 我的基因序列与标准序列为什么有差别?22. DNA片段很长,一个反应读不到头,怎么办?23. 测序完成后,测序样品和引物将如何处理(或保存)?24 怎样选择(设计)测序用引物?25. 觉得你给我的结果完全不是我需要的序列?26. 我的样品上有一个杂合位点,为什么在你们的报告上看不到杂合的信号?27. 超过6Kb的DNA片断如何进行测序?28. 全自动荧光测序的准确性如何?29. 用测序的方法检测点突变可靠吗?30. 我要求5’到3’正向测序,为什么你们给我的序列是反的?31. 我的样品在你们所说的可靠范围内有一处存在疑问,能否重新测一次?32. 我的样品你们已经测通了,但为什么在overlap区有这么多的错配,给出的全序列和单个报告也存在差异,我该相信谁?33. 你们为什么在primer walking时总将引物设计的那么靠前?34. 我有一个4KB的PCR片段,希望你们帮我测通。
引物设计常见问题与解答

引物设计常见问题与解答先看一下Tm的定义:Tm = Temperature at which 50% of a given oligonucleotide is hybridized to its complementary strand. In the absence of destabilizing agents, like formamide or urea, Tm will depend on 3 major parameters: The sequence: a GC-rich sequence has a higher melting temperature. The strand concentration: high oligonucleotide concentrations favor hybrid formation, which results in a higher melting temperature. The salt concentration: high ionic strength results in a higher Tm as cations stabilize the DNA duplexes.引物设计软件都可以给出Tm,引物长度,碱基组成,引物使用缓冲的离子强度有关。
长度为25mer以下的引物,Tm计算公式为:Tm = 4℃(G + C)+ 2℃(A + T)对于更长的寡聚核苷酸,Tm计算公式为:Tm = 81.5 + 16.6 x Log10[Na+] + 0.41 (%GC) – 600/size公式中,Size = 引物长度。
Tm叫溶解温度(melting temperature, Tm),即是DNA双链溶解所需的温度。
大家可以理解,这个温度是由互补的DNA区域决定的,而不互补的区域对DNA的溶解是没有作用的。
因此,对于引物的Tm,只有和模板互补的区域对Tm才有贡献。
测序常见问题分析的解答

测序常见问题分析主讲人: 张隽辉LOGO1. 序列结构对测序的影响图例:poly结构原因: 主要原因是在polyT结构后,测序酶容易在模板上 滑动,导致随后的峰型变乱 或者移码 解决办法: 此类样品通过对其反向互补序列进行测序,通过 拼接可以得到模板全长序列.图例:重复结构原因: 重复结构会导致测序复制框的滑移,另外测序试剂采用 了I, U 代替G, T, 与模板结合能力较弱, 测序酶很难延伸, 信号会迅速衰减或峰谱很乱 解决办法: 使用反向引物对模板进行测序,测到重复序列反向互 补位置时,即可完成模板全长的拼接,一般测A\C重复 序列比G\T要容易一些图例:回文结构位点94至137碱基前后互补, 形成回文结构,该结构导致 后面的信号衰减,出现错误的判读。
图例: 特殊结构现象: 信号在73bp(信号峰图2700处)突然中断, 测序PCR反应终止原因: 变性后的单链在该处很可能互补或在全序列中有大的 特殊结构,造成严重的二级结构,使dNTP 和 ddNTP不 能和模板结合, 测序酶无法正常延伸 解决方法: 酶切,做亚克隆测序2.测序常见图谱分析图例:双克隆1现象: T载体序列峰图正常, 连接位置处出现套峰原因: 1.有插入片段的载体和空载体同时存在 2.PCR产物用T载体进行连接时,其片段能以正反两个方向 插入T载体 解决办法: 重新挑取单克隆备注: 重新进行PCR反应或者酶切鉴定仅能证明该克隆含有插入片段, 并不足以证明模板的单一图例:双克隆2现象: 载体序列及插入片段前半部分峰图单一,套峰出现在插入片段中间原因: 1. pcr产物不纯,且产物的引物结合区以及峰图单一的序列 都一样 2. 部分模板碱基发生突变,双峰出现的位点是由碱基突变 的位置决定的,有时会一端正常一端双峰(插入片断超过一 个反应测序长度(800~900bp)), 或者两个反应都双峰 解决方法: 重新挑单克隆图例:非特异扩增现象: 峰图起始即双峰原因及解决方法: 1. 对于质粒样品, 引物在序列上有两个结合位点(载体上 和插入片段上),换一个引物测序 2.对于PCR产物,有两个模板,且两个模板都能和引物结合 扩增, 换引物或克隆测序图例:等位基因双模板现象:序列的80bp到120bp之间有两套峰存在,没有发生移码突变解决方法: 采取克隆的方法将两套模板分开,分别进行测序图例:碱基缺失碱基缺失常见在PCR产物中,特别是从基因组中扩增得到的PCR片段, 上图的186位缺失两个连续的T。
PCR测序常见问题
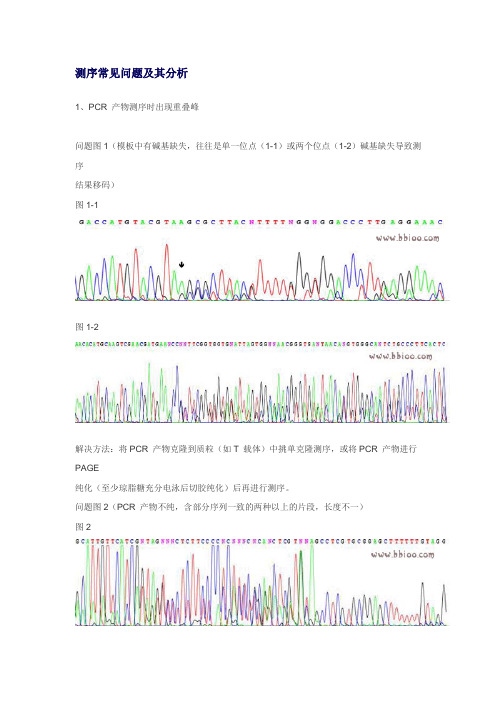
测序常见问题及其分析1、PCR 产物测序时出现重叠峰问题图1(模板中有碱基缺失,往往是单一位点(1-1)或两个位点(1-2)碱基缺失导致测序结果移码)图1-1图1-2解决方法:将PCR 产物克隆到质粒(如T 载体)中挑单克隆测序,或将PCR 产物进行PAGE纯化(至少琼脂糖充分电泳后切胶纯化)后再进行测序。
问题图2(PCR 产物不纯,含部分序列一致的两种以上的片段,长度不一)图2解决方法:主要原因是PCR 产物没有纯化,含有部分序列一致的两种以上长度不一的片段,将PCR 产物进行PAGE 纯化(至少琼脂糖充分电泳后切胶纯化)后再进行测序,便可解决。
问题图3(测序引物有碱基缺失)测序引物有碱基缺失(一般是引物的5'端缺失),和模板的碱基缺失即图1 有些类似,所不同的是模板碱基缺失一般是在一段正常测序序列后才出现移码,而引物碱基缺失的话,则从测序一开始就出现移码,表面在图形上便是一开始就是严重的峰形重叠。
解决方法:重新合成引物,或将引物进行PAGE 纯化2、克隆测序时出现峰形重叠原因:所挑选的重组子不是单克隆,所提供的测序用质粒中含有两种以上插入片段不同的质粒;或是是送测序的菌液污染解决方法:重新挑单克隆的菌落(划线分离单菌落),提质粒或送菌液再次测序。
3、样品有杂合/突变位点模板中有杂合型突变,也就说模板本身在这个位点出现突变;或者是从基因组中扩增出来的杂合位点。
如果模板有杂合(突变或缺失),那么测序图形中其他的位點一般都是單一的峰形,然后突然在某一個位點出現重叠峰(如图中箭頭所示)。
解决方法:建议将DNA 片段克隆到载体再测序。
4、polyA/T 和C/G cluster 导致的套峰和测序信号衰减图4-1图4-3图4-4RACE 测序时经常遇到图4-1 和图4-4 的情形,解决方法:从另一端测序;但如果这样的序列出现在中间,呵呵,目前还没有很好的解决方法,要看测序公司的本事了。
C/G cluster 在PrV 基因组测序时遇到过。
一代测序常见问题及解决策略知识分享
一代测序常见问题及解决策略知识分享测序常见问题及解决策略一、PCR常见问题1.假阴性,不出现扩增条带PCR出现假阴性结果,可从以下几个方面来寻找原因:1)模板:①模板中有杂蛋白;②模板中有Taq酶抑制剂;③在提取制备模板时丢失过多;④模板核酸变性不彻底。
2)酶:酶失活或反应时忘了加酶。
3)Mg2+浓度:Mg2+浓度过高可降低PCR扩增的特异性,浓度过低则影响PCR扩增产量甚至使PCR扩增失败而不出扩增条带。
4)反应条件:变性对PCR扩增来说相当重要,如变性温度低,变性时间短,极有可能出现假阴性;退火温度过低,可致非特异性扩增而降低特异性扩增效率退火温度过高影响引物与模板的结合而降低PCR扩增效率。
5)靶序列变异:靶序列发生突变或缺失,影响引物与模板特异性结合,或因靶序列某段缺失使引物与模板失去互补序列,其PCR扩增是不会成功的。
2.假阳性假阳性:出现的PCR扩增条带与目的靶序列条带一致,有时其条带更整齐,亮度更高。
常见原因有:1)引物设计不合适:选择的扩增序列与非目的扩增序列有同源性,因而在进行PCR扩增时,扩增出的PCR产物为非目的性的序列。
靶序列太短或引物太短,容易出现假阳性。
需重新设计引物。
2)靶序列或扩增产物的交叉污染:这种污染有两种原因:一是整个基因组或大片段的交叉污染,导致假阳性。
这种假阳性可用以下方法解决:操作时应小心轻柔,防止将靶序列吸入加样枪内或溅出离心管外。
二是空气中的小片段核酸污染,这些小片段比靶序列短,但有一定的同源性。
可互相拼接,与引物互补后,可扩增出PCR产物,而导致假阳性的产生,可用巢式PCR方法来减轻或消除。
3.出现非特异性扩增带PCR扩增后出现的条带与预计的大小不一致,或大或小,或者同时出现特异性扩增带与非特异性扩增带。
非特异性条带的出现,其原因:一是引物与靶序列不完全互补、或引物聚合形成二聚体。
二是Mg2+离子浓度过高、退火温度过低,及PCR循环次数过多有关。
PCR引物的相关问题

PCR引物的相关问题2008-3-12引物合成介绍1.引物是如何合成的?目前引物合成基本采用固相亚磷酰胺三酯法。
DNA合成仪有很多种, 主要都是由ABI/PE 公司生产,无论采用什么机器合成,合成的原理都相同,主要差别在于合成产率的高低,试剂消耗量的不同和单个循环用时的多少。
申能博彩公司采用的合成仪主要机型为ABI3900高通量合成仪,合成长链主要采用Beckman 1000M和PE8909 DNA合成仪,引物修饰和高OD数合成采用ABI394等。
亚磷酰胺三酯法合成DNA片段,具有高效、快速的偶联以及起始反应物比较稳定的特点。
亚磷酰胺三酯法是将DNA固定在固相载体上完成DNA链的合成的,合成的方向是由待合成引物的3'端向5'端合成的,相邻的核苷酸通过3'→5'磷酸二酯键连接。
??第一步是将预先连接在固相载体CPG上的活性基团被保护的核苷酸与三氯乙酸反应,脱去其5'-羟基的保护基团DMT,获得游离的5'-羟基;??第二步,合成DNA的原料,亚磷酰胺保护核苷酸单体,与活化剂四氮唑混合,得到核苷亚磷酸活化中间体,它的3'端被活化,5'-羟基仍然被DMT保护,与溶液中游离的5'-羟基发生缩合反应。
第三步,带帽(capping)反应,缩合反应中可能有极少数5'-羟基没有参加反应(少于2%),用乙酸酐和1-甲基咪唑终止其后继续发生反应,这种短片段可以在纯化时分离掉。
??第四步,在氧化剂碘的作用下,亚磷酰形式转变为更稳定的磷酸三酯。
经过以上四个步骤,一个脱氧核苷酸被连接到固相载体的核苷酸上。
再以三氯乙酸脱去它的5'-羟基上的保护基团DMT,重复以上步骤,直到所有要求合成的碱基被接上去。
合成过程中可以观察TCA处理阶段的颜色判定合成效率。
通过氨水高温处理,连接在CPG上的引物被切下来,通过OPC,PAGE等手段纯化引物,成品引物用C18浓缩,脱盐,沉淀。
一代测序常见问题及解决策略知识分享

学习资料测序常见问题及解决策略一.PCR常见问题1.假阴性,不出现扩增条带PCR出现假阴性结果,可从以下儿个方面来寻找原因:1)模板:①模板中有杂蛋口;②模板中有Taq酶抑制剂;③在提取制备模板时丢失过多;④模板核酸变性不彻底。
2)酶:酶失活或反应时忘了加酶。
2®浓度过高可降低PCR浓度:Mg3) Mg扩增的特异性,浓度过低则影响PCR 扩增产量甚至使PCR扩增失败而不岀扩增条带。
4)反应条件:变性对PCR扩增来说相当重要,如变性温度低,变性时间短,极有可能出现假阴性;退火温度过低,可致非特异性扩增而降低特异性扩增效率退火温度过髙影响引物与模板的结合而降低PCR扩增效率。
5)靶序列变异:靶序列发生突变或缺失,影响引物与模板特异性结合,或因靶序列某段缺失使引物与模板失去互补序列,其PCR扩增是不会成功的。
2.假阳性假阳性:出现的PCR扩增条带与口的靶序列条带一致,有时其条带更整齐,亮度更高。
常见原因有:1)引物设计不合适:选择的扩增序列与非口的扩增序列有同源性,因而在进行PCR扩增时,扩增出的PCR产物为非口的性的序列。
靶序列太短或引物太短,容易出现假阳性。
需重新设计引物。
2)靶序列或扩增产物的交义污染:这种污染有两种原因:一是整个基因组或大片段的交义污染,导致假阳性。
这种假阳性可用以下方法解决:操作时应小心轻柔,防止将靶序列吸入加样枪内或溅出离心管外。
二是空气中的小片段核酸污染,这些小片段比靶序列短,但有一定的同源性。
可互相拼接,与引物互补后,可扩增岀PCR产物,而导致假阳性的产生,可用巢式PCR方法来减轻或消除。
3.出现非特异性扩增带PCR扩增后出现的条带与预计的大小不一致,或大或小,或者同时出现仅供学习与参考.学习资料特异性扩增带与非特异性扩增带。
非特异性条带的出现,其原因:一是引物与靶序列不完全互补、或引物聚合形成二聚体。
二是Mg2+离子浓度过高・退火温度过低,及PCR循环次数过多有关。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1、我想问下:为什么引物不能退火之后跑胶验证纯度呢?
两条引物退火成功率达不到百分之百,里面会存在单链引物,引物退火后跑胶有杂链很正常,不能以此来判断单链引物纯度。
2、老师您好,我想自己设计测序引物,请问对于测序引物有什么要求吗?
在待测目的基因前面100bp左右设计引物,GC含量不能太高或太低,3’端不能有错配,引物的长度建议是18-20bp以内。
3、甲基化的建议反向测序,是什么意思?
正向测序有甲基化的结构,会导致测序峰图双峰或者严重的时候导致信号突然中断,就可以反向测序,然后正反双向测序结果拼接起来就可以得到完整的序列。
4、老师请问一个测序反应有效的大概有多长?
普通序列一个测序反应有效长度大概是800bp左右。
5、请问RNA最长能合多少呢
30bp以内。
6、稀释后引物保存时间多久?
稀释后的引物一般4℃保存3个月内是没有问题的
7、你们的积分的兑换时间有限制吗?
积分不清零的
8、测序为什么前70bp不准呢
一代测序,由于仪器的缺陷,所以前面会有几十bp是不准确的,严重的时候会有70bp左右不准确。
9、老师请问那引物最长能合多长呢?
引物最长能合139bp。
10、金开瑞引物合成与外面其他公司引物合成相比,优势有哪些?
我们公司技术服务内容比较多,内部和外部引物合成需求都是一起合成,效果和质量的反馈方面,我们综合有内部使用和外部使用反馈,更全面。
11、只做常规的pcr,用什么纯化方式的引物呢?
常规的pcr脱盐纯化就可以满足实验了。
12、引物测序时,单向、双向、测通之间的具体区别是什么呀?
这个主要是根据待测目的序列的长度来决定的,如果待测目的序列700bp以内的话可以选择正向或者反向都行;1500bp以内的序列一般双向测序就可以了;如果是待测目的序列大于1500bp就可以填测通,这边会根据测序结果自动安排测序反应,直到完全测通,我们会默认测通后拼接。
13、不同纯化方法,你们收费不一样是吗
不一样的,脱盐纯化<PEGA纯化<HPLC纯化。
14、金开瑞收到测序样品后一般最快多久出结果?
质粒和PCR产物24小时内出结果,菌液48小时内出结果。