The new PQP quality guideline
药品生产质量管理规范实施指南英文版

药品生产质量管理规范实施指南英文版Guidelines for the Implementation of Drug Production Quality Management StandardsIntroductionThe drug production industry plays a significant role in public health as it involves the development and manufacturing of essential medications. The quality of pharmaceutical products is crucial as it directly affects the safety and efficacy of treatments.To ensure the quality of drug production processes, various regulations and standards have been established. The implementation of these quality management standards is essential to ensure that pharmaceutical products meet the required safety and efficacy standards. In this document, we will outline guidelines for the implementation of drug production quality management standards.Setting Up Quality Management SystemsThe first step in implementing drug production quality management standards is to set up robust quality management systems in manufacturing facilities. This involves establishingquality control procedures, quality assurance systems, and documentation processes.Quality control procedures should include testing of raw materials, in-process samples, and finished products to ensure that they meet the required specifications. Quality assurance systems should involve regular audits of manufacturing processes and facilities to identify areas for improvement.Documentation processes should include the documentation of all processes and procedures related to drug production, including batch records, standard operating procedures, and validation reports. All documentation should be maintained in a controlled environment to ensure traceability and accountability.Training and EducationAnother key aspect of implementing drug production quality management standards is providing training and education to staff members. All personnel involved in drug production should receive training on Good Manufacturing Practices (GMP) and other relevant quality management standards.Training programs should be tailored to the specific needs of each employee, taking into account their roles and responsibilities within the organization. Regular refresher courses should also be provided to ensure that staff members are up-to-date with the latest regulations and standards.Quality Control and AssuranceQuality control and assurance are essential components of drug production quality management. Quality control measures should be implemented at every stage of the manufacturing process, from raw material procurement to finished product release.Quality assurance activities should involve regular monitoring of manufacturing processes to identify any deviations from established procedures. Corrective and preventive actions should be taken promptly to address any issues and prevent recurrence.Supplier QualificationThe quality of raw materials used in drug production is critical to the quality of the final product. It is essential to establish a supplier qualification program to ensure that all suppliers meet the required quality standards.Supplier audits should be conducted regularly to evaluate the quality management systems of suppliers and identify any areas for improvement. All suppliers should be required to provide documentation of their quality management processes and undergo periodic inspections.ConclusionImplementing drug production quality management standards is essential to ensure the safety and efficacy of pharmaceutical products. By setting up robust quality management systems, providing training and education to staff members, and implementing quality control and assurance measures, pharmaceutical companies can ensure that their products meet the required quality standards.By following the guidelines outlined in this document, drug production facilities can establish effective quality management systems that will help them maintain compliance with regulatory requirements and produce high-quality pharmaceutical products.。
Q10 药物质量体系20080604(中英文)

Q10 药物质量体系20080604(中英文)INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FORREGISTRATION OF PHARMACEUTICALS FOR HUMAN USEICH HARMONISED TRIPARTITE GUIDELINEICH三方协调指南PHARMACEUTICAL QUALITY SYSTEM药物质量体系Q10Current Step 4 versiondated 4 June 2008This Guideline has been developed by the appropriate ICH Expert Working Group and has been subject to consultation by the regulatory parties, in accordance with the ICH Process. At Step 4 of the Process the final draft is recommended for adoption to the regulatory bodies of the European Union, Japan and USA.Q10 Document History 文件历史1. PHARMACEUTICAL QUALITY SYSTEM (1)1.1 Introduction (1)1.2 Scope (1)1.3 Relationship of ICH Q10 to Regional GMP Requirements, ISO Standards and ICH Q7 (2)1.4 Relationship of ICH Q10 to Regulatory Approaches (2)1.5 ICH Q10 Objectives (2)1.5.1 Achieve Product Realisation (2)1.5.2 Establish and Maintain a State of Control (3)1.5.3 Facilitate Continual Improvement (3)1.6 Enablers: Knowledge Management and Quality Risk Management (3)1.6.1 Knowledge Management (3)1.6.2 Quality Risk Management (3)1.7 Design and Content Considerations (3)1.8 Quality Manual (4)2. MANAGEMENT RESPONSIBILITY (4)2.1 Management Commitment (4)2.2 Quality Policy (5)2.3 Quality Planning (5)2.4 Resource Management (5)2.5 Internal Communication (6)2.6 Management Review (6)2.7 Management of Outsourced Activities and Purchased Materials (6)2.8 Management of Change in Product Ownership (6)3. CONTINUAL IMPROVEMENT OF PROCESS PERFORMANCE AND PRODUCT QUALITY (7)3.1 Lifecycle Stage Goals (7)3.1.1 Pharmaceutical Development (7)3.1.2 Technology Transfer (7)3.1.3 Commercial Manufacturing (7)3.1.4 Product Discontinuation (7)3.2 Pharmaceutical Quality System Elements (7)3.2.1 Process Performance and Product Quality Monitoring System (8)3.2.2 Corrective Action and Preventive Action (CAPA) System (9)3.2.3 Change Management System (10)3.2.4 Management Review of Process Performance and Product Quality (11)4. CONTINUAL IMPROVEMENT OF THE PHARMACEUTICAL QUALITY SYSTEM (11)4.1 Management Review of the Pharmaceutical Quality System (11)4.2 Monitoring of Internal and External Factors Impacting the Pharmaceutical Quality System (12)4.3 Outcomes of Management Review and Monitoring (12)5. GLOSSARY (13)Annex 1:Potential Opportunities to Enhance Science and Risk Based Regulatory Approaches (16)Annex 2:Diagram of the ICH Q10 Pharmaceutical Quality System Model (17)1.制药质量体系1.1绪论1.2范围1.3ICHQ10与地方GMP要求,ISO标准与ICHQ7之间的关系1.4ICHQ10与法规方法间的关系1.5ICHQ10目的1.5.1产品实现1.5.2控制状态的建立和实现1.5.3 持续改进1.6支持者:知识管理和质量风险管理1.6.1知识管理1.6.2质量风险管理1.7设计和内容方面的考虑1.8质量手册2.管理职责2.1管理承诺2.2质量方针2.3质量策划2.4资源管理2.5内部沟通2.6管理评审2.7外包活动和物料采购的管理2.8产品所有权变更管理3.工艺性能和产品质量的持续改进3.1生命周期阶段目标3.1.1物料研发3.1.2技术转移3.1.3商业化生产3.1.4产品终止3.2制药质量体系原理3.2.1工艺性能和产品质量监控体系3.2.2纠正预防体系3.2.3变更管理体系3.2.4工艺性能和产品质量的管理评审4.制药质量体系的持续改进4.1制药质量体系的管理评审4.2制药质量体系的内外部影响因素的监控4.3管理评审和监控成果5.术语附件1:基于法规方法对科学和风险进行改进的潜在机会附件2:ICH Q10 制药质量体系模型图PHARMACEUTICAL QUALITY SYSTEM药物质量体系1. PHARMACEUTICAL QUALITY SYSTEM 药物质量体系1.1 Introduction 介绍This document establishes a new ICH tripartite guideline describing a model for an effective quality management system for the pharmaceutical industry, referred to as the Pharmaceutical Quality System. Throughout this guidelin e, the term “pharmaceutical quality system” refers to the ICH Q10 model.本文确立了新的ICH三方指南,叙述了制药工业有效质量管理体系的一个模型,被称之为制药质量体系。
新产品质量确保流程

新产品质量确保流程英文回答:New Product Quality Assurance Process.Introduction.A new product quality assurance process is a set of steps and procedures that are followed to ensure that a new product meets all of its quality requirements. This process typically begins with the development of a quality plan, which outlines the specific quality goals for the product and the steps that will be taken to achieve those goals. The process then continues through the design, development, and production phases of the product, with each phase including its own set of quality checks and inspections.Quality Planning.The first step in the new product quality assuranceprocess is to develop a quality plan. This plan should outline the specific quality goals for the product, as well as the steps that will be taken to achieve those goals. The quality plan should also include a list of the quality checks and inspections that will be performed throughout the product development process.Design.The design phase of the new product development process is when the product's specifications are finalized. During this phase, it is important to consider the quality of the materials that will be used to make the product, as well as the design of the product itself. The design should be reviewed by a quality engineer to ensure that it meets all of the quality requirements.Development.The development phase of the new product development process is when the product is actually built. During this phase, it is important to follow the quality plan and toperform all of the required quality checks and inspections. The product should also be tested to ensure that it meets all of its performance requirements.Production.The production phase of the new product development process is when the product is manufactured in large quantities. During this phase, it is important to maintain the same level of quality that was achieved during the development phase. The production process should be monitored closely to ensure that all of the quality checks and inspections are being performed correctly.Post-Production.Once the product has been manufactured, it is important to continue to monitor its quality. This can be done through customer feedback, warranty claims, and other data. The quality of the product should also be reviewed periodically by a quality engineer to ensure that it isstill meeting all of its quality requirements.Continuous Improvement.The new product quality assurance process should be continuously improved. This can be done by reviewing the quality data and identifying areas where the process can be improved. The process should also be updated as new technologies and techniques become available.Benefits of a New Product Quality Assurance Process.There are many benefits to implementing a new product quality assurance process. These benefits include:Reduced product defects.Improved customer satisfaction.Increased product sales.Reduced warranty claims.Improved brand reputation.中文回答:新产品质量保证流程。
Quality Inspection Guideline

Inspection Guidelines检验指南1. Sampling level and AQL requirement. 抽样标准和AQL要求。
1.1 Normal Inspection Level II, under ANSI/ASQC Z1.4, is the sampling plan Adopted by ProTVQuality assurance. 使用Level II抽样计划。
1.2 AQL – Critical 0 / Major2.5 / Minor 4.0.一般产品最终随机检验AQL标准是: AQL-严重0/主要1.0/次要4.0As for critical function, if the initial number is 1, another same number of samplesshould be drawn, the result will be pass if no critical function defective found.关于关键性的功能,如果第一次抽样的次品数是一,那么将第二次抽取同等的样板,如果没有关键性的功能项出现,结果为合格。
2. Terms of inspection 验货条件2.1 Inspection based on below status:and leave the factory after instruction.如果QC发现大货完成状态是”not ready”,需要打电话回办公室,在得到公司指令后离开工厂。
2.3 If factory couldn't offer a suitable environment or sufficient manpower to assist forInspection, QC needs to call back to office immediately for further instruction.如果工厂不能提供合适的验货环境或者没有足够的人手协助验货,QC要立即致电写字楼。
2-1 WHO-PQ认证指导原则及提交资料的格式要求 2013.06(英文)

• Prequalification of generic products approved by Stringent Regulatory Authorities (SRAs)
7
7
Prequalification Guidelines (Quality)
• Guidelines on active pharmaceutical ingredient master file procedure
15
15
CTD- triangle
Regional Admin Information Module 1
Not Part of the CTD
Clinical Overview
Module 2
Quality Overall Summary
Nonclinical Overview
The CTD
Nonclinical Summary
information –Reginon specific • • • Module 2 – Quality Overall Summary – QOS-PD Module 3 - Quality Module 5 – Bioequivalent study Reports(or biawaiver)
• Provide clear and transparent guidance to applicants for preparing and submitting these dossiers and facilitate the subsequent assessment;
国家药品计划抽验质量分析指导原则

英文回答:The National Drug Plan Quality Analysis Guidance Principles have been developed with the objective of establishing precise guidelines for the quality assessment of pharmaceutical products falling under the purview of the national drug plan. These principles are meticulously formulated to guarantee that the medications accessible to the populace adhere to stringent quality benchmarks and are deemed safe for consumption. The guidance principles epass a wide array of domains, including but not limited to, sampling methodologies, testing protocols, and criteria for quality assessment.制定了《国家药品计划质量分析指导原则》,目的是制定国家药品计划范围内药品质量评估的准确准则。
这些原则是精心制定的,以保证人民能够获得的药品符合严格的质量标准,并被认为可以安全消费。
指导原则涉及广泛的领域,包括但不限于取样方法、测试规程和质量评估标准。
It's super important for everyone involved in the national drug plan to stick to these guidance principles. This helps keep the program honest and effective. Following these principles means we can analyze pharmaceutical products in a consistent andthorough way, which leads to more accurate and reliable results. Basically, it helps keep the public safe by making sure they're getting high-quality drugs.凡是参与国家药品计划的,都要坚持这些指导性原则,这是极其重要的。
☆APQP英文培训教程

☆APQP英文培训教程APQP又叫做Advanced Product Quality Planning, 是新产品开发的一种高级质量规划技术。
这种技术的核心思想是在新产品的开发过程中,尽可能地发现并纠正问题,最大程度地提高产品的质量以及生产效率。
为了能够学会如何进行APQP,并为了在公司中实施该技术,我们需要进行APQP英文培训。
1. Introduction to APQPAdvanced Product Quality Planning (APQP) 是一种高级的质量规划技术,它是用于新产品开发的。
确保新产品的质量,并将产品生产和交付保持在要求的质量水平上。
APQP的目标是最大程度地减少产品生命周期中的成本,改善交付时间,提高产品质量,同时增强组织的竞争力。
2. Core Elements of APQPAPQP是基于五大核心元素的。
这些元素包括:- Plan and Define the Program在计划和定义项目时,APQP的目标是最大限度地满足顾客的需求,并确保产品质量和交付满足要求的目标指标。
从规定规范到开发项目经费以及资源,这个阶段的目标是确保确定可实现的计划。
- Design and Development在设计和开发阶段,重点是确保产品的功能性和性能。
为了做到这一点,需要确保必要的资源,设备,过程和技能,并根据项目方案实施相关的验证和测试。
- Validation在验证阶段,APQP的重点是验证产品是否符合各项指标。
为了做到这一点,需要根据验证计划执行相关的测试和确认。
- Production and Launch在生产和推出阶段,APQP的目标是确保产品可以按照要求的质量标准和时间表稳定地生产。
组织和资源的状况以及工艺设计的质量都是这一阶段的考虑因素。
- Feedback, Assessment and Improvement在回馈,评估和改进阶段,APQP的目标是检查产品在生命周期的不同阶段中的性能水平,收集数据并进行评估,以用于未来项目中的改进。
supplier evaluation guideline.doc
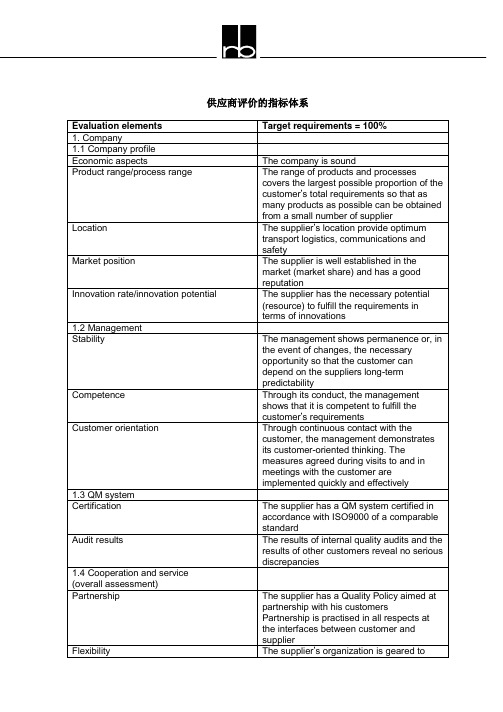
供应商评价的指标体系
情感语录
1.爱情合适就好,不要委屈将就,只要随意,彼此之间不要太大压力
2.时间会把最正确的人带到你身边,在此之前,你要做的,是好好的照顾自己
3.女人的眼泪是最无用的液体,但你让女人流泪说明你很无用
4.总有一天,你会遇上那个人,陪你看日出,直到你的人生落幕
5.最美的感动是我以为人去楼空的时候你依然在
6.我莫名其妙的地笑了,原来只因为想到了你
7.会离开的都是废品,能抢走的都是垃圾
8.其实你不知道,如果可以,我愿意把整颗心都刻满你的名字
9.女人谁不愿意青春永驻,但我愿意用来换一个疼我的你
10.我们和好吧,我想和你拌嘴吵架,想闹小脾气,想为了你哭鼻子,我想你了
11.如此情深,却难以启齿。
其实你若真爱一个人,内心酸涩,反而会说不出话来
12.生命中有一些人与我们擦肩了,却来不及遇见;遇见了,却来不及相识;相识了,却来不及熟悉,却还要是再见
13.对自己好点,因为一辈子不长;对身边的人好点,因为下辈子不一定能遇见
14.世上总有一颗心在期待、呼唤着另一颗心
15.离开之后,我想你不要忘记一件事:不要忘记想念我。
想念我的时候,不要忘记我也在想念你
16.有一种缘分叫钟情,有一种感觉叫曾经拥有,有一种结局叫命中注定,有一种心痛叫绵绵无期
17.冷战也好,委屈也罢,不管什么时候,只要你一句软话,一个微笑或者一个拥抱,我都能笑着原谅
18.不要等到秋天,才说春风曾经吹过;不要等到分别,才说彼此曾经爱过
19.从没想过,自己可以爱的这么卑微,卑微的只因为你的一句话就欣喜不已
20.当我为你掉眼泪时,你有没有心疼过。
工程质量保障体系-英文版

Quality Assurance SystemQuality Assurance SystemThe guideline and object of quality of projectThe guideline of quality: Employee is most important for company. And the quality of product and service is most important for company as well. We will provide honest service and standardize construction to satisfy all of clients.The object of quality: To reach the standard of state: ‹The Standard of Inspection Appraisal of Engineering Quality› or perfect engineering grade of Standard of Professional Inspection Appraisal.Arc welding machine, portable electric welding machine, lifting machine, trolley, and drillerThe plan of qualityProject manager:Carry out the contract strictly, carry out the responsibility of quality completely, and guaranty to realize the object of quality of engineering. Take full responsibility for quality of engineering directly.Ensure all of documents of quality system be implemented properly in project team, and take responsibility for validity of implement.To organize the construction reasonable, carry out the technical documents and technical standard strictly, control the whole process of construction, organize technical staff to establish the instruction documents of keyprocedures and special procedures, and supervise the implement as well. In charge of supervise staff of Management Department to establish the plan of quality of project and technical document of project, ensure the synchronization and reality of construction and quality certificate, carry out the rejected comments of Quality Department.To organize analyze and handle the normal quality accident, avoid the problems happen again.To organize the trial run and handover, ensure the quality of engineering fulfill contract requirements.Engineer of quality inspection (chief of Quality Inspection Team)In charge to implement the inspection and test plan, take full responsibility for this inspection and test plan directly.Take part in the inspection for key section of quality control point, and confirm in addition.Feedback the information of this inspection and test to superintend, provide the situation information of implement regularly.For any act against the plan of inspection and test, have the veto power for it.Site professional engineerInstruct construction team to perform the construction according to the design drawing, construction acceptance criterion, operation criterion, construction procedure and technical documents. Establish the measuresand requirements of quality guaranty, especially for key link, section and factor of quality control, must prepare it in writing; take responsibility for controlled engineering quality.In control of the situation of construction quality of each team, adopt necessary measures of quality control promptly, to correct the common fault and solve out the problems. Be in charge of carry out the correction work, which requested by quality inspection department and report the result to quality inspection department as well.Inspect and supervise all of teams to do self-check, check each other and check of handover. Attend the quality appraisal work of subentry of engineering and subsection of engineering. Be in charge of pack up the information of quality appraisal of engineering, and take responsibility for its reality, validity and integrality. Support the quality inspection work of quality inspection officer.For the quality problems caused by breach the rules or lose control, be entitled to order the team or the operator to redo it again or stop the operation.Quality inspection officer take full responsibility for quality inspection work in controlled area.Should pay special attention on the process handover of material inspection officer, hidden engineering inspection and subentry engineering appraisal sample.Check on all of record of construction and sign it as well, take responsibility for the reality of signed quality materials.If find the problems such as quality lose control, should insist the principle, repair, redo it again or stop the operation. And report to quality inspection engineer, functioned with veto of quality.Take part in the analysis and investigation of engineering quality and accident.Construction teamTake full responsibility for quality of construction engineering.Take full responsibility for control the factor, which formed during the quality control process, and be ready for quality inspection and record all of self-checks.Ensure only qualified materials and equipments will be used by, unqualified process not allowed to handover.Have the authority to reject all of uncompleted technical materials. For the quality requirements not mentioned in criterion standard of engineering and drawings, have the authority to get the supporting document in writing. There is the duty to cooperate with quality supervise department, report the situation of quality control, when quality modification notice be received, should carry out promptly as well as take responsibility for the result of modification.Management of inspection and testThe quality management and quality guaranty of this engineering will perform according to GB/T19002-ISO9002 1994 ‹The quality guaranty model of production, installation and services of quality systems›. Operate carefully, ensure the quality, comply the contract, provide honest service. All of the engineering project which we been done, be inspected and controlled strictly, all of our staff, all of stations will work for the quality requirement of owner, rely on the scientific management, distribute source reasonable, standardize the performance, strengthen the management, ensure the quality of engineering reach perfect grade. Establish and sound the structure of quality management, carry out the system of job responsibility. The full time quality management staff will be assigned to play the role of supervision, and have the authority to reject. Main measures of inspection and test managementCheck on the drawings carefully, all of the construction performance will follow the drawings exactly. The drawings for this project have special characteristic as it is concerning foreign affairs project. To understand the drawing and design intent is precondition of correct construction. It is the discipline to do the work according to the drawings.Prepare quality plan, clear the quality requirement, prepare detailed quality plan before the construction commencement. Mainly include:The technical condition (drawing, standard, criterion)The setting of quality control pointThe construction working procedure and technical requirements of special procedure and key procedure, as well as the method of inspection of quality control (and large-scale equipment hoisting etc.)The above information should inform construction team in writing document, make operator understand the specialty of engineering, technical requirements, quality standard, construction techniques and operation method.Determine the quality control point according to the drawings, the requirements of technical criterion and acceptance standard, which provided by owner, the inspection for quality control point should confirm by quality engineer and supervision engineer, the next working procedure can be performed after this confirmation.When installation finish, should adjust all of system according to the regulation and requirement. Before trial run, should establish the operation scheme according to design drawing and concerned criterion requirement, the scheme will carry out as soon as approved by owner.Management of quality of material: Full time material management staff will be assigned, strength the inspection of material, such as: inspection for specification and type, inspection for quality, inspection for quantity, inspection for certificate and quality guaranty documents. During the transportation, prevent the material properly, and loading, unloading carefully, strength the storage management of materials and equipments.The control of disqualified working procedure:The disqualified working procedure found by self-check, engineer will record it and mark it.The disqualified working procedure found by owner and supervision staff, engineer would record it and mark it.The disqualified subentry, subsection found during quality inspection appraisal, quality inspection officer would record it and mark it.The disqualified product found by quality inspection officer during the inspection, quality inspection officer would mark it according to the record.The quality inspection officer of project department should setup an engineering record.The sketch map of procedure of quality control:。
GMP Quality Manual质量手册

GMP Quality Manual质量手册This quality manual for Good Manufacturing Practices is based on th e ISO 9001 standard, and addresses the requirements of FDA's medical d evice regulations.此GMP质量手册是基于ISO 9001标准,叙述FAD医用仪器的法规要求。
Table of Contents 名目Parent Category 上级分类POLICY 方针Quality Policy质量方针We are dedicated to achieving customer satisfaction through an unco mpromising commitment to quality. 我们致力于让客户中意,承诺对质量永不妥协。
(保证产品质量,让用户中意)These high standards are realized through the following practices:通过下列实践达到高标准An open door policy that enables all personnel to bring to manageme nt's attention any concerns regarding quality achievement and customer sat isfaction.保证产品质量,让用户中意应成为每一职员的工作关注点。
Personnel performing their work in a manner that ensures products a nd services meet all quality requirements.职员的操作方式、行为规范要以满足所有质量要求为差不多准则。
PQAP_零部件质量保证计划培训

阶段 1. 计划
阶段 1.3 项目计划
号 阶段 码
1.3 项目计划
关键步骤
责任人 附件
1.提供给供应商机加工计划。 2.供应商提供项目计划和报价。 3.审核供应商的项目计划是否覆盖关键节
点和活动:是否有组织管理项目的能 力:
-工具制造 -设备安装 -零件检测(例如测量和试验设备时间表)
9.不要关注本工序分析, 下道工序错误追溯到上道 工序,不是在上道工序分 析。
Volvo Construction Equipment
18 2021/8/15
阶段 2. 准备
阶段 2.2 控制计划
号 阶段 码
关键步骤
2.3 控制计划 1.在FMEA完成后制作
2.获得CP并分析考虑:
-覆盖工序中的关键和人为因素(例如 构成要素,热处理和清理)。
2 2021/8/15
PQAP 有什么优点?
零部件质量保证计划
Volvo Construction Equipment
3 2021/8/15
互动
• 假设RSV现在是一个主机厂,您是RSV的产品开发部经理,现在RSV需 要到供应商A去开发一个斗杆,您会怎么进行您的工作?
Volvo Construction Equipment
-定义和控制所有的生产工序。
-测量的频率和方法
责任人 王启
附件 备注
供应商控制计划中的典型缺点: 1.没有和FMEA联系在一起, FMEA中定义的风险和控制措 施没有体现。 2.过多的关注产品控制而不是 过程控制。 3.作业指导书太简单。 供应商不需要把作业指导书 发给我们,这和老的版本不一 样。 反应得到操作者的认可。
阶段 2.1 RTS 完结
药品使用质量管理技术指南最新版

英文回答:Technical guidance on quality management of drug use is a set of technical standards and norms for quality management in the production and use of drugs。
The guide emphasizes management requirements and technical specifications for the production, storage, transport, sale and use of medicines。
With regard to the production of medicines, enterprises must ensure the security and stability of the quality of medicines by strictlyplying with the national quality control norms for their production。
Storage and transport links require well—developed management systems that strictly control environmental humidity and prevent the deterioration of medicines。
In terms of marketing and use, enterprises must put in place sound management systems to ensurepliance with the requirements of national laws and regulations and to ensure the safety of patients。
eu guideline for good manufacturing practice

eu guideline for good manufacturing practiceThe EU guideline for good manufacturing practice (GMP) is a set of standards that ensure production processes are consistent and controlled in order to ensure the quality, safety, and efficacy of pharmaceutical products. The guideline applies to all stages of pharmaceutical manufacturing including sourcing of materials, processing, packaging, labeling, and storage.The key elements of EU GMP include:1. Quality Management System: This includes quality control, quality assurance, and quality risk management.2. Personnel: Adequate training, qualification, and experience of personnel involved in manufacturing process.3. Facilities and Equipment: Buildings, equipment, and utilities used for manufacturing should be fit for their purpose.4. Materials: Raw materials, primary and secondary packaging materials must be fully traceable.5. Production: Manufacturing process must be well-defined and controlled with appropriate documentation to ensure reproducibility and consistency.6. Quality Control: Testing of products at various stages of manufacturing to ensure that quality standards are met.7. Documentation and Records: All documentation regarding themanufacturing process should be accurate, complete, and retained for a specified period.8. Complaints and Product Recall: Adequate processes should be in place to address customer complaints and for recall of products. By adhering to EU GMP guidelines, pharmaceutical manufacturing companies can assure their products are of high quality, consistency, and efficacy.。
品质工程 试点工作的意见

品质工程试点工作的意见English Answer.Quality Engineering Pilot Work.1. Purpose.> The purpose of this quality engineering pilot work is to improve the quality of our products and services. We will do this by identifying and eliminating the root causes of defects and by developing and implementing new processes and procedures that will prevent defects from occurring in the future.2. Scope.> This pilot work will be conducted in the following areas:Product development.Manufacturing.Customer service.3. Objectives.> The objectives of this pilot work are to:Reduce the number of defects in our products and services.Improve the quality of our products and services.Increase customer satisfaction.Improve our profitability.4. Methodology.> We will use the Six Sigma methodology to conduct this pilot work. Six Sigma is a data-driven approach toquality improvement that focuses on identifying and eliminating the root causes of defects.5. Timeline.> We will conduct this pilot work over a period of six months.6. Resources.> We will need the following resources to conduct this pilot work:Funding.Personnel.Training.Equipment.7. Evaluation.> We will evaluate the success of this pilot work by measuring the following metrics:The number of defects in our products and services.The quality of our products and services.Customer satisfaction.Our profitability.Chinese Answer.质量工程试点工作。
PPQP PPQP supplier operation guideline

供应商质量保证书的填写及签署
Fill in SUPPLIER WARRANT and sign-off
填写零件递交状态
对于差错纠正的 零件可以在补充 说明中注明
1~5项中的任何一项如果回答为“否”或“未按 计划进行”的都必须填写整改计划
供应商质量保证书的填写及签署
Fill in SUPPLIER WARRANT and sign-off
流程简介
Process Introduction
应用范围
Scope
PPQP应用于所有提供PPO制造所需的试制零件的供应商
PPQP applies to all internal and external Suppliers of Pre-Production parts to PPO Build shops
PPQP将试用范围内的试制零件分为两个部分进行定义:
The PPQP classifies parts that are being used in Pre-Production builds into three categories:
a) 试制关键零件 Prototype/Pre-production Critical parts b) 试制非关键零件 Prototype/Pre-production Non-Critical parts
在产品开发、PPO及供应商间建立一个统一合作的流程
Unify associated processes across Product Development, PPO and Suppliers globally
保证每个零件在发运及安装之前都能够被批准
Establish methods to assure that all parts are approved before shipment and installation
PQS制药企业的同行学习质量管理体系吧

ISO9001中管理者的责任:
现在是GMP的要求;(与其他ISO要求一样)
EU GMP Part I Chapter 1
之前,EU GMP没有明确要求建立CAPA制度。 现在: 1.4(XV) 和1.8 (VII) 明确规定:
应建立CAPA制度,应用质量风险管理原则找出 缺陷的根源。
EU GMP Part I Chapter 1
质量手册没有纳入正式文件体系 没有明确各部门职责 没有质量目标
PQS:检查常见缺陷
高层管理:
责任与义务没有明确 没有强调或没有意识 没有定期审核,没有规定具体频率 没有审核记录 没有评审意见
没有结论 是否满意 没有从高层的角度提出改进建议
PQS:核心
知识与科学 Knowledge and Science based
医药质量体系:PQS
1.1-1.7 1.4 PQS应能保证:
通过设计、计划、执行和持续改进,输出合格产品 产品生命周期内全面管理产品与工艺 药品开发与设计应考虑GMP要求 制定生产与控制的清晰规程,执行GMP 明确规定管理职责 生产、供应与使用正确的起始物料和包装材料;供应商选择与监督; 每批货物来自批准的供应链; 确保外包工作的管理 工艺与产品质量受控,使用有效的监督与控制体系 产品放行:产品与工艺监控结果,偏差调查,避免今后再次发生的 预防措施 中间体、中间控制,验证 随着产品知识与现行工艺水平的进步,持续改进,并有措施
医药质量体系:PQS
1.1-1.7 1.4 PQS应能保证:
建立措施,前瞻性评估计划性变更,批准前通知法规部门或等待法 规部门批准 变更执行后的评估:确认达到质量目标,不影响产品质量 根源分析:偏差调查、产品缺陷、其他问题;质量风险管理的应用;
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Regional Admin Information
Module 1
Module 2
Quality Overall Summary
Nonclinical Overview Clinical Overview Nonclinical Summary Nonclinical Study Reports Clinical Summary Clinical Study Reports
8|
Lynda Paleshnuik | January 2011
Guideline development process
Consultation process with PQP senior assessors → new draft External consultation process (formal EC circulation) → final draft Presentation to EC on Specifications for Pharmaceutical Preparations Currently: guidelines provisionally accepted for pilot use in PQP
4|
Lynda Paleshnuik | January 2011
Background
Previous generic guideline: “Guideline on Submission of Guideline Documentation for Prequalification of Multi Multi-source (Generic) Finished Pharmaceutical Products (FPPs) Used in the Treatment of HIV/AIDS, Malaria and Tuberculosis Tuberculosis” Published in 2005: policy/approaches to assessment change continually over time due to harmonization efforts, scientific advances, development of approaches - e.g. process validation (≈2002), pharmaceutical development 2002), approaches have changed dramatically over the past 10 years
6|
Lynda Paleshnuik | January 2011
Background
This common format for submitting dossiers to agencies (CTD) affects: - the assessment report – based on the dossier template (QOS) the -the dossier data, enabling the logical organization of data and a single dossier to be submitted to multiple agencies A guideline updated according to current requirements, and adopting CTD format, was n | January 2011
Key changes from the previous guideline
CTD format adopted Updating of requirements Elaboration of how to meet quality requirements, including full elaboration on the three ways to submit API data: - CEP - APIMF - full API data provided in the dossier
10 |
Lynda Paleshnuik | January 2011
Introduction to the two documents I: the preparation guideline
11 |
Lynda Paleshnuik | January 2011
Adapting the CTD-NDS (new drug) NDS to CTD-ANDS (generic) ANDS
2|
Lynda Paleshnuik | January 2011
Overview Continued
Key changes from the previous PQP quality guideline Questions raised
3|
Lynda Paleshnuik | January 2011
5|
Lynda Paleshnuik | January 2011
Background
ICH: International Conference on Harmonization ► joint regulatory-industry initiative to harmonise industry regulatory requirements ► issued the Common Technical Document ( (CTD) Quality (ICH M4Q) guideline resulting in considerable harmonization on the organization of the Quality Module of the registration documents ► CTD format has become widely accepted by regulatory authorities within and beyond the ICH regions.
The CTD
Quality
Module 3
Module 4
Module 5
12 |
Lynda Paleshnuik | January 2011
Introduction to the two documents I: the preparation guideline
Provides guidance on the location of regional information (Module 1) and other general data requirements. Primarily addresses the organization of the information to be presented in PDs for multisource products. It is not intended to indicate what studies are required. It indicates an appropriate format for the data that have been acquired.
15 |
Lynda Paleshnuik | January 2011
Key changes from the previous guideline
New guidelines
“Preparation” guideline: 10.375: Guideline on submission of documentation for a multisource (generic) finished pharmaceutical product (FPP): Preparation of product dossiers (PDs) in Common Technical Document (CTD) Format; “Quality” guideline: 10.373: Guideline on submission of documentation for a multisource (generic) finished pharmaceutical product (FPP): Quality part
7|
Lynda Paleshnuik | January 2011
Guideline development process
guideline drafted in CTD format (crafting of preparation document, plus formatting of quality document) → draft guideline populated with quality technical guidance - updated according to current practice - including additional information on how to meet the requirements → new draft Clarity and transparency of requirements significantly improves the quality of dossiers
Assessment Workshop Copenhagen – January 2011
The new PQP quality guideline
1|
Lynda Paleshnuik | January 2011
Overview
Background (PQP quality guidelines) Guideline development process Introduction to the two documents: I: the preparation guideline II: the quality guideline